

Abstract
Serine protease inhibitor Kazal (SPIK) is an inflammatory protein whose levels are elevated in numerous cancers. However, the role of this protein in cancer development is unknown. We have recently found that SPIK suppresses serine protease-dependent cell apoptosis. Here, we report that anti-SPIK antibodies can co-immmunoprecipitate serine protease granzyme A (GzmA), a cytolytic granule secreted by cytotoxic T lymphocytes and natural killer cells during immune surveillance, and that SPIK suppresses GzmA-induced cell apoptosis. Deletion studies show that the C3–C4 region of SPIK is critical for this suppression. These studies suggest that over-expression of SPIK may prevent GzmA-mediated immune-killing, thereby establishing the tolerance of cancer cells to the body's immune surveillance system. Suppression of over-expressed SPIK can restore the susceptibility of these cells to apoptotic death triggered by GzmA. This finding implies that it is possible to overcome tolerance of cancer cells to the body's immune surveillance system and restore the GzmA-mediated immune-killing by suppressing the over-expression of SPIK.
Keywords: apoptosis, cancer, granzyme A, inflammation, serine protease inhibitor Kazal
Introduction
Serine protease inhibitor Kazal (SPIK/SPINK-1), also known as TATI (tumour-associated trypsin inhibitor) and PSTI (pancreas secretory trypsin inhibitor), is a 79-amino-acid protein. It was first discovered in the pancreas as an inhibitor of the autoactivation of trypsinogen. 1,2 The expression of SPIK is elevated in numerous cancers such as colorectal tumours, renal cell carcinoma, gastric carcinoma, hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma.3–8 It is known that SPIK can be activated as a reactant during inflammation.9–11 For example, SPIK was activated in rat liver cells to counter turpentine-induced liver inflammation12 and in response to inflammatory cytokines during human viral hepatitis.13 Our recent findings showed that replication of the hepatitis B virus and hepatitis C virus, two main causes of chronic hepatitis, can up-regulate the expression of SPIK.14 Interestingly, a high level of SPIK transcripts was correlated with cancer progression and recurrence after surgical resection.7,8,15 Furthermore, the highest levels of SPIK are often associated with the latest stages of cancer, probably implying a cumulative, dose-dependent effect of SPIK on cell transformation.7,8,16 Together, these studies suggest that in addition to, or perhaps because of, its role as an inflammatory protein, SPIK may play an important role in the formation and development of cancer.
It has been strongly suggested that the progression of cancer could, at least in part, be because of the tolerance of cancer cells to the immune response and immune-mediated clearance. This tolerance stems from the body's inability to induce apoptosis in these cells, resulting in uncontrolled cell growth.17,18 One mechanism for blocking immune-mediated apoptosis would be to inhibit apoptosis induced by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells by suppression of apoptotic cytolytic granules such as granzyme A (GzmA) and granzyme B (GzmB), which are secreted by activated CTLs and NK cells to kill target cells during immune clearance. Because both GzmA and GzmB are cytotoxic serine proteases, the suppression of GzmA-/GzmB-induced apoptosis may be caused by the activity of a serine protease inhibitor.19,20 We have demonstrated that SPIK inhibits apoptosis, suppressing serine protease-dependent cell apoptosis (SPDCA), and therefore it is possible that cancer cells evade immune killing induced by CTLs and NK cells through the inability of serine proteases such as GzmA and GzmB to withstand the effects of the elevated levels of SPIK.21,22 This idea is supported by the observation that rat SPIK can bind GzmA and inhibit its ability to hydrolyse substrates such as N-α-benzyloxycarbonyl-l-lysine thiobenzyl ester.23 Low nanomolar concentrations of GzmA trigger a pro-inflammatory effect,24,25 whereas high nanomolar concentrations of GzmA induce SPDCA. 26,27 It is likely that suppression of GzmA-induced apoptosis by over-expression of SPIK would result in the escape of cancer cells from immune clearance and even suppress the immune response.21,22 This idea is supported by the observation that high levels of SPIK are closely associated with early recurrence of HCC and intrahepatic cholangiocarcinoma in patients following surgical resection.7,8 Because recurrence of cancer often implies an inability of the immune system to clear lingering oncogenic cells, early recurrence of HCC and intrahepatic cholangiocarcinoma in patients with high levels of SPIK raises the possibility that the over-expression of SPIK interferes with the elimination of lingering oncogenic cells by the immune system. Uncontrolled expansion of these lingering cells triggers cancer recurrence.
Here, we provide direct evidence to show that SPIK can interact with GzmA and suppress GzmA-induced cell apoptotic death. We also show that the C3–C4 region of SPIK is critical for its function. Finally, we show that suppression of over-expressed SPIK can restore GzmA-mediated cell killing. All of these studies can help illuminate the complex process of cancer formation and contribute to the development of a new class of anticancer drugs, which can over-ride the tolerance of cancer cells to immune surveillance by suppressing over-expressed levels of SPIK.
Material and methods
Cell lines and plasmids
Cell lines S2-3, SP23 and G54, derived from the Huh7 cell line, were created in our laboratory and described in detail in our publications.14,28 HeLa cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured and maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum. Pancreatic cell line Panc1 was purchased from ATCC and cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum plus 4·5 g/l glucose, sodium pyruvate and l-glutamine. All of the plasmids, including wild-type (PWT) and its deletion mutants PD1, PD2 and PD3, were constructed using pCMV-Script Vector (Stratagene, La Jolla, CA). A myc tag with the sequence MEQKLISEEDL was placed in the N-terminus to replace the first nine amino acids of SPIK in PWT to allow antibody recognition. All of the deletion mutants, PD1, PD2 and PD3, which lacked C2 to C3 (CTKIYDPV), C3 to C4 (CGTDGNTYPNE), or 12 amino acids (QTSILIQKSGPC) in the C-terminus of SPIK, respectively, were constructed based on PWT. SPIK small interfering RNA (siRNA) L71 and L183 have been previously described.14,28
Northern blot assay
Samples of mRNA (20 μm) from different human tissues were resolved in 1% denatured agarose gel and transferred to a nylon membrane, which was purchased from Zyagen (San Diego, CA). The membrane was then hybridized with a SPIK-specific probe, which was labelled with [32P]dCTP using SPIK-template DNA derived from PCR amplification. After the hybridization process was completed, the bands were visualized using a Phosphorimager (Amersham Biotech–Molecular Dynamics, Sunnyvale, CA), and the accompanying graphics software package was used to estimate the intensity of the bands. Equal sample loading was confirmed in each experiment by ethidium bromide-stained ribosomal RNA. The SPIK mRNA in HCC from a patient was used for comparison.
Western blot analysis
The level of SPIK protein was examined by Western blot using an anti-SPIK (SPINK) monoclonal antibody (Abcam, Cambridge, MA). Either 15 μl culture medium or 15 μl cell lysate was run on a 4–12% SDS–PAGE gradient gel (Invitrogen, Carlsbad, CA). After being transferred to a PVDF membrane, SPIK protein was visualized by staining with a mouse monoclonal anti-SPIK antibody at a 1 : 1000 dilution and then with a standard anti-mouse horseradish peroxidase secondary antibody at 1 : 7500. An ECL Advance Western Blotting Detection Kit (GE Healthcare, Piscataway, NJ; formerly Amersham) was used to visualize the image, which was captured using a Gel Logic 1500 Molecular Imaging System and Kodak Imaging software (Carestream Health Inc, Rochester NY). Membranes were then stripped for subsequent Western blot analyses using a standard antibody stripping protocol consisting of 20 min incubation with 65 mm Tris–HCl buffer, pH 6·7, 2% SDS, 100 mm 2-mercaptoethonal, at 50°. Anti-GzmA antibody and anti-myc antibody (Abcam) were used to subsequently detect their respective proteins.
Co-immunoprecipitating GzmA with anti-SPIK antibody
A total of 50 μl medium from S2–3, G54, and pancreatic cells was incubated at 4° overnight with magnetic agarose beads covalently linked with monoclonal anti-SPIK antibody or anti-GzmA antibody as described by the manufacturer (Calbiochem, San Diego, CA), either with or without the addition of 200 ng recombinant human GzmA (Calbiochem). After the beads were washed, the captured proteins were released from the beads by treatment with glycine–HCl buffer, 0·01m, pH 2·5 for 1 min. The proteins were then resolved on a 4–12% SDS–PAGE gradient gel. After transfer to a PVDF membrane, SPIK protein and GzmA protein were visualized by staining the membrane with mouse monoclonal anti-SPIK or anti-GzmA antibodies, respectively. The image was captured using a Gel Logic 1500 molecular imaging system and Kodak Imaging software (Carestream Health Inc) after incubation with a standard anti-mouse horseradish peroxidase secondary antibody.
Transfection
For transfection, 105 cells were seeded in 60-mm dishes. After overnight incubation, 2 μg plasmid, such as PWT and its deletion mutants, was transfected with 6 μl FuGENE 6 as described by the manufacturer (Roche Diagnostics, Indianapolis IN). Mock transfection, consisting of transfection of the relevant vector lacking the gene of interest, was used as a transfection control. The transfection efficiency was determined by co-transfection and expression of green fluorescent protein. After 3 days, the cells were split into two daughter 60-mm dishes and cultured for an additional day. Half of the cells from one dish were lysed with 0·05m Tris–HCl pH 7·5, 0·15 m NaCl, 0·005 m MgCl2, and 0·2% Nonidet P-40 for Western blot analysis. The remaining half of the cells was released by limited treatment with trypsin for apoptosis analysis. For silencing study, SPIK siRNA plasmids L71 and L183 were transfected into S2–3 cells as described previously.14,28
Induction and examination of cell apoptosis
To induce cell apoptosis, the cells released by digestion with trypsin were incubated with 2 μm recombinant GzmA and 254 ng/ml perforin (PFR; Abnova, Taipei, Taiwan) at 37° for 20 hr. Cell apoptosis was evaluated either by Hoechst staining or by flow cytometry after staining with annexin V-FITC. For Hoechst staining, the cells were washed once with water and then incubated with 100 μg/ml dye at room temperature for 5 min. The cells were then washed twice more with water, and the condensed nuclei, indicative of apoptotic cells, were visualized with a fluorescent microscope. For annexin V-FITC staining, apoptotic cells were washed with PBS and incubated with annexin V-FITC for 25 min at room temperature and in the dark, in 100 μl binding buffer per the manufacturer's instructions (Biosource International, Camarillo, CA). In the case of double staining with annexin V-FITC and propidium iodide (PI), 0·5 μg PI was added 3 min before the end of the incubation with annexin. The cells were then resuspended in 400 μl binding buffer; apoptosis was quantified using a Guava EasyCyte Plus (Guava Technologies, Billerica, MA) or Becton Dickinson FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The cell debris was gated out in flow cytometry analysis.
Results
SPIK expression is limited or inactivated outside the pancreas but abundant in liver cancer
Because SPIK is a pancreatic protein,2 we wanted to determine if the SPIK gene is active in other tissues. We therefore examined SPIK expression in a variety of human tissues such as the liver, colon, lung, spleen and prostate etc. Northern blot analysis of total RNA (Fig. 1) showed that the expression of SPIK in normal human tissues was limited or inactivated outside the pancreas. A very low level of SPIK is seen in normal liver tissue but is not repeatable (Fig. 1, lane 2), however, a large increase in SPIK expression could be seen in liver cancer taken from patients with HCC, which were used as a positive control (Fig. 1, HCC lane). This finding was in agreement with our previous observation that the number of SPIK transcripts was increased more than 1600-fold in HCC cells compared with the number in normal human liver cells, which has been confirmed by others.8,29 This result also suggests that the activation of SPIK expression may result in the transformation of cells.
Figure 1.
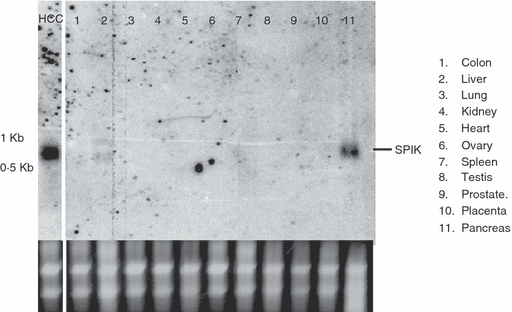
Serine protease inhibitor Kazal (SPIK) gene expression is limited or inactivated outside the pancreas; 20 μm mRNA samples from different human tissues were resolved in 1% denatured agarose gel and transferred to a nylon membrane for Northern blot analysis. The membrane was then hybridized with a SPIK-specific probe. The SPIK mRNA in hepatocellular carcinoma (HCC) cells is presented for comparison. Ribosomal RNA is used as loading control.
SPIK secreted from pancreatic cells differs from that secreted from cancer cells by cleavage of a nine-amino-acid fragment from the N-terminus
Because SPIK expression increased in HCC cells, we were interested in determining if the structure and function of SPIK from cancer cells differed from SPIK normally secreted by pancreatic cells. We used two cell lines: a stable cell line, S2–3, and the hepatitis C virus (HCV) replicon cell line G54, both of which express high levels of SPIK by either integration of the SPIK gene into the host DNA or through induction by replicating HCV. Both of these cell lines are derived from an HCC cell line.14,28 The differences between SPIK generated by these cells and that generated by pancreatic cells were examined and compared by Western blot with an anti-SPIK monoclonal antibody. Figure 2 indicates that although pancreatic cells make less SPIK, all three cell types produce SPIK protein of about the same size, around 8,500 Dalton molecular weight, indicating the correct molecular weight for full length SPIK (Fig. 2a, Cell Lysate). This is in agreement with our previous results that showed no difference of SPIK gene existing in these cells.14,28 Analysis of secreted protein, however, showed that the SPIK secreted from pancreatic cells is smaller than its counterpart from the cell lines S2–3 and G54 (Fig. 2a, Medium). An attenuated SPIK with 56 amino acids has been reported in pancreatic fluid,30 which, along with our findings, suggests that pancreatic SPIK is proteolytically cleaved upon secretion. However, this proteolytic incision does not occur during SPIK secretion from S2–3 and G54 cells (Fig. 2a, medium, Compare Panc-1 to S2–3 and G54). Edman N-terminal analysis suggested that a nine-amino-acid fragment with the sequence MKVTGIFLL was removed from the N-terminus of SPIK secreted from the pancreas (Fig. 2b, Edman N-terminal Analysis; Fig. 2c, Sequence of SPIK Protein). The reason that HCC cells secrete uncleaved SPIK protein is unclear. Whether secretion of uncleaved SPIK is a characteristic of all cancer cells is also unclear and is under investigation.
Figure 2.
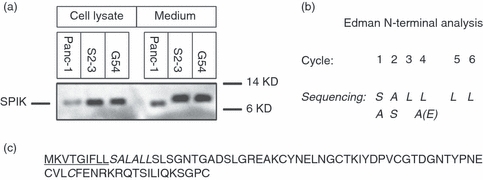
Serine protease inhibitor Kazal (SPIK) secreted by hepatocellular carcinoma (HCC) cells has nine extra amino acids in the N-terminus. Culture medium or cell lysate from S2–3 and G54 cells or human pancreatic cells (Panc-1) was run on an SDS–PAGE gel. After transfer to a PVDF membrane, SPIK protein was visualized by staining with a monoclonal anti-SPIK antibody and then with an anti-mouse-horseradish peroxidase secondary antibody. An ECL Advance kit was used to visualize the image. (a) The Western blot used to detect SPIK either in cell lysates or in culture medium. (b) Edman degradation, used to determine the N-terminus sequence of pancreatic SPIK. The N-terminal sequence of SPIK secreted from pancreatic cells was analysed by Edman N-terminal degrading analysis, which was performed by Alphalyse Inc. (Palo Alto, CA) after excision of the pancreatic SPIK band from the PVDF membrane. The sequence predicted by Edman degradation in the N-terminal of secreted pancreatic SPIK is bold and italicized. (c) The sequence of intact SPIK. The missing sequence in secreted pancreatic SPIK is underlined.
Anti-SPIK antibody can co-precipitate GzmA
In addition to preventing autoactivation of trypsinogen, as a serine protease inhibitor, SPIK can also suppress SPDCA.28 To study the role of SPIK in the prevention of cell apoptosis, and in particular its role in GzmA-mediated apoptosis, the ability of SPIK to bind GzmA was analysed by co-immunoprecipitation. Cell culture medium collected from pancreatic cells as well as S2–3 and G54 cells was incubated with recombinant GzmA and then immmunoprecipitated using anti-SPIK or anti-GzmA coated magnetic agarose beads. Western blot analysis was then performed on the captured proteins and visualized by staining with monoclonal anti-GzmA or anti-SPIK antibodies (Fig. 3a).
Figure 3.
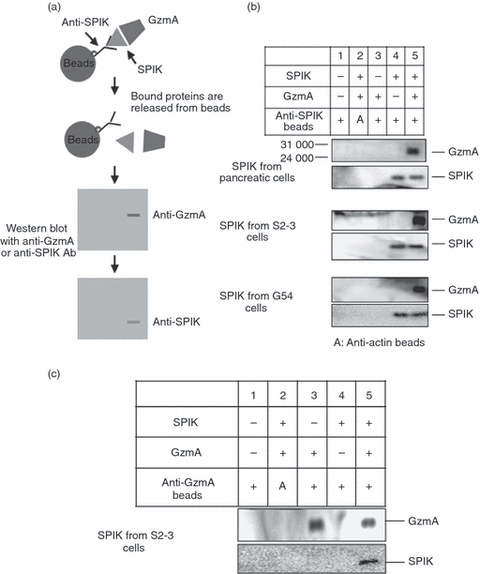
Granzyme A (GzmA) was co-immmunoprecipitated with serine protease inhibitor Kazal (SPIK). The medium from cells expressing high levels of SPIK, such as pancreatic cells, S2–3 cells, and G54 cells, was incubated with 200 ng recombinant GzmA and then incubated at 4° overnight with magnetic beads covalently linked with anti-SPIK monoclonal antibody. SPIK and binding proteins were released from beads by treatment with pH 2·5 buffer and resolved in SDS–PAGE. The proteins were then transferred to the membrane and analysed by Western blot with monoclonal anti-GzmA and anti-SPIK antibodies, respectively. (a) Experimental procedure. (b) Western blots showing SPIK and binding proteins after immune co-precipitation with anti-SPIK antibody. GzmA and SPIK are indicated on blots. (c) Immune co-precipitation with anti-GzmA antibody.
Anti-SPIK antibodies can precipitate SPIK (Fig. 3b, lane 4, SPIK staining) but cannot precipitate GzmA (Fig. 3b, lane 3, GzmA staining). However, when GzmA was incubated with SPIK before immunoprecipitation, a GzmA/SPIK complex could be precipitated using either anti-SPIK or anti-GzmA coated beads (Fig. 3b,c, lane 5, GzmA and SPIK staining). This co-precipitation was specific, as beads coated with antibody against β-actin did not precipitate SPIK, GzmA, or their complex (Fig. 3b,c, lane 2, SPIK and GzmA staining). These findings indicate that SPIK directly binds GzmA. It is important to note that despite the N-terminal cleavage, pancreatic SPIK was able to bind GzmA, similar to SPIK secreted from S2–3 and G54 cells (Fig. 3b, pancreatic cells). This result suggested that, although the nine-amino-acid fragments were cut, the function of SPIK was not affected.
SPIK prevents GzmA-induced cell apoptosis
The finding that SPIK directly binds to GzmA strongly suggests that SPIK may be a GzmA inhibitor, resulting in suppression of GzmA-induced apoptosis. We therefore wanted to determine if cells that express high levels of SPIK could prevent GzmA-induced cell apoptosis. To do this, we used the stable cell line S2–3, which expresses and secretes high levels of uncleaved SPIK (Fig. 2a) 28 and its complementary cell line, SP23, which expresses and secretes only background levels of SPIK and is susceptible to SPDCA induced by brefeldin A and cycloheximide.28 The GzmA-mediated apoptosis was assessed by incubating cells with 2 μm recombinant GzmA and 254 ng/ml PFR, a protein that is secreted along with granzymes by CTLs and NK cells to create pores in the target cell membrane and that is necessary for GzmA entry into target cells.31 The dose of PFR was adjusted to maximize GzmA entry without cytotoxicity according to the methods of Martinvalet et al.26 (see Supplementary material, Fig. S1). To determine levels of apoptosis, cells were stained with Hoechst dye and analysed for condensed nuclei 20 hr post-treatment. These bright blue dots, a hallmark of apoptosis, were not visible in cells treated with GzmA or PRF alone, indicating that cell death was not the result of cytotoxicity caused by either of the proteins (Fig. 4a, bottom panels, SP23 with GzmA or PFR). Combined treatment with both GzmA and PFR, however, caused obvious nuclear condensation in SP23 cells (Fig. 4a, upper right panel, SP23 with GzmA/PFR), indicating apoptotic death in these cells. In contrast, we found no obvious staining in S2–3 cells under any of the three treatments, including the combined GzmA/PRF treatment (Fig. 4a, upper left panel, S2–3, GzmA/PFR), suggesting that over-expression of SPIK in these cells resulted in resistance to GzmA-mediated apoptotic death. To ensure apoptosis was a specific result of GzmA treatment and not of PFR treatment, a decreased concentration of GzmA was used without changing the concentration of PRF. An obvious reduction of apoptosis was seen in these treated cells (see Supplementary material, Fig. S2). In addition, the fact that neither treatment of GzmA nor of PFR alone can induce apoptosis further supports the specificity of treatment (Fig. 4a).
Figure 4.
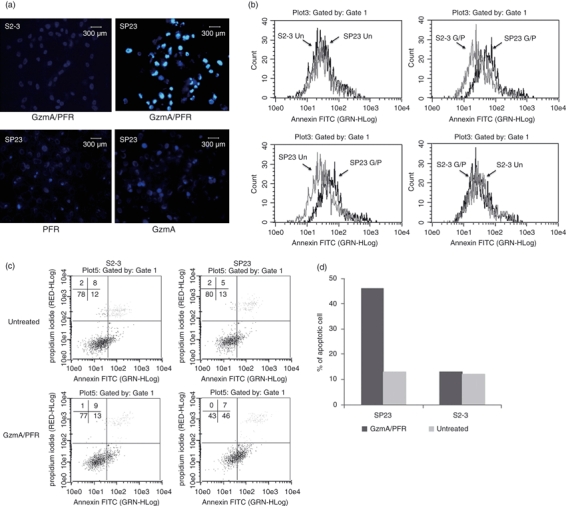
Over-expression of serine protease inhibitor Kazal (SPIK) results in cellular resistance to Granzyme A (GzmA) -induced apoptosis. S2–3 and SP23 cells were incubated with GzmA and perforin (PFR), individually or combined. Apoptosis was assessed. (a) Hoechst staining of apoptotic cells. (b) Cell apoptosis was analysed by flow cytometry after staining with annexin V-FITC. (c) Cell apoptosis was quantified by flow cytometry after double staining with propidium iodide (PI) and annexin V-FITC. The lower-right quadrant indicates the apoptotic cells. (d) Percentage of apoptotic cells from flow cytometry with double staining.
Fluorescence-labelled annexin V, which identifies cell surface changes in the early stages of apoptosis, was also used to quantify cellular apoptosis by flow cytometry. After treatment with GzmA/PFR, the cells were stained with annexin V-FITC. The fluorescent shift to the right in the annexin peak indicates apoptotic cell death. Fig. 4b showed that the background levels of apoptosis in both S2–3 and SP23 cells were similar, which was indicated by the overlapping of the respective annexin peaks (Fig. 4b, upper left). GzmA/PFR treatment, however, clearly resulted in a rightward shift of the annexin peak in the SP23 cells (Fig. 4b, bottom left, compared with untreated cells), suggesting an increased number of cells undergoing apoptosis. In contrast, the same conditions caused no visible shift of the annexin peak in S2–3 cells (Fig. 4b bottom right), suggesting that the expression of SPIK resulted in cellular resistance to GzmA-induced apoptosis. Importantly, double staining of cells with PI and annexin V-FITC showed that treatment with GzmA plus PFR caused 46% of SP23 cells to undergo apoptosis (Fig. 4c,d. SP23, GzmA/PFR), whereas it only caused 13% of S2–3 cells to undergo apoptosis (Fig. 4c,d S2–3). In contrast, there was no detectable difference between untreated SP23 and S2–3 cells (Fig. 4c,d, untreated). These results were consistent among multiple replicates and were statistically significant (data not shown). They were also consistent with our previous findings describing SPIK as an apoptosis inhibitor, preventing SPDCA.14,28
Suppression of SPIK expression restores the sensitivity of cells to GzmA-induced apoptosis
After showing that over-expression of SPIK could lead to the resistance of cells to GzmA-induced cell death, we wanted to know if reducing SPIK expression could restore GzmA-induced killing. To repress over-expressed SPIK, S2–3 cells were transfected with plasmids expressing SPIK siRNA. Two SPIK siRNAs, L71 and L183, were transfected into S2–3 cells,14 and the levels of SPIK were then detected by Northern blot assay. Small interfering RNA for woodchuck hepatitis B virus (WHBV) was used as a control for silencing specificity.
Figure 5(a) shows that compared with mock transfection (S2–3), transfection of both L71 and L183 clearly reduced the expression of SPIK, though not completely to the background level in SP23 cells (Fig. 5a, SP23). No suppression of SPIK expression by WHBV siRNA was observed, suggesting that the silencing was specific. A Western blot analysis showed that transfection of L71 and L183 obviously reduced SPIK protein levels (Fig. 5b), but transfection of WHBV siRNA had no effect on the level of SPIK protein.
Figure 5.
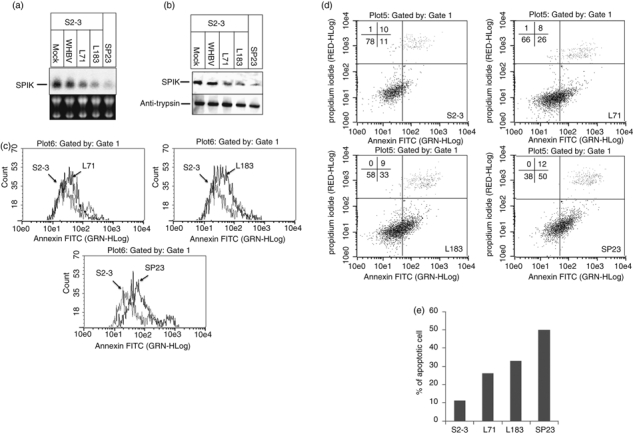
Silencing of over-expressed serine protease inhibitor Kazal (SPIK) in S2–3 cells restores cell sensitivity to Granzyme A (GzmA) -induced cell apoptosis. S2–3 cells were transfected with SPIK small interfering (si) RNA plasmids L71 and L183 or with their vector for mock transfection. The plasmid for woodchuck hepatitis B virus (WHBV) siRNA was used as a control for specificity. SP23 cells, expressing only background levels of SPIK, were used as a control. Cell apoptosis was induced by treatment of cells with GzmA/PFR 3 days after transfection. (a) The SPIK RNA levels were examined by Northern blot with the probe specified for SPIK. (b) SPIK protein was examined by Western blot with monoclonal anti-SPIK antibody. (c) Cell apoptosis was analysed by flow cytometry after staining with annexin V-FITC. (d) Cell apoptosis was quantified by flow cytometry after double staining with propidium iodide (PI) and annexin V-FITC. The lower-right quadrant indicates the apoptotic cells. (e) Percentage of apoptotic cells from flow cytometry with double staining.
Successful silencing of SPIK was able to restore sensitivity of the cells to GzmA-induced apoptosis. Figure 5(c) showed that, compared with mock-transfected S2–3 cells, incubation of GzmA/PFR with cells transfected with SPIK siRNA L71 and L183 resulted in an observable rightward shift of the annexin peak, indicating more cells undergoing apoptosis (Fig. 5c, L71 and L183). Double staining with annexin V-FITC and PI showed that GzmA/PFR treatment led to an increase in cell death from 11% to 33% in L183-transfected, and to 26% in L71-transfected S2–3 cells (Fig. 5d,e). The sensitivity of cells to GzmA/PFR-induced apoptosis was not completely restored to background levels (compared with 50% apoptotic death in the control SP23 cells, Fig. 5d,e), but this is probably because of the inability to completely silence SPIK in the S2–3 cells (Fig. 5a,b).
The C3–C4 region of SPIK is critical for SPIK function
After demonstrating that SPIK can directly bind GzmA, thereby preventing GzmA-induced cell death, we next investigated the critical functional region of SPIK. A series of deletion mutants, PD1, PD2 and PD3, that lacked, respectively, the C2–C3 (CTKIYDPV) region, C3–C4 (CGTDGNTYPNE) region, or 12 amino acids (QTSILIQKSGPC) from the C-terminus of SPIK, were constructed (Fig. 6a). To account for possible loss of the antibody binding affinity to SPIK, a 10-amino-acid segment with a myc tag was added to the N-terminus of SPIK to replace the segment containing the first nine-amino-acid fragment, which is cleaved in secreted pancreatic SPIK. This nine-amino-acid segment has been shown not to be a requirement for the binding of SPIK to GzmA (Fig. 2 and 3b). We therefore could use an anti-myc antibody to identify the mutated SPIK. To reduce the background level of SPIK, we used HeLa cells, a cell line in which SPIK expression is undetectable.28
Figure 6.
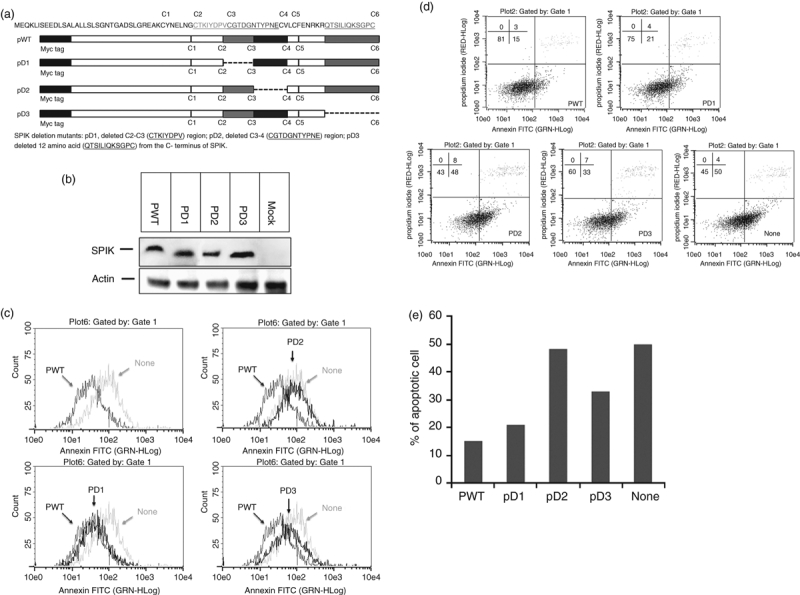
The C3–C4 region is critical for the ability of serine protease inhibitor Kazal (SPIK) to suppress Granzyme A (GzmA) -induced apoptosis. Wild-type SPIK plasmid PWT was constructed with pCMVscript vector with a myc tag sequence at the N-terminus. Three mutants PD1, PD2 and PD3 were constructed based on PWT. (a). The structure of PWT and its deletion mutants. (b). HeLa cells were transfected with either wild-type plasmid or its mutants (PD1, PD2 and PD3) or mock transfected with vector as a control (none). Cellular SPIK levels were assessed with Western blot using anti-myc antibody. Actin was used as loading control. (c) Cell apoptosis was analysed by flow cytometry after staining with annexin V-FITC. (d) Cell apoptosis was quantified by flow cytometry after double staining with propidium iodide (PI) and annexin V-FITC. The lower-right quadrant indicates the apoptotic cells. (e) Percentage of apoptotic cells from flow cytometry with double staining.
HeLa cells were transfected with either wild-type SPIK, mutant SPIK, or mock transfected with the pCMV-Script vector. It has previously been reported that a SPIK mutation, particularly a change in the conserved C3–C4 region, resulted in decreased levels of secreted SPIK but did not affect protein synthesis.32 Therefore, the level of intracellular SPIK from transfected cells was examined by Western blot using an anti-myc antibody. Figure 6(b) shows that although the deletion yielded SPIK with a lower molecular weight, as expected (Fig. 6b, compared with the wild-type), all deletion mutants produced a level of SPIK protein similar to that of the wild-type, suggesting that the deletions did not affect the synthesis of the SPIK protein. As we have reported previously, suppression of SPDCA is dependent on the SPIK protein but not on the transcripts,28 suppression of GzmA-induced cell apoptosis is also dependent on SPIK protein. It was shown that wild-type SPIK with a mutation in the start codon AUG no longer inhibited GzmA-induced cell apoptosis, and this result supports this conclusion (see Supplementary material, Fig. S3).
To determine which region of SPIK is responsible for the inhibition of GzmA function, we induced GzmA-mediated apoptosis in cells transfected with wild-type SPIK or the deletion mutants. As expected, transfection of wild-type SPIK (PWT) resulted in decreased numbers of apoptotic cells compared with the mock-transfected cells (none) after incubation with GzmA/PFR (Fig. 6c, upper left panel, PWT and none). Significant overlap between the annexin peak for the cells transfected with PD1 or with PWT suggests that deletion of the C2–C3 region of SPIK did not affect the resistance of cells to GzmA-induced apoptosis (Fig. 6c, lower left panel, PWT and PD1). In combination with the finding that the N-terminal, nine-amino-acid fragment was unnecessary for SPIK to bind to GzmA, these results suggest that the N-terminus of SPIK may be non-essential for proper SPIK function. On the other hand, the annexin peak of the cells transfected with PD2 almost completely overlapped with the peak of the mock-transfected cells (Fig. 6c, up right panel, none and PD2), indicating that the deletion of C3–C4 almost completely eradicated the cellular resistance to GzmA-induced apoptosis. This result implies that the C3–C4 region of SPIK is critical in suppressing GzmA-induced cell apoptosis. Transfection of PD3, in which 12 amino acids have been deleted from the C-terminal end of SPIK, only caused a slight shift of the annexin peak away from the PWT peak, implying that the C-terminus of SPIK may be only partially involved in the binding of SPIK and GzmA (Fig. 6c, lower right panel, PWT, none and PD3).
Double staining with annexin V-FITC and PI also confirmed these results. Around 50% of the mock-transfected cells underwent apoptosis (Fig. 6d, lower right panel, none and Fig. 6e), whereas only 15% of wild-type SPIK transfected cells were apoptotic (Fig. 6d, upper left panel, PWT and Fig. 6e). Transfection of PD1 did not noticeably change the resistance of the cells to GzmA-mediated cell death, with only about 21% of these cells undergoing apoptosis (Fig. 6d, upper right panel, PD1 and Fig. 6e). In contrast, deletion of the C3–C4 region of SPIK resulted in the complete eradication of cellular resistance to GzmA-induced apoptosis. In this case, 48% of cells underwent apoptosis, similar to the levels seen in the mock transfection (50%, Fig. 6d,e). Thirty-three per cent of treated cells transfected with PD3 underwent apoptosis after incubation with GzmA/PFR, further supporting the conclusion that the C-terminus of SPIK may be partially involved in suppression of GzmA-induced cell apoptosis of SPIK.
Discussion
The protein SPIK was first discovered in the pancreas as a protease inhibitor, functioning to prevent autoactivation of trypsinogen.1,2 Multiple additional biological functions have been gradually revealed by recent studies. For example, SPIK was shown to function as an anti-inflammatory reactant9–11 and has been shown to inhibit apoptosis, suppressing SPDCA induced by brefeldin A/cycloheximide.14,28 In addition, these current studies show that SPIK can bind GzmA, preventing cell death triggered by GzmA-induced apoptosis. These results further confirm the multiple biological functions of SPIK. Because GzmA triggers both a pro-inflammatory effect at low concentrations 24,25 and induces SPDCA at high concentrations,26,27 the binding of SPIK to GzmA strongly supports the possible double-sided function of SPIK in GzmA-related anti-inflammation and anti-apoptosis.
GzmA is a cytotoxic serine protease secreted by activated CTLs and NK cells to kill target cells during immune surveillance.26,33 However, the body's immune system can also use additional pathways to eliminate malignant cells, including the tumour necrosis factor (TNF) dependent Fas-FDD-induced cell death pathway and the GzmB-induced cell apoptotic pathway. This makes the situation in vivo more complex. Evidence shows that the TNF and GzmB-dependent pathways induce apoptosis in a caspase-dependent manner known as caspase-dependent cell apoptosis (CDCA), and our previous work has shown that SPIK is unable to prevent CDCA.14,28 Our unpublished data further suggest that, unlike GzmA, SPIK may be unable to bind GzmB and, because of this, will not prevent its induction of apoptosis. Therefore, the role of SPIK under more physiological conditions may need further study. To begin to address this issue, suppression studies of both GzmA and GzmB by SPIK under physiological conditions are currently ongoing. Although the role of SPIK in vivo is currently being investigated, the role of GzmA-induced apoptosis in CTL-mediated and NK-cell-mediated immune removal of malignant cells, such as tumour precursor/tumour germ cells, has been previously shown. 21,22,27,34 The discovery that SPIK can bind GzmA and suppress its function implies that SPIK may contribute to the resistance of these malignant cells to escape from immune clearance. Considering that over-expression of SPIK occurs in numerous cancers, including colorectal tumours, renal cell carcinoma, gastric carcinoma, HCC and intrahepatic cholangiocarcinoma,3–8 it is possible that the development of cancer could, at least in part, be the result of the ability of malignant cells to evade immune clearance because of their increased levels of SPIK. Our study did not directly address the role of SPIK in vivo. However, both SPIK and GzmA are secreted proteins, therefore, it is possible that the secreted SPIK directly interacts with GzmA, perhaps at the immunological synapse (micro-environment created by the lymphocyte–cancer cell contact zone) and prevents its entry into target cells, ultimately blocking apoptosis. However, it is more likely that intracellular SPIK (Fig. 2a) is able to inhibit GzmA-induced cell apoptosis by blocking GzmA once it enters the target cell. The ability of SPIK to function intracellularly to suppress SPDCA induced by brefeldin A and cycloheximide supports this hypothesis. 14,28 While these studies do not take into account the role of GzmA as an inducer of inflammation at low concentrations24,25, it would be interesting to know whether SPIK also functions to inhibit this pathway and act as an anti-inflammatory protein in vivo. This may be clarified by determining whether SPIK can inhibit pro-inflammatory cytokine levels in cells treated with GzmA.
Functionally, compared with those in the C-terminus, the N-terminal amino acids of SPIK appear to have a minimal effect on the inhibition of GzmA activity. This finding is in agreement with the observation that the active form of SPIK in pancreatic secretions is attenuated. Bartelt et al.30 reported that a 23-amino-acid fragment thought to be a signal peptide in the N-terminus of activated pancreatic SPIK was cut during secretion. Our Edman degradation study suggested that a nine-amino-acid fragment in the N-terminus was attenuated in the SPIK secreted by cultured pancreatic cells; the difference between our observation and the observation by Bartelt et al.30 may be because of our use of more recent and advanced biotechnological methods compared with those used 30 years ago (before 1980). However, we cannot exclude the possibility that more amino acids are cut in vivo than in vitro during secretion of pancreatic SPIK. Our functional analyses also suggested that the conserved C3–C4 region of SPIK was critical to the inhibition of GzmA-induced apoptosis. Furthermore, the C-terminus of SPIK seemed to be partially associated with the SPIK function. This finding is supported by additional mutation studies, which showed that amino acids G48, D50 and Y54, all of which are in the C3–C4 region, are critical to SPIK function. R65 and R67, which are in the C-terminus of SPIK, have been shown to be functionally indispensable for SPIK.32 Interestingly, our study found that liver cancer cells S2–3 and G54 secreted intact, uncleaved SPIK. Why liver cancer cells secrete only uncleaved SPIK is unknown. It would be interesting to determine whether the secretion of uncleaved SPIK is a liver-specific or a cancer-specific characteristic. It would also be of interest to know whether the secretion of uncleaved SPIK is a common characteristic of cancer cells, because SPIK levels are elevated in other cancers. More studies are needed to clarify these issues. Our study suggests that suppressing over-expressed SPIK in HCC-derived S2–3 cells can restore sensitivity to GzmA-induced death and may overcome the tolerance of cancer cells to the CTL-mediated and NK-cell-mediated immune responses via GzmA. Ideally, this would restore the immune system's ability to clear these cancer cells from the body. Ultimately, using this information would allow us to develop a new class of anti-cancer drugs that can restore the susceptibility of cancer cells to the body's natural immune surveillance system. This is different from existing anticancer drugs, which use toxins to kill the cells as they divide, or in some cases, during a specific phase of the cell cycle. Not only would this new class of drugs be more specifically targeted, but it would also reduce the toxicity to the patient as it uses the body's existing defences to destroy the malignant cells.
Acknowledgments
This work was supported by an appropriation from the Commonwealth of Pennsylvania; the Hepatitis B Foundation, USA, ImCare Biotech LLC and National Cancer Institute, NIH. We thank Ms Pamela Fried, Academic Publishing Services, Drexel University College of Medicine, for her critical reading of the manuscript.
Glossary
Abbreviation
- CDCA
caspase-dependent cell apoptosis
- CTL
cytotoxic T lymphocyte
- GzmA
granzyme A
- GzmB
granzyme B
- HCC
hepatocellular carcinoma
- NK
natural killer
- PFR
perforin
- PI
propidium iodide
- siRNA
small interfering RNA
- SPDCA
serine protease-dependent cell apoptosis
- SPIK
serine protease inhibitor Kazal
- WHBV
woodchuck hepatitis B virus
Disclosures
X.L receives research support from ImCare Biotech.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. The optimal sublytic concentration of perforin (PFR) for granzyme A (GzmA) to induce cell apoptosis.
Figure S2. The level of apoptosis depends on the concentration of Granzyme A (GzmA).
Figure S3. Serine protease inhibitor Kazal (SPIK) protein is responsible for cellular resistance to Granzyme A (GzmA) -induced apoptosis.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Stenman UH. Tumor-associated trypsin inhibitor. Clin Chem. 2002;48:1206–9. [PubMed] [Google Scholar]
- 2.Greene LJ. Pancreatic exocrine secretory proteins. J Surg Oncol. 1975;7:151–4. doi: 10.1002/jso.2930070211. [DOI] [PubMed] [Google Scholar]
- 3.Higashiyama M, Monden T, Tomita N, et al. Expression of pancreatic secretory trypsin inhibitor (PSTI) in colorectal cancer. Br J Cancer. 1990;62:954–8. doi: 10.1038/bjc.1990.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomita N, Doi S, Higashiyama M, et al. Expression of pancreatic secretory trypsin inhibitor gene in human colorectal tumor. Cancer. 1990;66:2144–9. doi: 10.1002/1097-0142(19901115)66:10<2144::aid-cncr2820661017>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Ohmachi Y, Murata A, Matsuura N, et al. Specific expression of the pancreatic-secretory-trypsin-inhibitor (PSTI) gene in hepatocellular carcinoma. Int J Cancer. 1993;55:728–34. doi: 10.1002/ijc.2910550505. [DOI] [PubMed] [Google Scholar]
- 6.Lukkonen A, Lintula S, von Boguslawski K, et al. Tumor-associated trypsin inhibitor in normal and malignant renal tissue and in serum of renal-cell carcinoma patients. Int J Cancer. 1999;83:486–90. doi: 10.1002/(sici)1097-0215(19991112)83:4<486::aid-ijc9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 7.Tonouchi A, Ohtsuka M, Ito H, et al. Relationship between pancreatic secretory trypsin inhibitor and early recurrence of intrahepatic cholangiocarcinoma following surgical resection. Am J Gastroenterol. 2006;101:1601–10. doi: 10.1111/j.1572-0241.2006.00612.x. [DOI] [PubMed] [Google Scholar]
- 8.Lee YC, Pan HW, Peng SY, et al. Overexpression of tumour-associated trypsin inhibitor (TATI) enhances tumour growth and is associated with portal vein invasion, early recurrence and a stage-independent prognostic factor of hepatocellular carcinoma. Eur J Cancer. 2007;43:736–44. doi: 10.1016/j.ejca.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa M, Shibata T, Niinobu T, Uda K, Takata N, Mori T. Serum pancreatic secretory trypsin inhibitor (PSTI) in patients with inflammatory diseases. Adv Exp Med Biol. 1988;240:505–8. doi: 10.1007/978-1-4613-1057-0_63. [DOI] [PubMed] [Google Scholar]
- 10.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–6. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 11.Cavestro GM, Zuppardo RA, Bertolini S, et al. Connections between genetics and clinical data: role of MCP-1, CFTR, and SPINK-1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am J Gastroenterol. 2010;105:199–206. doi: 10.1038/ajg.2009.611. [DOI] [PubMed] [Google Scholar]
- 12.Uda K-I, Murata A, Nishijima J-I, et al. Elevation of circulating monitor peptide/pancreatic secretory trypsin inhibitor-I (PSTI-61) after turpentine-induced inflammation in rats: hepatocytes produce it as an acute phase reactant. J Surg Res. 1994;57:563–8. doi: 10.1006/jsre.1994.1183. [DOI] [PubMed] [Google Scholar]
- 13.Greene LJ, Pubols MH, Bartelt DC. Human pancreatic secretory trypsin inhibitor. Methods Enzymol. 1976;45:813–25. doi: 10.1016/s0076-6879(76)45075-9. [DOI] [PubMed] [Google Scholar]
- 14.Lamontagne J, Pinkerton M, Block TM, Lu X. Hepatitis B and hepatitis C virus replication upregulates serine protease inhibitor Kazal, resulting in cellular resistance to serine protease-dependent apoptosis. J Virol. 2010;84:907–17. doi: 10.1128/JVI.01249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiksten JP, Lundin J, Nordling S, Kokkola A, Stenman UH, Haglund C. High tissue expression of tumour-associated trypsin inhibitor (TATI) associates with a more favourable prognosis in gastric cancer. Histopathology. 2005;46:380–8. doi: 10.1111/j.1365-2559.2005.02073.x. [DOI] [PubMed] [Google Scholar]
- 16.Gaber A, Nodin B, Hotakainen K, et al. Increased serum levels of tumour-associated trypsin inhibitor independently predict a poor prognosis in colorectal cancer patients. BMC Cancer. 2010;10:498. doi: 10.1186/1471-2407-10-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99:1472–7. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 19.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–33. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–26. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 21.Pardo J, Balkow S, Anel A, Simon MM. Granzymes are essential for natural killer cell-mediated and perf-facilitated tumor control. Eur J Immunol. 2002;32:2881–7. doi: 10.1002/1521-4141(2002010)32:10<2881::AID-IMMU2881>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 22.Pardo J, Aguilo JI, Anel A, et al. The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes Infect. 2009;11:452–9. doi: 10.1016/j.micinf.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Tsuzuki S, Kokado Y, Satomi S, et al. Purification and identification of a binding protein for pancreatic secretory trypsin inhibitor: a novel role of the inhibitor as an anti-granzyme A. Biochem J. 2003;1:227–33. doi: 10.1042/BJ20021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardo J, Simon MM, Froelich CJ. Granzyme A is a proinflammatory protease. Blood. 2009;114 doi: 10.1182/blood-2009-07-231027. author reply 3969–70. [DOI] [PubMed] [Google Scholar]
- 25.Metkar SS, Menaa C, Pardo J, et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity. 2008;29:720–33. doi: 10.1016/j.immuni.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–70. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010;235:93–104. doi: 10.1111/j.0105-2896.2010.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu X, Lamontagne J, Lu F, Block T. Tumor-associated protein SPIK/TATI suppresses serine protease dependent cell apoptosis. Apoptosis. 2008;13:483–94. doi: 10.1007/s10495-008-0193-x. [DOI] [PubMed] [Google Scholar]
- 29.Lu X, Block T. Study of the early steps of the hepatitis B virus life cycle. Int J Med Sci. 2004;1:21–33. doi: 10.7150/ijms.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartelt DC, Shapanka R, Greene LJ. The primary structure of the human pancreatic secretory trypsin inhibitor. Amino acid sequence of the reduced S-aminoethylated protein. Arch Biochem Biophys. 1977;179:189–99. doi: 10.1016/0003-9861(77)90103-5. [DOI] [PubMed] [Google Scholar]
- 31.Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–47. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- 32.Boulling A, Le Marechal C, Trouve P, Raguenes O, Chen J-M, Ferec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur J Hum Genet. 2007;15:936–42. doi: 10.1038/sj.ejhg.5201873. [DOI] [PubMed] [Google Scholar]
- 33.Lieberman J, Fan Z. Nuclear war: the granzyme A-bomb. Curr Opin Immunol. 2003;15:553–9. doi: 10.1016/s0952-7915(03)00108-0. [DOI] [PubMed] [Google Scholar]
- 34.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17:616–23. doi: 10.1038/cdd.2009.206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.