

Abstract
Regulatory T (Treg) cells represent one of the main mechanisms of regulating self-reactive immune cells. Treg cells are thought to play a role in down-regulating immune responses to self or allogeneic antigens in the periphery. Although the function of Treg cells has been demonstrated in many experimental settings, the precise mechanisms and antigen specificity often remain unclear. In a hepatitis B e antigen–T-cell receptor (HBeAg-TCR) double transgenic mouse model, we observed a phenotypically unique (TCR+ CD4−/CD8− CD25+/− GITRhigh PD-1high FoxP3−) HBeAg-specific population that demonstrates immune regulatory function. This HBeAg-specific double-negative regulatory cell population proliferates vigorously in vitro, in contrast to any other known regulatory population, in an interleukin-2-independent manner.
Keywords: double-negative regulatory T cells, hepatitis B e antigen, hepatitis B virus infection, tolerance
Introduction
The primary function of the immune system is to protect the self from pathogens. A highly effective and dynamic cellular network has evolved to signal the presence of pathogens and initiate a response that is specific for the invading pathogen while maintaining tolerance to self. Distinguishing between self and non-self is a fundamental property of the immune system and is accomplished by a variety of mechanisms. A function of regulatory T (Treg) cells is to prevent self-reactive immune cells from damaging self. The Treg cells, particularly CD4+ CD25+ conventional Treg (cTreg) cells, are thought to play a role in down-regulating immune responses to self or allogeneic antigens in the periphery.1–4 Although the function of Treg cells has been shown in a number of in vivo models of autoimmunity and transplantation, the precise mechanism and antigen specificity often remains unclear.5
In 1971 it was first suggested that Treg cells had the ability to transfer antigen-specific tolerance to naive animals.6 Even though a role for regulatory cells during an immune response was widely accepted, the existence of Treg cells was controversial until a specific surface marker was described by Sakaguchi et al.7 Conventional Treg cells constitutively express a variety of cell markers, such as CD4, CD25, CD45RBlow, CD62 ligand (CD62L), CD103, as well as cytotoxic T-lymphocyte antigen 4 (CTLA-4) and glucocorticoid-induced tumour necrosis factor receptor-related protein (GITR).7–14 Although cTreg cells express CD4+ CD25+, CD25 is not a specific marker for cTreg cells. Other cell markers (i.e. CTLA-4, GITR and CD103) are also not exclusive markers for Treg cells, because in most cases they are up-regulated on effector T cells upon activation. The transcription factor Forkhead box P3 (FoxP3) is predominantly expressed on Treg cells and appears to be expressed at the thymic CD4+/CD8+ stage.15–18 In contrast to the cell surface markers mentioned above, FoxP3 is not observed in non-Treg cells upon activation or differentiation into T helper type 1/ type 2 cells, nor in natural killer T cells. Furthermore, retroviral gene transfer of FoxP3 converts naive T cells to a regulatory T-cell phenotype similar to cTreg cells.17 However, some Treg cell populations (i.e. Tr1 and Th3) do not express FoxP3. Taken together, FoxP3 is also not an exclusive marker for, but rather is a specific control gene for the development and function of cTreg cells. To determine a possible therapeutic application for the use of Treg cells, it is extremely important to know the detailed phenotype of a Treg cell population.
Recently, Zhang et al.19 and others reported the presence of MHC class I restricted CD4− CD8− double-negative (DN) T cells with a unique phenotype for a Treg cell population (i.e. TCR+ CD4− CD8− CD25+ CD28−). The DN T cells comprise 1–3% of peripheral T lymphocytes in the mouse.20–22 The DN Treg cells isolated from mice that have permanently accepted allografts or xenografts can specifically suppress and kill syngeneic anti-donor CD4+ and CD8+ T cells in vitro.20,23–25 Upon expansion in vitro with allogeneic donor lymphocytes, the DN Treg cells can specifically suppress proliferation of syngeneic CD4+ and CD8+ T cells in vitro and prolong donor-specific allogeneic skin graft survival when infused into syngeneic naive mice. Recent studies suggest that this immune suppressive function is mediated by suppression of antigen-presenting cell (APC) function.26,27 Also, adoptively transferred DN Treg cells augment recipient Treg cell accumulation and enhance long-term cardiac allograft survival.28
We have produced a number of T-cell receptor (TCR) transgenic (Tg) mice specific for the hepatitis B core (HBcAg) and precore (HBeAg) antigens, which share significant amino acid homology.29 When the TCR-Tg lineage 7/16-5 is bred with Tg mice that secrete HBeAg in the serum, the resulting double-Tg (dbl-Tg) mice demonstrate T-cell tolerance.29,30 The degree and nature of tolerance is dependent on the nature of the hepatitis B virus (HBV) antigen. For example, in HBeAg × 7/16-5 dbl-Tg mice, in vitro interleukin-2 (IL-2) production is significantly reduced compared with 7/16-5 single TCR-Tg mice and the dbl-Tg mice do not spontaneously produce anti-HBe antibodies in vivo. In contrast, in 7/16-5 TCR-Tg mice bred with HBcAg-Tg mice, which express the HBcAg intracellularly in the liver, the resulting dbl-Tg mice undergo spontaneous anti-HBc seroconversion between 4 and 6 weeks of age and are significantly less tolerant at the T-cell level than HBeAg-expressing dbl-Tg mice.30 Furthermore, in triple-Tg mice expressing the 7/16-5 TCR, secreted HBeAg, and intracellular HBcAg spontaneous anti-HBc seroconversion is suppressed. These studies demonstrated that the secreted HBeAg functions as a tolerogen in a TCR-Tg system in which the intracellular HBcAg is an immunogen and the presence of HBeAg as a serum protein can regulate the immune response to the HBcAg.30,31 Because HBeAg-specific tolerance is not mediated by clonal deletion of 7/16-5 TCR-Tg T cells in HBeAg × 7/16-5 dbl-Tg mice,30 we explored the possibility that HBeAg-specific Treg cells may be present.
In this study, we demonstrate an HBeAg-specific Treg cell population in the TCR × HBeAg-dbl-Tg mouse model that possesses a unique DN phenotype (i.e. TCR+ CD4− CD8− CD25+/− GITRhigh PD-1high FoxP3−). Most strikingly, these HBeAg-specific DN T cells exhibit extremely efficient regulatory function compared with other Treg cells in vitro. As a result of its vigorous proliferation in vitro, suppressive effects and unique phenotype, the HBeAg-specific DN T-cell population described herein may represent a distinct Treg cell subset.
Materials and methods
Transgenic mice
The 7/16-5 transgenic TCR (Vβ11+-Vα5+) is specific for residues 120–140 of HBc/HBeAgs, is restricted by the I-Ab MHC class II molecule, is expressed on 53% of CD4+ T cells,29,30 and is uniquely expressed on a high proportion of CD8+ T cells (unpublished data). Transgenic mice engineered to express relatively high levels of HBeAg in the serum (4–10 μg/ml) and HBcAg in the liver (0·2–2 μg/mg protein) through the use of the liver-specific major urinary protein promoter have been described.32,33 All Tg mice were bred onto a C57BL/10 background. The mice designated as HBcAg or HBeAg-Tg were hemizygous for the transgenes, as were the 7/16-5 TCR-Tg mice. Ovalbumin-specific OT-II Tg mice, MHC class I knockout (KO) mice, and TCR α-chain KO mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All animal care was performed according to the National Institutes of Health standards as set forth in the Guide for the Care and Use of Laboratory Animals.
Recombinant proteins and synthetic peptides
Recombinant HBcAg of the ayw subtype was produced in Escherichia coli and purified as described elsewhere.34 A recombinant HBeAg corresponding in sequence to serum-derived HBeAg encompassing the 10 precore amino acids remaining after cleavage of the precursor and residues 1–149 of HBcAg was produced as described previously.34 The presence of the 10 precore amino acids prevents particle assembly, and HBeAg is recognized efficiently by HBeAg-specific monoclonal antibodies (mAbs) but displays little HBc antigenicity. Peptides were synthesized by the simultaneous multiple peptide synthesis method.35 The HBe/HBcAg-derived synthetic peptide representing the recognition site for the 7/16-5 TCR was designated from the N-terminus of HBcAg: 120–140, VSFGVWIRTPPAYRPPNAPIL. OVA (323–339) peptides were purchased from Anaspec (Fremont, CA).
Antibodies and reagents
The following antibodies were all purchased from eBioscience (San Diego, CA): Fluorescence- or biotin-labelled anti-CD4, anti-CD8, anti-Vβ11, anti-CD25, anti-CD11c, anti-CD11b, anti-CD49b, anti-B220, anti-GITR, anti-FAS, anti-FASL, anti-IL-15R, anti-CTLA-4, anti-PD-1 and Foxp3 intracellular staining. Cell separation apparatus and reagents used were purchased from Miltenyi Biotech (Auburn, CA).
Cell preparation
Five- to 10-week-old HBeAg × 7/16-5 TCR dbl-Tg mice were used as a DN T-cell source. Spleen cells were cultured in Dulbecco's modified Eagle's medium supplemented with nutrients, 4% fetal calf serum, 100 μg/ml streptomycin and 50 μm 2-mercaptoethanol. Single cell suspensions from spleen or cultured cells were labelled with biotinylated anti-CD4, anti-CD8, anti-B220, anti-CD11c, anti-CD11b and Gr1 for negative depletion. Cells were then magnetized with streptavidin-Microbeads (Miltenyi Biotech), and passed through the LS column to collect the flow through as DN T cells. For cultured cells, dead cell removal was performed before negative depletion using the dead cell removal kit from Miltenyi Biotech. Briefly, 4-day-cultured splenocytes from HBeAg × 7/16-5 dbl-Tg were harvested and labelled with propidium iodide, and subsequently mixed with anti-propidium iodide-Microbeads. Propidium iodide-labelled dead cells were subsequently removed by magnetic selection. B cells were purified by positive selection with B220-microbeads. For DN progenitor depletion, cells were positively selected by biotinylated anti-CD4, anti-CD8, anti-B220, anti-CD11c, anti-CD11b and anti-Gr1 and were subsequently magnetized with streptavidin-Microbeads to exclude DN T-cell progenitors.
Cell culture
For antigen-specific stimulation, 4 × 105 splenocytes from 7/16-5 TCR Tg mice or HBeAg × 7/16-5 dbl-Tg mice were cultured in 4% fetal calf serum supplemented with Dulbecco's modified Eagle's medium in the presence of truncated HBcAg149, or the HBcAg-derived peptide p120–140 at concentrations of 0·2–2 μg/ml. Cells were placed in flat-bottom 96-well-plates for 1, 2, 3 or 4 days for further analysis. At day 2 and day 4, supernatants were collected for cytokine analysis. For the DN T-cell suppression assay, 4 × 105/well of naive 7/16-5 TCR-Tg splenocytes were used as target cells and 4-day cultured HBeAg-specific DN T cells were separated by negative enrichment as described above, and were added to the culture at given numbers.
Flow cytometry analysis
To analyse T-cell activation, antigen-specific T cells (Vβ11+ CD4 T cells) were stained for CD25 as well as CD69 at given time points. For the surface staining, cells were incubated in 3% fetal bovine serum in PBS containing 10 μg/ml 2.4G2 to block FcR binding followed by the addition of fluorochrome-conjugated antibodies. All fluorochrome-conjugated antibodies were obtained from eBioscience as described above. For the detection of CTLA-4, we performed intracellular staining and surface staining because of the nature of CTLA-4 expression. Foxp3 intracellular staining was performed using a commercially available kit from eBioscience. Flow cytometry was performed on an LSRII flow cytometer (Becton Dickinson, CA) available through the CCMI at the Salk Institute (La Jolla, San Diego, CA). Data were analysed using FlowJo software (Tree Star, Ashland, OR).
In vitro cytokine analysis and proliferation assay
Spleen cells from either unprimed or primed TCR-Tg, TCR × antigen dbl-Tg or wild-type mice were cultured (4 × 105/well) with various concentrations of a series of antigens. Culture supernatants were harvested at 48 hr for IL-2 determination and at 96 hr for interferon-γ (IFN-γ) determination. Cytokines were measured by two-site ELISA with pairs of cytokine-specific mAbs. One unlabelled mAb was adsorbed to the microtitre plate well and used as a capture antibody, and the other labelled mAb served as the probe. For the multicytokine assay, we have used Multiflex Biomarker Immunoassay (Millipore, Billerica, MA). To determine T-cell proliferation, TCR-Tg spleen T cells were labelled in vitro with the intracellular dye carboxyfluorescein succinimidyl ester (CFSE) by using the Vybrant CFSE SE tracer kit (Molecular Probes, Eugene, OR.). Carboxyfluorescein succinimidyl ester (CFSE; 0·5 mm) was added to the cell suspensions, and the mixture was incubated for 10 min at 37°. The labelling reaction was stopped by repetitive washing with ice-cold RPMI-1640 medium containing 10% fetal calf serum. Labelled cells (4 × 106/ml) were cultured with various concentrations of antigen for 2, 3 or 4 days. Cultured cells were harvested, washed and labelled with anti-TCR Vβ11 antibodies. The number of cell divisions was determined by dilution of the intracellular dye CFSE in TCR Vβ11+ gated T cells by flow cytometry. In some experiments, Bodipy-FL (Invitrogen, Carlsbad, CA) was used for the cell proliferation assay instead of CFSE.
Results
HBeAg is tolerogenic in HBeAg × 7/16-5 TCR-dbl-Tg mice
To measure the effect of HBeAg on effector T-cell activation in vitro, we cultured splenocytes from naive 7/16-5 TCR-Tg and 7/16-5 × HBeAg dbl-Tg mice in the presence of 1 μg/ml of the HBeAg-derived peptide p120–140 and analysed CD69 expression by FACS. There was a dramatic difference between T-cell activation in 7/16-5 TCR-Tg mice versus 7/16-5 × HBeAg dbl-Tg mice. A high percentage (59·30%) of T cells derived from 7/16-5 TCR-Tg mice expressed CD69 after 3 days in culture, which was sustained after 6 days. In contrast, only 18·56% of T cells derived from HBeAg × 7/16-5 dbl-Tg mice expressed CD69 at 3 days of culture and the expression of CD69 remained low (11·17%) at day 6 (Fig. 1a). Similarly, in vitro IL-2 production by 7/16-5 × HBeAg dbl-Tg splenic T cells cultured with either HBeAg or HBcAg was significantly reduced compared with T cells from 7/16-5 single TCR-Tg mice (Fig. 1b). It is notable that the tolerance exhibited in 7/16-5 × HBeAg dbl-Tg mice is not the result of clonal deletion of 7/16-5 TCR-Tg T cells either in the thymus or in the spleen (data not shown,30).
Figure 1.
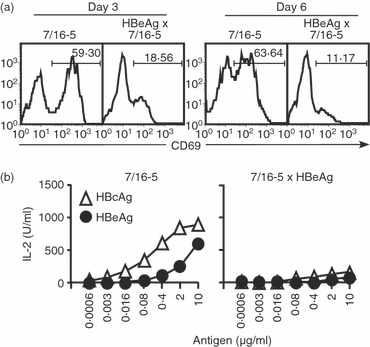
Evidence of T-cell tolerance in in vitro cultures of splenocytes from naive hepatitis B virus e antigen (HBeAg) × 7/16-5 T-cell receptor (TCR) double transgenic (dbl-Tg) mice. Splenocytes from 7/16-5 TCR-Tg mice and HBeAg × 7/16-5 dbl-Tg mice were collected and (a) 4 × 105 splenocytes/well were cultured in the presence of 1 μg/ml of the HBeAg-derived peptide 120–140 and CD69 expression was analysed by FACS at day 3 and day 6. (b) Supernatants of pooled splenocytes from 7/16-5-Tg and HBeAg × 7/16-5 dbl-Tg mice cultured with various concentrations of the HBc/HBe antigens were collected at day 2 and interleukin-2 (IL-2) was measured by ELISA. The data shown are representative of three independent experiments.
Expansion of an HBeAg-specific TCR+, CD4−/CD8− (DN) population in vitro
CFSE-labelled splenocytes from 7/16-5 TCR-Tg and 7/16-5 × HBeAg dbl-Tg mice and 7/16-5 × HBcAg dbl-Tg mice were cultured in vitro in the presence of HBeAg peptide p120–140 to examine CD4+ and CD8+ effector cell proliferation. As shown in Fig. 2, after 4 days in culture both CD4+ and CD8+ Vβ11+ TCR-Tg T cells from 7/16-5 mice and 7/16-5 × HBc dbl-Tg mice proliferated. In contrast, the vast majority of the proliferating Vβ11+ TCR-Tg T cells from 7/16-5 × HBeAg dbl-Tg mice were neither CD4+ cells nor CD8+ cells (Fig. 2, middle column) and represented a DN, Vβ11+ population. Interestingly, this predominant Vβ11+/DN T-cell population can be expanded only from 7/16-5 TCR-Tg mice also expressing the HBeAg, but not from HBcAg-expressing 7/16-5 TCR-dbl-Tg mice or single 7/16-5 TCR-Tg mice (Fig. 2). This indicates the absolute requirement for the presence of HBeAg in vivo for the development of HBeAg-specific DN T cells in the TCR-Tg model. To determine if the proliferation of DN T cells was MHC class II restricted, we added anti-MHC class II and anti-MHC class I antibodies in the culture compared with an isotype control. Anti-MHC class II antibodies (anti-I-Ab) completely inhibit the proliferation of DN T cells, whereas anti-MHC class I antibodies had no effect (data not shown). Therefore, DN T cells proliferate in an MHC class II-restricted manner.
Figure 2.
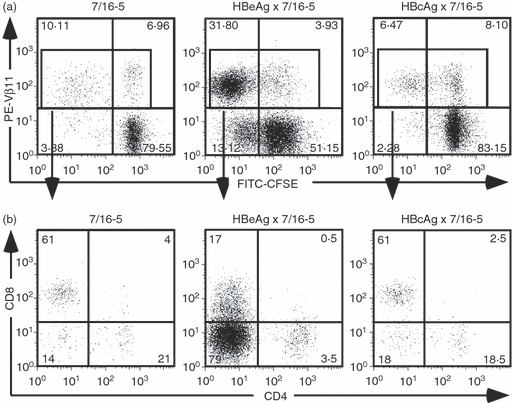
Efficient proliferation of hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T cells. (a) Splenocytes from 7/16-5 transgenic (Tg), HBeAg × 7/16-5 double (dbl) -Tg and HBcAg × 7/16-5 dbl-Tg mice were harvested and labelled with CFSE then cultured in the presence of 1 μg/ml of HBeAg peptide 120–140 for 4 days. (b) Proliferating T-cell receptor (TCR) -Tg (Vβ11+) T cells were gated and further analysed for CD4 and CD8 expression. Data shown are representative of more than three independent experiments.
DN T cells are a phenotypically distinct population
We next examined the cell surface markers of DN T cells. Cells were harvested from a 4-day spleen culture of 7/16-5 × HBeAg dbl-Tg mice, then negatively depleted of CD4+, CD8+, B220+, CD11c+ and Gr-1+ cells. The majority of cells were harvested as flow through, and these cells were collected as purified DN T cells. As expected from the FACS analysis, approximately 50% of total cells harvested were DN T cells. The subsequent FACS analysis revealed that the Vβ11+ DN T cells were Thy-1.2+ (data not shown), B220−, PD-1+, GITRhigh and CD25low (Fig. 3a), and CD49b (DX-5)− (data not shown). Interestingly, the CD25 expression on DN T cells was very low, but PD-1, which is known as an inhibitory co-stimulatory molecule, was highly expressed (51·49%). Therefore, autocrine consumption of IL-2 in the culture environment may not be the mechanism driving the proliferation of DN T cells. A DN Treg cell phenotype has been reported previously;19,21,36 however, the previously reported DN Treg cells highly expressed CD25 and produced IL-2 and IFN-γ, whereas the HBeAg-specific, Vβ11+, DN T cells have low expression of CD25 and no detectable IL-2 and IFN-γ production after in vitro activation (see below and Fig. 4). In addition to this unique phenotype, HBeAg-specific DN T cells proliferate in vitro very efficiently compared with the anergic status of most Treg cells in vitro (see Fig. 2).
Figure 3.
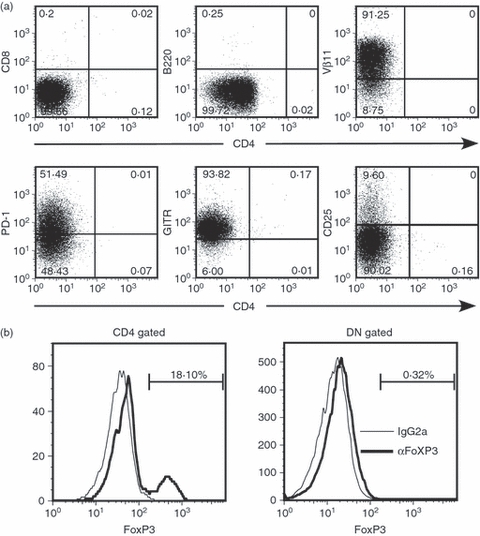
Surface phenotype of hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T cells. (a) After 4 days of culture with 1 μg/ml HBeAg peptide 120–140, splenocytes from HBeAg × 7/16-5 dbl-Tg mice were collected and incubated with biotinylated anti-CD4, -CD8, -B220, -CD11c, -CD11b and -Gr-1 followed by incubation with streptavidin-Microbeads (Miltenyi Biotec) for negative depletion. The indicated cell surface markers were analysed on the purified HBeAg-specific DN T-cell population by FACS, and revealed > 98% purity. (b) Intracellular staining was performed for FoxP3 detection. CD4 cells and DN cells were gated respectively and FoxP3-positive cells were compared with the isotype control.
Figure 4.
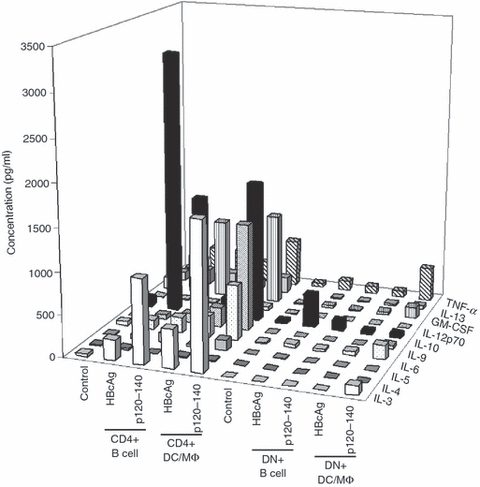
Cytokine profile of hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T cells. HBeAg-specific DN T-cell progenitor cells were harvested from HBeAg × 7/16-5 double transgenic (dbl-Tg) mice as described in the Materials and methods. HBeAg-specific DN T-cell progenitor cells were cultured with B cells or dendritic cell/ macrophages (DC/MΦ) in the presence of HBcAg or p120–140 for 4 days. Supernatants were analysed for the indicated cytokines using Multiflex Biomarker Immunoassay (Millipore, MA). HBeAg-specific CD4+ T cells from 7/16-5-Tg mice were cultured identically for comparison.
CTLA-4 is often expressed by cTreg cells and may play an important role in the suppressive function of Treg cells.14,37–39 However, HBeAg-specific Vβ11+ DN T cells do not express CTLA-4 (data not shown). Conventional Treg cells also express FoxP3 in the cytoplasm, which can represent a specific marker for cTreg cells. FoxP3 can also be involved in the generation of Treg cells as shown in an FoxP3 expression model in vitro.17 To investigate the expression of FoxP3 in DN cells, intracellular FACS staining was performed, however, no detectable FoxP3 was observed in HBeAg-specific, Vβ11+ DN T cells (Fig. 3b).
Cytokine profile of HBeAg-specific DN cells at day 4 of culture
Because cytokines other than IL-2 may be involved in the proliferation of T cells, we have examined the cytokine production profile of in vitro cultured HBeAg-specific DN T cells, using the Multiflex Biomarker Immunoassay (Fig. 4). After 4 days of culture with p120–140, DN T cells derived from 7/16-5 × HBeAg dbl-Tg mice were compared with 7/16-5 TCR-Tg-derived CD4 T cells. Although a variety of cytokines were produced by 7/16-5 CD4+ T cells after in vitro culture with both HBcAg and p120–140 peptide presented by either B cells or dendritic cell (DC)/macrophage (MΦ) APCs, no significant production of cytokines was detected in the culture of HBeAg-specific DN T cells.
DN cells inhibit the cytokine production of HBeAg-specific and HBeAg-non-specific effector cells
Because HBeAg-specific DN T cells predominate in a 4-day culture and are only observed in HBeAg-expressing dbl-Tg mice, we examined the possibility that the DN T cells possessed regulatory activity. In previous unpublished experiments, total spleen cells from 7/16-5 × HBeAg dbl-Tg mice inhibited the HBeAg-specific production of cytokines by 7/16-5 effector cells, whereas, fractionated CD4+, CD8+ or both did not inhibit the activation of effector cells. Therefore, we fractionated the DN T cells from 4-day HBeAg-specific cultures and co-cultured the DN, Vβ11+ T cells with 7/16-5 effector T cells in the presence of p120–140 and measured antigen-specific expansion and cytokine (i.e. IL-2 and IFN-γ) production by the 7/16-5 T cells. As shown in Fig. 5(a), the cytokine production of the 7/16-5 effector T cells was dramatically suppressed by the DN T cells, and the proliferation of the CD4+, Vβ11+ effector T cells was also inhibited even at an effector cell : Treg cell ratio as low as 32 : 1. This is a very low ratio of Treg cells to effector cells and indicates potent regulatory activity by the DN T cells. Further studies will be needed to clarify the precise mechanism of suppression. These data indicate that the DN T cells are HBeAg-specific, highly proliferative and effective suppressors, which defines a unique population of HBeAg-induced Treg cells in 7/16-5 × HBeAg dbl-Tg mice.
Figure 5.
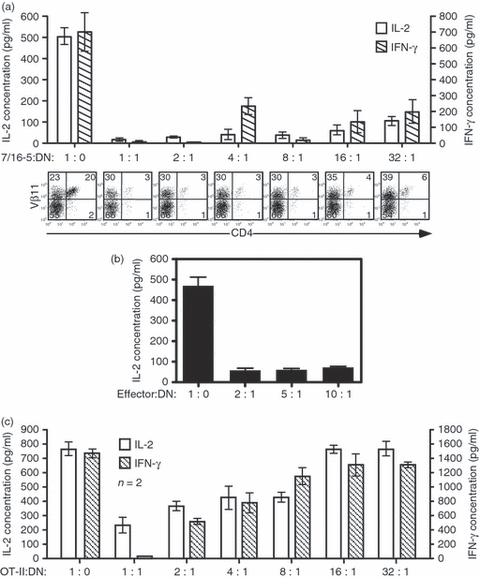
The hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T-cell population inhibits cytokine production and proliferation of HBeAg-specific CD4+ effector T cells. (a) After 4 days of culture with p120–140, DN T cells were purified by negative selection and added to naïve 7/16-5-transgenic (Tg) spleen cells (effector T-cell culture) at the indicated ratios and p120–140 was added. Supernatants from cultured wells were collected at day 2 and day 4 for interleukin-2 (IL-2) and interferon-γ (IFN-γ) analysis, respectively. Cytokines were measured by ELISA. Remaining cells were analysed by FACS for CD4 and Vβ11 as an indicator of effector cell proliferation. (b) Spleen cells from HBeAg-immunized B10 wild-type (WT) mice were collected and cultured in vitro in the presence of 5 μg/ml of HBeAg and varying numbers of DN T cells. Supernatants were collected at day 2 for IL-2 analysis. (c) Spleen cells from ovalbumin-specific OT-II mice were collected and cultured with class II-restricted ovalbumin (323–339) peptide and in vitro activated DN T cells as described in (a). Supernatants from cultured wells were collected at day 2 and day 4 for IL-2 and IFN-γ analysis, respectively. Cytokines were measured by ELISA. Data are expressed as mean value ± SD from three independent experiments.
To investigate whether this suppression by DN T cells is only specific for the 7/16-5 Tg-TCR, we investigated the inhibitory effect of DN T cells on a polyclonal HBeAg-specific T-cell population. We immunized B10 mice with 20 μg HBeAg to prime polyclonal HBeAg-specific T cells, and harvested spleen cells after 10 days and restimulated the spleen cells in the presence of HBeAg and the indicated numbers of DN T cells. As shown in Fig. 5(b), even at a 10 : 1 effector : DN T-cell ratio, IL-2 production was effectively suppressed indicating that the Treg cell activity is functional for a polyclonal HBeAg-specific CD4+ T-cell response, and is not restricted to 7/16-5 Tg-TCR-bearing effector cells. Furthermore, to confirm whether this inhibitory effect is HBeAg specific or not, we investigated the inhibitory effect of DN T cells on cytokine production in an unrelated MHC class II-restricted TCR-Tg lineage, OT-II (Fig. 5c). The DN T cells activated in vitro inhibited the production of IL-2 from OT-II effector cells at a ratio of 8 : 1 (effector cell : regulatory cell) at day 2. Similar inhibitory effects were observed in IFN-γ production at day 4. Therefore, while activation of the DN T cells is HBeAg-specific, once activated the suppressive function is non-specific. However, it is also notable that the inhibitory effect of DN T cells in an antigen-specific setting is superior to non-specific inhibition.
The DN T-cell population originates from DN progenitor T cells
As a result of the vigorous HBeAg-specific proliferative property of the DN T-cell population during in vitro culture, it is possible that the DN cells are derived from HBeAg-specific CD4+, CD8+ or from an independent DN progenitor population. To determine the origin of the DN T-cell population, depletion of T-cell subpopulations from total spleen of HBeAg × 7/16-5 dbl-Tg mice was performed and the remaining cells were cultured in vitro with p120–140 for 4 days and compared with total spleen cells. As shown in Fig. 6, CD4+ and CD8+ T-cell depletion from total spleen did not affect the generation of the DN T-cell population in the culture (i.e. 40–48%). However, negative depletion of DN T cells before culture prevented the generation of the HBeAg-specific DN T-cell population in the 4-day culture (i.e. 8%). Hence, HBeAg-specific DN T cells exist in the periphery and are not generated from CD4+ or CD8+ T cells in the periphery. It is notable that without DN T cells, CD4+ T cells from HBeAg × 7/16-5 dbl-Tg mice demonstrate robust proliferation and cytokine production in vitro (data not shown).
Figure 6.
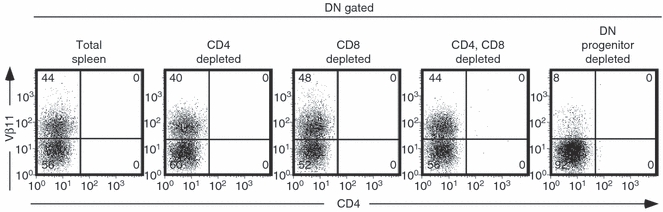
The hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T-cell population originates from DN progenitor T cells. HBeAg × 7/16-5 double transgenic (dbl-Tg) splenocytes were harvested and prepared as single cell suspensions. The CD4+, CD8+ or both CD4+/CD8+ T cells were depleted, respectively, using magnetic beads, and flow through from each preparation was cultured. For the progenitor DN-depleted culture, positively purified CD4+ and CD8+ cells were cultured with antigen-presenting cells. The HBeAg-specific DN progenitor cells were depleted as described in the Materials and methods. Each preparation of cells was cultured in the presence of 2 μg/ml of p120–140 for 4 days. Cells were gated for DN (CD4− and CD8−) and further analysed for Vβ11 positivity.
The DN T cells expand in vivo in the presence of high concentrations of exogenous p120–140
The frequency of Vβ11+ DN T cells in thymus and spleen ex vivo was measured. The Vβ11+ DN T cells in thymus of 7/16-5 × HBeAg dbl-Tg mice were present at a slightly higher frequency (5% higher) than in the thymus of 7/16-5 × HBcAg dbl-Tg mice. There was also a 20% higher frequency of Vβ11+ DN T cells in the ex vivo spleens of 7/16-5 × HBeAg compared with the spleens of 7/16-5 single TCR-Tg mice (data not shown). However, in absolute terms DN Vβ11+ T cells are present in low frequency in situ in HBeAg × 7/16-5 dbl-Tg mice (i.e. 3–5%) and require antigen stimulation for expansion. It is not clear if DN T cells can proliferate and be activated in vivo. To determine the capability of DN T cells to expand in vivo, we injected HBeAg-derived p120–140 (250 μg) into 7/16-5 × HBeAg-dbl Tg mice. As shown in Fig. 7, at least a twofold increase in the DN T-cell frequency in vivo was observed 1 and 2 weeks after injection, whereas in control, 7/16-5 mice no evidence of expansion of DN T cells occurred. Although HBeAg-specific Treg cells appear quiescent in vivo in HBeAg × 7/16-5 dbl-Tg mice, these cells are capable of being activated in vivo, in this case by exogenous antigen. The ability to activate DN T cells in vivo will permit further studies of their in vivo function.
Figure 7.
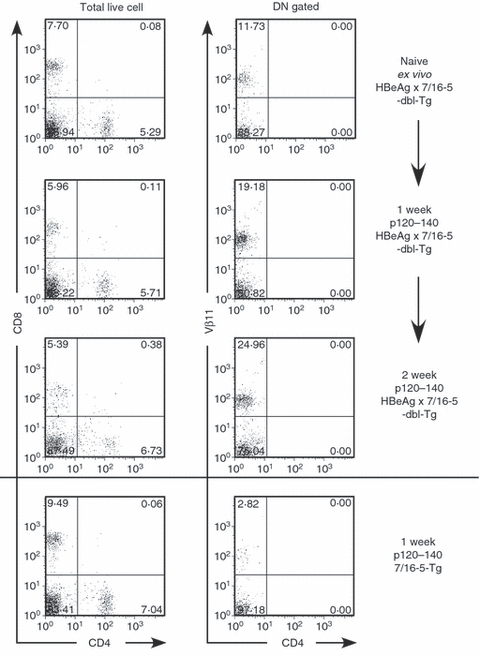
Injection of of hepatitis B virus e antigen (HBeAg) -derived p120–140 peptide triggers in vivo expansion of double-negative (DN) T cells. HBeAg × 7/16-5 double transgenic (dbl-Tg) mice were injected with 250 μg of HBeAg-derived p120–140 at the indicated times. One week and 2 weeks after peptide injection, mice were killed and splenocytes were analysed for the DN T-cell population by FACS analysis. The parallel experiment was performed in 7/16-5 Tg mice (bottom panel).
MHC class I molecules and mature CD8+ T cells and/or endogenous TCR α-chains do not directly affect the presence of DN T cells in the periphery
To further pursue the origins of DN T cells, we bred 7/16-5 × HBeAg dbl-Tg mice onto MHC class I KO, and TCR α-chain KO backgrounds. Because the 7/16-5 TCR is surprisingly expressed on CD8+ as well as CD4+ T cells in the thymus and the periphery, it was important to determine if expression of CD8 was necessary for selection of the DN T-cell population. The absence of MHC class I molecules and mature CD8+ T cells did not affect the presence of DN T cells in the periphery (Fig. 8a). Furthermore, because it is possible that endogenous TCR α-chains are necessary for DN T-cell selection or function and because we cannot monitor the frequency of the TCR-Tg Vα5 chain by FACS (mAb against Vα5 is not commercially available), we also bred 7/16-5 × HBeAg dbl-Tg mice on to a TCR α-chain KO background. The absence of endogenous TCR α-chains did not affect the presence of DN T cells in the periphery (Fig. 8b). This demonstrates that TCR-Tg Vα5 is sufficient to confer the DN T-cell phenotype.
Figure 8.
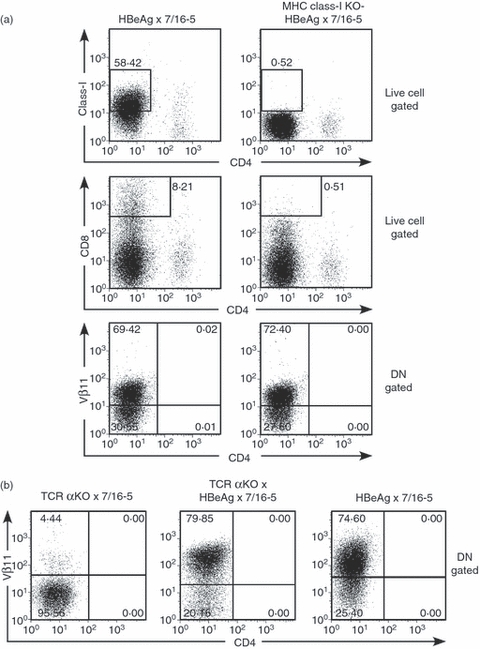
Absence of MHC class I molecules and mature CD8+ T cells and/or endogenous T-cell receptor (TCR) α-chains does not affect the presence of double-negative (DN) T cells in the periphery. (a) Splenocytes from hepatitis B virus e antigen (HBeAg) × 7/16-5 dbl-Tg mice and MHC class I knockout (KO) × HBeAg × 7/16-5 mice were cultured in the presence of HBeAg-derived peptide p120–140 for 4 days. Cells were collected and analysed for class I, CD8 and Vβ11 expression from the DN gated population by FACS. (b) Splenocytes from TCR α-chain KO × HBeAg × 7/16-5 mice and TCR α-chain KO × 7/16-5 and HBeAg × 7/16-5 mice were cultured in the presence of HBeAg-derived peptide p120–140 for 4 days. Cells were collected and analysed for Vβ11 from the DN gated population by FACS.
DN T cells require IL-15 for proliferation that may be produced by DC/MΦ
To examine the APC requirement for DN T-cell expansion, DN progenitor T cells from 7/16-5 × HBeAg dbl-Tg mice were fractionated, and cultured with different APC populations in the presence of HBeAg peptide p120–140. As shown in Fig. 9(a), fractionated B cells do not support the proliferation or survival of DN T cells even with a relatively high concentration of antigen. This is surprising because B cells are the primary APCs for HBcAg and present p120–140 and HBc/HBeAgs efficiently to HBc/HBeAg-specific CD4+ T cells.40,41 To examine non-B-cell APC function, an APC fraction from B-cell KO (μMT) mice was used for co-culture with DN T-cell progenitors. Non-B cells supported the proliferation of DN T cells efficiently even at low concentrations (0·2 μg/ml) of p120–140 peptide (Fig. 9a). It was also interesting that APCs from μMT mice support the survival of DN T cells even in the absence of antigen. It appeared that in the induction phase of DN T-cell expansion, specific soluble factors or surface co-stimulatory molecules from DC or MΦ, but not from B cells specifically support the survival and proliferation of DN T cells.
Figure 9.
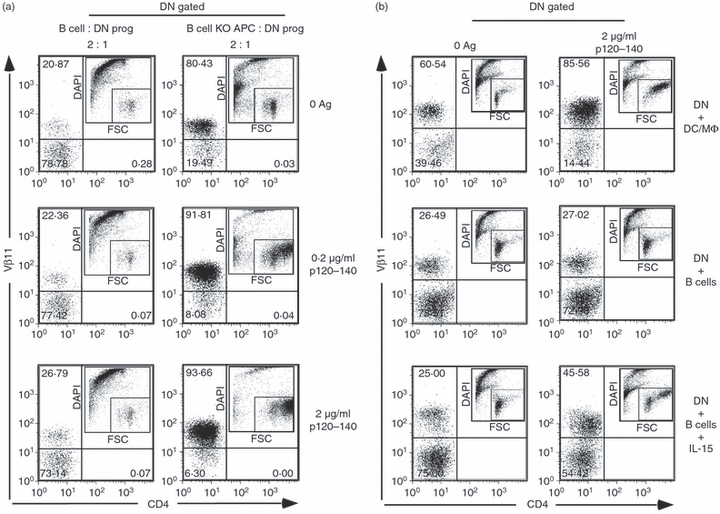
Non-B cells act as antigen-presenting cells (APCs) for hepatitis B virus e antigen (HBeAg) -specific double-negative (DN) T cells, and interleukin-15 (IL-15) is crucial for proliferation. (a) HBeAg-specific DN T cells were purified by negative depletion as described and cultured with purified B cells from B10 mice or T-cell-depleted splenocytes from B-cell knockout (KO) mice in the presence of HBeAg-specific peptide p120–140 at the indicated concentrations. Fluorescently labelled cells were collected by flow cytometry and gated as DN (CD4− and CD8−) and analysed for Vβ11 positivity. The ancestry plot indicates the activation of cells. (b) Recombinant IL-15 (5 μg/ml) was added to HBeAg-specific DN T cells cultured with B-cell APCs, which do not support proliferation, and compared with dendritic cells/macrophages (DC/MΦ) APCs. The ancestry plot indicates the activation of cells. Data shown are representative of multiple independent experiments.
Although IL-2 is not necessary for the proliferation of DN T cells, there are several other soluble factors involved in the proliferation of T cells. Notably, IL-7 and IL-15 are prominent candidates for the induction of IL-2-independent proliferation. Interleukin-7 is known as a regulator of proliferation of T cells, in IL-2-dependent and IL-2-independent circumstances.42 Both IL-15 and IL-7 are also known to mediate homeostatic proliferation of naive T cells.42,43 Additionally, IL-15 is produced by DCs and can have IL-2-like function. To test the effect of IL-7 and IL-15 on the proliferation of DN T cells, we cultured purified DN progenitor cells with different APCs in the presence of antigen and cytokines. As observed in the previous experiment, DC/MΦ supported the proliferation of DN T-cell progenitor cells, whereas B cells did not, even at the higher concentration of antigen. However, when exogenous IL-15 was added to the B-cell APC culture, it rescued the proliferation of DN T-cell progenitor cells in the presence of p120–140 (Fig. 9b). This result suggests that IL-15 produced by DC/MΦ may play an important role in the proliferation of DN T cells. The DN T cells express high levels of IL-15R on their surface, whereas DN gated splenocytes from control mice do not express IL-15R (Fig. 10). IL-7 showed no significant induction of proliferation (data not shown).
Figure 10.
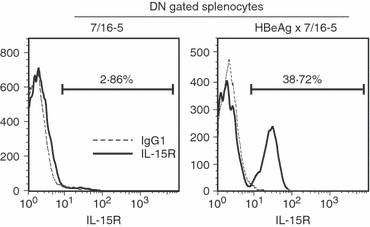
Double-negative (DN) T cells express high levels of interleukin-15 receptor (IL-15R); 4 × 105 splenocytes/well from 7/16-5 and hepatitis B virus e antigen (HBeAg) × 7/16-5 were cultured in 96-well plates for 72 hr with 2 μg/ml of the HBeAg-derived peptide 120–140, and cells were harvested and analysed for surface expression of IL-15R by FACS.
Discussion
Herein, we demonstrated that Vβ11+/Vα5+ DN T cells derived from TCR × HBeAg dbl-Tg mice represent a unique population that possesses a distinctive cell surface marker phenotype, (i.e. TCR+, Thy-1.2+, CD4−, CD8−, CD25low, GITR+, PD-1+, FoxP3−). Furthermore, our data directly show that this DN T-cell population possesses suppressive function against effector T cells specific for the same HBeAg specificity as well as non-specific T cells. In contrast to cTreg cells, the Vβ11+ DN T cells defined in this model system possess a vigorous proliferative capacity upon in vitro antigenic stimulation and represent as much as 70% (it varies between 50 and 70%) of the cells remaining after 4 days of in vitro culture. Those characteristics are unique and a similar Treg cell population has not been previously reported. We therefore refer to this unique population as DN Treg cells. Considering that this DN Treg cell population is only observed in TCR-Tg mice, which also express the secreted HBeAg and their strong suppressive effect, HBeAg-specific DN Treg cells may play a role in tolerance induction by HBeAg in the murine model system. We do not know if an identical DN Treg cell population may exist in chronically infected humans; however, in the mouse model the HBeAg, but not the HBcAg, has the potential to elicit Treg cells in vivo. Therefore, the induction of HBeAg-specific Treg cells may be added to the repertoire of mechanisms by which the secreted HBeAg mediates T-cell tolerance. Recent publications have suggested that Treg cells may contribute to impaired immune function in an HBV-Tg mouse model 44 and in chronic HBV patients.45–47 Furthermore, in one study, in which the T-cell response to HBcAg was studied, an increase in Treg cell frequency and function was observed in HBeAg-positive compared with HBeAg-negative patients, suggesting a role for HBeAg.46 The previous studies of Treg cells in either an HBV-Tg mouse model or HBV patients have concentrated exclusively on CD25+ Treg cells or cTreg cells. The HBeAg-specific DN Treg cells observed in the 7/16-5 × HBeAg dbl-Tg mouse model may serve as a useful tool to study the functional characteristics of HBeAg-specific Treg cells in general such as clonal expansion and mechanisms of suppression, which may have implications for viral persistence during natural HBV infection. However, to relate the presence and function of DN Treg cells to T-cell tolerance and chronicity in HBV infection will require further studies.
In contrast to anergic cTreg cells that lack efficient in vitro expansion, HBeAg-specific DN Treg cells proliferate vigorously in vitro, suggesting that this DN Treg cell population may be a useful tool to elucidate the proliferative potential of Treg cells in general. The mechanism by which HBeAg-specific DN Treg cells proliferate is not yet clear; however, preliminary data suggest that IL-15 produced by DC/MΦ plays an important role in the proliferation of DN Treg cells. There may be other possible factors that promote the proliferation of DN Treg cells in combination with IL-15, possibly other cytokines or co-stimulatory molecules that deliver signals to DN Treg cells. This is the subject of ongoing investigations.
The function of Treg cells has been described, both in vitro and in vivo. It has been proposed that Treg cells function as modulators of autoimmune responses because of their suppressive effect on autoreactive lymphocytes. Furthermore, this suppressive function can be transferred by injecting Treg cells into autoimmune animal model systems.7 The Treg cells have also been shown to function in many non-autoimmune models such as graft-versus-host disease and allergy.48–51 In contrast, Treg cells can interrupt the activation of effector T cells responding to tumour cells and infectious pathogens.46 However, clinical applications using Treg cell suppressive function have been limited because of the hypoproliferative property and polyclonal nature of Treg cells. In vitro studies using cTreg cells show that only a relatively high ratio of Treg : effector cells can suppress the effector cells (i.e. 5 : 1 to 1 : 1). As a result of this inefficient in vitro suppression, the therapeutic potential of Treg cells has been critically limited. However, HBeAg-specific DN Treg cells demonstrate superior suppressive effects on effector cells at effector cell : Treg cell ratios as low as 32 : 1 (see Fig. 5).
The multiple mechanisms of suppression used by Treg cells is an ongoing subject of research and remains somewhat controversial. The suppressive effects of cTreg cells in vitro have been reported mostly on CD4+ and CD8+ effector cells, but have also been found to act directly on APCs and natural killer cells.52–56 Inhibitory cytokines, IL-10 and transforming growth factor(TGF)-β are known to be produced by cTreg cells and thought to be a part of the mechanism of Treg cells.57,58 According to our preliminary data in a transwell system, IL-10 and TGF-β are not candidates as the primary mediators of suppression demonstrated by HBeAg-specific DN Treg cells (data not shown). Another report showed that the regulatory function of Treg cells is serine protease granzyme-B (GZ-B)-dependent using GZ-B−/− mice.59 Other suppressive mechanisms have been suggested to function via cell–cell contact. CTLA-4, FAS–FASL, GITR and CD103 have also been suggested to play a role in the function of Treg cells. Recently, the inhibitory function of Treg cells has been demonstrated to be mediated through the exoenzymes CD73/CD39.60–62 Interestingly, a high frequency of HBeAg-specific DN Treg cells are CD73+/CD39+ after activation (Fig. 11). We are investigating whether this pathway may explain the efficient immunoregulation mediated by HBeAg-specific DN Treg cells.
Figure 11.
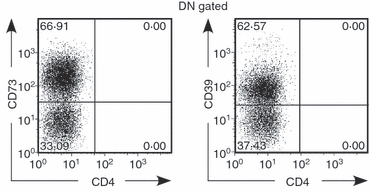
Double-negative (DN) T cells express high levels of ectoenzyme CD73 and CD39 4 × 105 splenocytes/well from hepatitis B virus e antigen (HBeAg) × 7/16-5 were cultured in 96-well plates for 72 hr with 2 μg/ml of HBeAg-derived peptide 120–140, and cells were harvested and analysed for surface expression of CD73 and CD39 by FACS.
It is likely that HBeAg-specific DN Treg cells result from positive selection in the thymus or as a result of continuous exposure to the HBeAg in the periphery but are not derived from CD4+ or CD8+ HBeAg-specific T cells in the periphery. It is interesting that the 7/16-5 TCR is expressed on CD8+ T cells as well as CD4+ T cells although both CD4+ and CD8+ T cells are specific for p120–140 in the context of MHC class II molecules (I-Ab). It is possible that the 7/16-5 TCR may also recognize a self-peptide in the context of MHC class I molecules in the thymus with sufficient affinity to be selected on MHC class I. To address this question, we bred 7/16-5 × HBeAg dbl-Tg mice on a MHC class I negative background. While HBeAg × 7/16-5 dbl-Tg mice on a MHC class I KO background do not produce mature CD8+ T cells in the periphery, HBeAg-specific DN T cells are produced, and are, therefore, not dependent on MHC class I or CD8 expression. Endogenous TCR-α chains also do not affect the presence of DN T cells in the periphery.
At present, we have no direct evidence to address whether this DN Treg cell population is unique to this model or not. The frequency of this population is low in situ in 7/16-5 × HBeAg dbl-Tg mice and their presence in other systems may be difficult to detect. The 7/16-5 × HBeAg dbl-Tg mice may be a useful model for low-affinity self-reactive T cells that escape deletion in the thymus and are quiescent in the periphery until activated (i.e. tissue injury, mimicked here by high concentrations of peptide in vitro or in vivo). Most dbl-Tg mice are models of high-affinity self-reactive T cells, which are largely deleted in vivo. It is anticipated that further characterization of this low-affinity DN Treg cell population may yield a phenotypic marker that would allow identification in other systems.
Recent publications have suggested that Treg cells may contribute to impaired immune function in an HBV-Tg mouse model 44 and in patients with chronic HBV.45–47 Furthermore, in one study, in which the T-cell response to HBcAg was studied, an increase in Treg cell frequency and function was observed in HBeAg-positive patients compared with HBeAg-negative patients, suggesting a role for HBeAg.46 Previous studies of Treg cells in either an HBV-Tg mouse model or HBV-infected patients have concentrated exclusively on CD25+ Treg cells or cTreg cells. The HBeAg-specific DN Treg cells observed in the 7/16-5 × HBeAg dbl-Tg mouse model may serve as a useful tool to study functional characteristics of HBeAg-specific Treg cells in general such as clonal expansion and mechanisms of suppression, which may have implications for viral persistence during natural HBV infection.
Acknowledgments
We thank David Chambers and Jonna Barrie for operating the Salk Institute Flow Cytometry facility, Darrell Peterson (Virginia Commonwealth University) for providing recombinant HBcAg and Frank Chisari (The Scripps Research Institute) for providing HBc/HBeAg-Tg mice. This work was supported by the National Institutes of Health grants AI 20720-28, and AI 049730-08.
Glossary
Abbreviations
- APC
antigen-presenting cell
- CD62L
CD62 ligand
- CTLA-4
cytotoxic T-lymphocyte antigen-4
- DC
dendritic cell
- DN
double-negative
- FoxP3
Forkhead box P3
- GITR
glucocorticoid-induced tumour necrosis factor receptor-related protein
- HBcAg
hepatitis B core antigen
- HBeAg
hepatitis B e antigen
- HBV
hepatitis B virus
- IL-2
interleukin-2
- IFN
interferon
- mAb
monoclonal antibody
- MΦ
macrophage
- TCR
T-cell receptor
- Tg
transgenic
- Treg cell
regulatory T cell
Disclosures
The authors have no conflicts of interests to declare.
References
- 1.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 3.von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol. 2003;3:223–32. doi: 10.1038/nri1029. [DOI] [PubMed] [Google Scholar]
- 4.O'Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10:801–5. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 5.Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3:756–63. doi: 10.1038/ni816. [DOI] [PubMed] [Google Scholar]
- 6.Gershon RK, Kondo K. Infectious immunological tolerance. Immunology. 1971;21:903–14. [PMC free article] [PubMed] [Google Scholar]
- 7.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 8.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–53. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+ CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–42. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 10.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4+ CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 11.Lehmann J, Huehn J, de la Rosa M, et al. Expression of the integrin αEβ7 identifies unique subsets of CD25+ as well as CD25– regulatory T cells. Proc Natl Acad Sci U S A. 2002;99:13031–6. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+ CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruder D, Probst-Kepper M, Westendorf AM, et al. Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol. 2004;34:623–30. doi: 10.1002/eji.200324799. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005;202:901–6. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 18.Yagi H, Nomura T, Nakamura K, et al. Crucial role of FOXP3 in the development and function of human CD25+ CD4+ regulatory T cells. Int Immunol. 2004;16:1643–56. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med. 2000;6:782–9. doi: 10.1038/77513. [DOI] [PubMed] [Google Scholar]
- 20.Chen W, Zhou D, Torrealba JR, Waddell TK, Grant D, Zhang L. Donor lymphocyte infusion induces long-term donor-specific cardiac xenograft survival through activation of recipient double-negative regulatory T cells. J Immunol. 2005;175:3409–16. doi: 10.4049/jimmunol.175.5.3409. [DOI] [PubMed] [Google Scholar]
- 21.Fischer K, Voelkl S, Heymann J, et al. Isolation and characterization of human antigen-specific TCR αβ+ CD4– CD8– double-negative regulatory T cells. Blood. 2005;105:2828–35. doi: 10.1182/blood-2004-07-2583. [DOI] [PubMed] [Google Scholar]
- 22.Lee BP, Mansfield E, Hsieh SC, et al. Expression profiling of murine double-negative regulatory T cells suggest mechanisms for prolonged cardiac allograft survival. J Immunol. 2005;174:4535–44. doi: 10.4049/jimmunol.174.8.4535. [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Ford MS, Young KJ, Cybulsky MI, Zhang L. Role of double-negative regulatory T cells in long-term cardiac xenograft survival. J Immunol. 2003;170:1846–53. doi: 10.4049/jimmunol.170.4.1846. [DOI] [PubMed] [Google Scholar]
- 24.Ma Y, He KM, Garcia B, Min W, Jevnikar A, Zhang ZX. Adoptive transfer of double negative T regulatory cells induces B-cell death in vivo and alters rejection pattern of rat-to-mouse heart transplantation. Xenotransplantation. 2008;15:56–63. doi: 10.1111/j.1399-3089.2008.00444.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang ZX, Ma Y, Wang H, et al. Double-negative T cells, activated by xenoantigen, lyse autologous B and T cells using a perforin/granzyme-dependent, Fas-Fas ligand-independent pathway. J Immunol. 2006;177:6920–9. doi: 10.4049/jimmunol.177.10.6920. [DOI] [PubMed] [Google Scholar]
- 26.Ford McIntyre MS, Gao JF, Li X, Naeini BM, Zhang L. Consequences of double negative regulatory T cell and antigen presenting cell interaction on immune response suppression. Int Immunopharmacol. 2011;11:597–603. doi: 10.1016/j.intimp.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 27.Gao JF, McIntyre MS, Juvet SC, et al. Regulation of antigen-expressing dendritic cells by double negative regulatory T cells. Eur J Immunol. 2011 doi: 10.1002/eji.201141428. EPub ahead of print. [DOI] [PubMed] [Google Scholar]
- 28.Zhang ZX, Lian D, Huang X, et al. Adoptive transfer of DNT cells induces long-term cardiac allograft survival and augments recipient CD4+ Foxp3+ Treg cell accumulation. Transpl Immunol. 24:119–26. doi: 10.1016/j.trim.2010.11.003. ???? [DOI] [PubMed] [Google Scholar]
- 29.Chen M, Sallberg M, Thung SN, Hughes J, Jones J, Milich DR. Nondeletional T-cell receptor transgenic mice: model for the CD4+ T-cell repertoire in chronic hepatitis B virus infection. J Virol. 2000;74:7587–99. doi: 10.1128/jvi.74.16.7587-7599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, Sallberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol. 2005;79:3016–27. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen MT, Billaud JN, Sallberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A. 2004;101:14913–8. doi: 10.1073/pnas.0406282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guidotti LG, Martinez V, Loh YT, Rogler CE, Chisari FV. Hepatitis B virus nucleocapsid particles do not cross the hepatocyte nuclear membrane in transgenic mice. J Virol. 1994;68:5469–75. doi: 10.1128/jvi.68.9.5469-5475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guidotti LG, Matzke B, Pasquinelli C, Shoenberger JM, Rogler CE, Chisari FV. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. J Virol. 1996;70:7056–61. doi: 10.1128/jvi.70.10.7056-7061.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schodel F, Peterson D, Zheng J, Jones JE, Hughes JL, Milich DR. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem. 1993;268:1332–7. [PubMed] [Google Scholar]
- 35.Sallberg M, Ruden U, Magnius LO, Norrby E, Wahren B. Rapid “tea-bag” peptide synthesis using 9-fluorenylmethoxycarbonyl (Fmoc) protected amino acids applied for antigenic mapping of viral proteins. Immunol Lett. 1991;30:59–68. doi: 10.1016/0165-2478(91)90090-w. [DOI] [PubMed] [Google Scholar]
- 36.Priatel JJ, Utting O, Teh HS. TCR/self-antigen interactions drive double-negative T cell peripheral expansion and differentiation into suppressor cells. J Immunol. 2001;167:6188–94. doi: 10.4049/jimmunol.167.11.6188. [DOI] [PubMed] [Google Scholar]
- 37.Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-β in CD4+ CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 38.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+ CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–83. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manzotti CN, Tipping H, Perry LC, Mead KI, Blair PJ, Zheng Y, Sansom DM. Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol. 2002;32:2888–96. doi: 10.1002/1521-4141(2002010)32:10<2888::AID-IMMU2888>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 40.Milich DR, Chen M, Schodel F, Peterson DL, Jones JE, Hughes JL. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci U S A. 1997;94:14648–53. doi: 10.1073/pnas.94.26.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee BO, Tucker A, Frelin L, et al. Interaction of the hepatitis B core antigen and the innate immune system. J Immunol. 2009;182:6670–81. doi: 10.4049/jimmunol.0803683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A. 2001;98:8732–7. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–32. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roh S, Kim K. Overcoming tolerance in hepatitis B virus transgenic mice: a possible involvement of regulatory T cells. Microbiol Immunol. 2003;47:453–60. doi: 10.1111/j.1348-0421.2003.tb03370.x. [DOI] [PubMed] [Google Scholar]
- 45.Franzese O, Kennedy PT, Gehring AJ, Gotto J, Williams R, Maini MK, Bertoletti A. Modulation of the CD8+ T-cell response by CD4+ CD25+ regulatory T cells in patients with hepatitis B virus infection. J Virol. 2005;79:3322–8. doi: 10.1128/JVI.79.6.3322-3328.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, Janssen HL. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology. 2005;41:771–8. doi: 10.1002/hep.20649. [DOI] [PubMed] [Google Scholar]
- 47.Barboza L, Salmen S, Goncalves L, et al. Antigen-induced regulatory T cells in HBV chronically infected patients. Virology. 2007;368:41–9. doi: 10.1016/j.virol.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 48.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+ CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–50. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 49.Ermann J, Hoffmann P, Edinger M, et al. Only the CD62L+ subpopulation of CD4+ CD25+ regulatory T cells protects from lethal acute GVHD. Blood. 2005;105:2220–6. doi: 10.1182/blood-2004-05-2044. [DOI] [PubMed] [Google Scholar]
- 50.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4+ CD25+ regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389–99. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann P, Ermann J, Edinger M. CD4+ CD25+ regulatory T cells in hematopoietic stem cell transplantation. Curr Top Microbiol Immunol. 2005;293:265–85. doi: 10.1007/3-540-27702-1_12. [DOI] [PubMed] [Google Scholar]
- 52.Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol. 2005;175:4180–3. doi: 10.4049/jimmunol.175.7.4180. [DOI] [PubMed] [Google Scholar]
- 53.Smyth MJ, Teng MW, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+ CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol. 2006;176:1582–7. doi: 10.4049/jimmunol.176.3.1582. [DOI] [PubMed] [Google Scholar]
- 54.Azuma T, Takahashi T, Kunisato A, Kitamura T, Hirai H. Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res. 2003;63:4516–20. [PubMed] [Google Scholar]
- 55.Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+ CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol. 2004;172:4676–80. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 56.Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macrophage function by human CD4+ CD25+ regulatory T cells. Hum Immunol. 2005;66:222–30. doi: 10.1016/j.humimm.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groux H. Type 1 T-regulatory cells: their role in the control of immune responses. Transplantation. 2003;75:8S–12S. doi: 10.1097/01.TP.0000067944.90241.BD. [DOI] [PubMed] [Google Scholar]
- 58.Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 59.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+ CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–6. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 60.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 61.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–32. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]