

Abstract
BACKGROUND AND PURPOSE
Depolarization-induced suppression of inhibition (DSI) and excitation (DSE) are two forms of cannabinoid CB1 receptor-mediated inhibition of synaptic transmission, whose durations are regulated by endocannabinoid (eCB) degradation. We have recently shown that in cultured hippocampal neurons monoacylglycerol lipase (MGL) controls the duration of DSE, while DSI duration is determined by both MGL and COX-2. This latter result suggests that DSE might be attenuated, and excitatory transmission enhanced, during inflammation and in other settings where COX-2 expression is up-regulated.
EXPERIMENTAL APPROACH
To investigate whether it is possible to control the duration of eCB-mediated synaptic plasticity by varied expression of eCB-degrading enzymes, we transfected excitatory autaptic hippocampal neurons with putative 2-AG metabolizing enzymes: COX-2, fatty acid amide hydrolase (FAAH), α/β hydrolase domain 6 (ABHD6), α/β hydrolase domain 12 (ABHD12) or MGL.
KEY RESULTS
We found that overexpression of either COX-2 or FAAH shortens the duration of DSE while ABHD6 or ABHD12 do not. In contrast, genetic deletion (MGL−/−) and overexpression of MGL both radically altered eCB-mediated synaptic plasticity.
CONCLUSIONS AND IMPLICATIONS
We conclude that both FAAH and COX-2 can be trafficked to neuronal sites where they are able to degrade eCBs to modulate DSE duration and, by extension, net endocannabinoid signalling at a given synapse. The results for COX-2, which is often up-regulated under pathological conditions, are of particular note in that they offer a mechanism by which up-regulated COX-2 may promote neuronal excitation by suppressing DSE while enhancing conversion of 2-AG to PGE2-glycerol ester under pathological conditions.
Keywords: endocannabinoid, COX-2, ABHD6, ABHD12, 2-AG, DSE, retrograde transmission
Introduction
Inflammation features prominently in numerous neuropathologies and is a focus of intense scientific scrutiny in the hope of developing more effective therapeutic agents (Dantzer et al., 1998). The COXs, particularly COX-2, are prominent mediators of inflammatory stimuli (McAdam et al., 2000). While COX-2 is constitutively expressed in the CNS (Burian and Geisslinger, 2005), dramatic CNS up-regulation of COX-2 has been found in a striking variety of pathological conditions including transient brain ischaemia (Planas et al., 1995), kainate-induced seizures (Tocco et al., 1997), glutamate toxicity (Tocco et al., 1997), glucocorticoid exposure (Yamagata et al., 1993), Alzheimer's disease (Ho et al., 1999), Parkinson's disease (Teismann et al., 2003) and mechanical pain hypersensitivity (Vardeh et al., 2009). The extent to which this is due to up-regulation of neuronal COX-2 is of great interest; in the case of mechanical pain hypersensitivity the pathology is very likely mediated by COX-2 up-regulation in CNS neurons (Vardeh et al., 2009). The role of COX-2 in inflammation has been viewed largely through the prism of prostaglandin production and their presumed action at prostaglandin receptors. However, COX-2 also effectively metabolizes the components of another endogenous signalling system, the endocannabinoids (eCBs) (Kozak et al., 2004; Jhaveri et al., 2007). Indeed, we and others have shown that COX-2 regulates key aspects of cannabinoid signalling (Kim and Alger, 2004; Hashimotodani et al., 2007; Straiker and Mackie, 2009), raising the possibility that some of the actions of neuronal COX-2 may be due to its metabolism of eCBs.
The eCB signalling system functions throughout the CNS. It consists of cannabinoid receptors [CB1 and CB2 (Matsuda et al., 1990; Munro et al., 1993)], eCBs (Devane et al., 1992; Stella et al., 1997) and an assortment of proteins to produce, transport and break down eCBs. It is this system that is engaged by exogenous cannabinoids such as the psychoactive ingredients of marijuana and hashish (Gaoni and Mechoulam, 1964). eCBs have a prominent role in the modulation of neurotransmission; one form that this modulation takes is depolarization-induced suppression of excitation or inhibition (DSE/DSI) (Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001). This retrograde inhibition is observed in many regions of the CNS and is probably mediated by 2-arachidonylglycerol (2-AG), an endogenous ligand of cannabinoid receptors (Trettel and Levine, 2003; Yanovsky et al., 2003; Melis et al., 2004; Di et al., 2005). We have previously characterized DSE and DSI in autaptically cultured hippocampal neurons (Straiker and Mackie, 2005; 2009;). In autaptic neurons, as a result of growth on a limited permissive substrate, neurons synapse (or ‘autapse’) onto themselves forming an architecturally simple and tractable model of synaptic transmission to evaluate molecular events associated with this process (Bekkers and Stevens, 1991). The duration of DSE/DSI in these neurons is probably determined by the rate of 2-AG degradation (Straiker et al., 2009; Straiker and Mackie, 2009).
To date, five enzymes have received serious attention as possible mediators of 2-AG hydrolysis. The enzyme that accounts for the majority of 2-AG breakdown in brain homogenates is monoacylglycerol lipase (MAG lipase, MGL) (Blankman et al., 2007). Anatomical studies have established the presence of MGL in presynaptic terminals, making it well-sited to break down 2-AG, terminating DSE/DSI (Dinh et al., 2002; Gulyas et al., 2004; Straiker et al., 2009). Fatty acid amide hydrolase (FAAH) can break down 2-AG in vitro. However, while the other major eCB anandamide (AEA) and other acyl amide levels are dramatically increased in FAAH−/− mice, 2-AG levels are unchanged, suggesting that FAAH's role is primarily to degrade acyl amides (Cravatt et al., 2001). COX-2 (Cravatt et al., 1996; Kozak et al., 2000; Kozak et al., 2004) can also metabolize 2-AG and appears to have a clearer physiologically relevant role, since its inhibition prolongs DSI in hippocampal slices (Kim and Alger, 2004) and in cultured neurons (Hashimotodani et al., 2007; Straiker and Mackie, 2009). In addition, PGE2–glycerol ester (PGE2-G), the product of 2-AG oxidation by COX-2, is itself an endogenous bioactive metabolite that enhances neuronal activity and modulates pain and inflammation (Sang et al., 2006; Hu et al., 2008). Lastly, ABHD6 and ABHD12 each account for a portion of 2-AG hydrolysis in brain homogenates (Blankman et al., 2007). The role of ABHD12 in modulating eCB plasticity remains elusive, but blockade of ABHD6 has been found to increase 2-AG levels and signalling by several measures, including facilitation of long-term depression in mouse cortex (Marrs et al., 2010).
Depending on the subcellular localization of these enzymes, it is possible that an enzyme of relatively low abundance as measured in a homogenate assay may terminate 2-AG signalling in a specific set of synapses, particularly if it is localized close to the site of action of 2-AG. An enzyme's relative contribution may vary among neuronal populations, or within a given neuron, or may even be shared in some manner as it appears to be in one form of autaptic DSI (Straiker and Mackie, 2009) and other forms of hippocampal DSI (Kim and Alger, 2004; Hashimotodani et al., 2007). An enzyme's relative contribution may also increase if it is up-regulated. This is particularly relevant to the case of COX-2, which is known to be up-regulated in neurons in response to synaptic activation (Adams et al., 1996) as well as during a variety of pathological conditions as outlined above. The potential inducibility of putative 2-AG-metabolizing enzymes has motivated us to systematically evaluate the role of specific enzymes in the termination of eCB signalling at the synaptic level. Cultured excitatory autaptic hippocampal neurons express functional MGL but not detectable FAAH, ABHD6 or COX-2 activity (Straiker and Mackie, 2005; Straiker et al., 2009). This gives us the opportunity to explore whether any of these proteins can act co-operatively with MGL to more rapidly hydrolyze 2-AG and shorten the duration of DSE. We have addressed this question by transfecting autaptic hippocampal neurons with FAAH, ABHD6, ABHD12, COX-2 or MGL and measuring the sensitivity and duration of DSE.
Methods
Culture preparation
All procedures used in this study were approved by the Animal Care Committee of Indiana University and conform to the Guidelines of the National Institutes of Health on the Care and Use of Animals. Mouse [CD1 strain, or mixed 129SvEv/C57Bl/6J (in the case of MGL−/− and MGL+/+) strain] hippocampal neurons isolated from the CA1-CA3 region were cultured on micro-islands as described previously (Furshpan et al., 1976; Bekkers and Stevens, 1991). Neurons were obtained from animals (age postnatal day 0–2) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously (Levison and McCarthy, 1991). Cultures were grown in high-glucose (20 mM) medium containing 10% horse serum, without mitotic inhibitors and used for recordings after 8 days in culture and for no more than 3 h after removal from culture medium.
Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses – or ‘autapses’– onto itself. All experiments were performed on isolated autaptic neurons. Whole-cell voltage-clamp recordings from autaptic neurons were carried out at room temperature using Axopatch 200A amplifier (Axon Instruments, Burlingame, CA). The extracellular solution contained (in mM) 119 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgCl2, 30 glucose, and 20 HEPES. Continuous flow of solution through the bath chamber (∼2 mL·min−1) ensured rapid drug application and clearance. Drugs were typically prepared as stocks then diluted into extracellular solution at their final concentration and used on the same day.
Recording pipettes of 1.8–3 MΩ were filled with (in mM) 121.5 Kgluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 MgATP and 0.5 LiGTP. Access resistance and holding current were monitored, and only cells with both stable access resistance and holding current were included for data analysis. Conventional stimulus protocol: the membrane potential was held at −70 mV, and excitatory postsynaptic currents (epscs) were evoked every 20 s by triggering an unclamped action current with a 1.0 ms depolarizing step. The resultant-evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower epsc. The size of the recorded epscs was calculated by integrating the evoked current to yield a charge value (in pC). Calculating the charge value in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
DSE stimuli: after establishment of a 10–20 s 0.5 Hz baseline, DSE was evoked by depolarizing to 0 mV for 50 ms, 100 ms, 300 ms, 500 ms, 1 s, 3 s and 10 s, followed in each case by resumption of a 0.5 Hz stimulus protocol for 20 to >80 s, allowing epscs to recover to baseline values. This approach allowed us to determine the sensitivity of the synapses to DSE induction. Three-second depolarizations were used for the purpose of assessing recovery time courses.
The half-life (t1/2) of DSE recovery was calculated as follows. A decay constant (K) was calculated by fitting a single exponential to the first 50 s of the DSE recovery time course [∼80% recovery, presented with 95% confidence intervals (95% CI)]. For multiple comparisons, a Bonferroni multiple comparison correction was applied to compare the four Ks and their variances to the control values. For ease of interpretation, K-values were converted to t1/2s and are reported in s with 95% CIs.
Transfection of autaptic hippocampal cultures
We transfected neurons using a calcium phosphate-based method adapted from a previously published method (Jiang et al., 2004). Briefly, plasmids for the protein of interest and for the fluorescent marker EYFP or mCherry (2 µg·per well) were combined with 2 M CaCl2, water and gradually added to HBS; the mixture was added to the serum-free neuronal media. Coverslips were incubated with this mixture in a separate well for 2.5 h, while extra medium was placed in a 10% CO2 incubator to induce acidification. At the end of 2.5 h, the reaction mixture was replaced with acidified serum-free media for 20 min. Cells were then returned to their home wells containing the original media.
Rat FAAH and MGL plasmids were obtained as described previously (Waleh et al., 2002; Straiker et al., 2009). Human Cox2pCMV6-AC plasmid was obtained from Origene Technologies (Cat no. SC323764, Rockville, MD). Mouse ABHD6 and ABHD12 cDNA was purchased from Open Biosystems (Huntsville, AL, USA) (Cat nos., MMM1013-7512548 and MMM1013-63276 respectively). The genes were amplified and BamHI and NotI restriction sites introduced using specific primers: ABHD6 (5′-atcggatcc GATCTCGATGTGGTTAACATGTTT GTG, 3′-atcgcggccgcTCAGTTCAGCTTC TTGTT GTCTGT), ABHD12 (5′-atcggatccACGTATG ACGCACTCCATGTTTTTG, 3′-atcgcgg ccgcTCA GTGCTGGCGTTCCGG). Amplicons were cut, gel purified and ligated into a pcDNA3.0 expression vector previously altered to contain a Kozak start site followed by an HA11 epitope tag between HindIII and KpnI restriction sites on the multiple cloning sites. All inserts were confirmed via fluorescent sequencing.
Staining of autaptic cultures
Coverslips with autaptic cultured neurons (grown 8–15 days) were fixed and washed in PBS (Straiker et al., 2009). Cells were incubated with a neuron blocking solution (5% milk in 0.1 M PBS + 0.3% Triton-X) for 30 min at room temperature to reduce non-specific staining. Neurons were next incubated with a mouse monoclonal antibody against the axonal marker 2H3 (Developmental Studies Hybridoma Bank, University of Iowa; 1:300), the glial marker GFAP (Covance, Princeton NJ; 1:500), or the dendritic marker MAP2 (Millipore, Billerica MA; 1:1000) overnight at 4°C and then washed six times with 0.1 M PBS. Antibodies against FAAH (Bracey et al., 2002;, 1:100), CB1 (Bodor et al., 2005; 1:300) and the HA tag (1:500; Covance) were also used. Cells were next incubated with anti-mouse or anti-rabbit Texas Red- or FITC-conjugated donkey secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA; 1:200) for 1.5 h at room temperature. Finally, cover slips were washed, dried and mounted as above. Images were acquired with a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany) using Leica LAS AF software and a 63× oil objective. Images were processed using ImageJ (available at http://rsbweb.nih.gov/ij/) and/or Photoshop (Adobe Inc., San Jose, CA). Images were modified only in terms of brightness and contrast.
Our drug/molecular target nomenclature (e.g. receptors, ion channels, etc.) conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Results
Overexpression of COX-2 with endogenous MGL shortens the duration of DSE
Pharmacological experiments using blockers for MGL, COX-2, ABHD6 and FAAH suggest that DSE in excitatory autaptic hippocampal neurons is chiefly regulated by MGL (Straiker and Mackie, 2005; 2009; Straiker et al., 2009). Previously, we have shown, using a pharmacological approach, that a population of cultured inhibitory neurons express both MGL and COX-2 – this results in an unusually brief form of DSI (Straiker and Mackie, 2009). The implication from this finding is that co-expression of COX-2 with MGL results in more rapid clearance of 2-AG from the synaptic region, shortening the duration of DSI. If so, it should be possible to induce a more rapid DSE time course by overexpressing COX-2 in excitatory neurons. If overexpressed COX-2 were appropriately localized to accelerate the breakdown of 2-AG, one would predict a more rapid termination of DSE. We tested if this was the case by transfecting excitatory autaptic neurons with COX-2 and then examining DSE. Since we have previously shown MGL is expressed in these neurons, in these experiments we examined the combined effect of COX-2 and MGL on DSE. We first collected a ‘dose’–response curve for neurons expressing COX-2 to determine if expression of COX-2 changed the sensitivity of DSE induction. By depolarizing neurons for longer durations (50 ms, 100 ms, 300 ms, 500 ms, 1 s, 3 s, 10 s) and measuring the resulting CB1-dependent inhibition we were able to assess whether neurons expressing COX-2 (or other proteins) retain their characteristic eCB signalling properties (Figure 1A). In the case of COX-2, the dose–response curve closely resembled that for wild-type neurons (P > 0.05, two-way Anova with Bonferroni post hoc test). However, when examining the duration of DSE, we found that the DSE recovery rates were substantially faster as measured by their t1/2 (Figure 1B and C; t1/2 control = 40 s, 95% CI, 31–57 s, n = 11; t1/2 COX-2 = 20 s, 95% CI, 18–24, n = 6, non-overlapping CIs).
Figure 1.
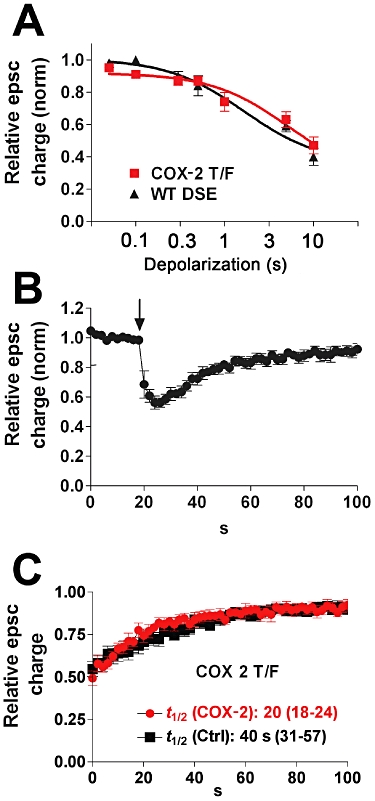
Overexpression of COX-2 with endogenous MGL shortens the duration of DSE. (A) ‘Dose’–response for DSE using a range of depolarizations from 50 ms to 10 s. The wild-type DSE dose–response is shown for comparison. (B) Averaged DSE time courses for neurons transfected with COX-2. Arrow indicates the point at which the cell is depolarized for 3 s. (C) Recovery time courses for COX-2-transfected and control cells. t1/2-values with 95% CIs are listed below the curves.
Overexpression of FAAH with endogenous MGL also shortens the duration of DSE
Substantially elevated AEA levels were seen in FAAH knockout mice, suggesting that FAAH plays a major role in the breakdown of AEA (Cravatt et al., 1996). However, it is possible that FAAH, which is capable of hydrolyzing 2-AG (Cravatt et al., 1996; Di Marzo et al., 1998) in vitro, also plays a role in synaptic 2-AG breakdown under specific circumstances. To test whether FAAH, could, in principle, serve this role in an intact endogenous cannabinoid signalling module, we transfected neurons with FAAH. Over-expression of FAAH did not alter the sensitivity of the synapses to DSE (Figure 2A, P > 0.05, two-way anova with Bonferroni post hoc test). Interestingly, however, FAAH clearly sped up the rate of DSE recovery, shortening the duration of DSE (Figure 2B and C; t1/2 FAAH = 13 s, 95% CI, 12–15, n = 9, non-overlapping CIs vs. WT time course). To test whether this change in time course is indeed FAAH-dependent, we repeated the experiments in the presence of the FAAH blocker URB597 (250nM; Figure 2B, n = 8). URB597 does not alter the time course of DSE in autaptic neurons (Straiker and Mackie, 2005) but clearly slowed the time course of DSE in FAAH-transfected neurons.
Figure 2.
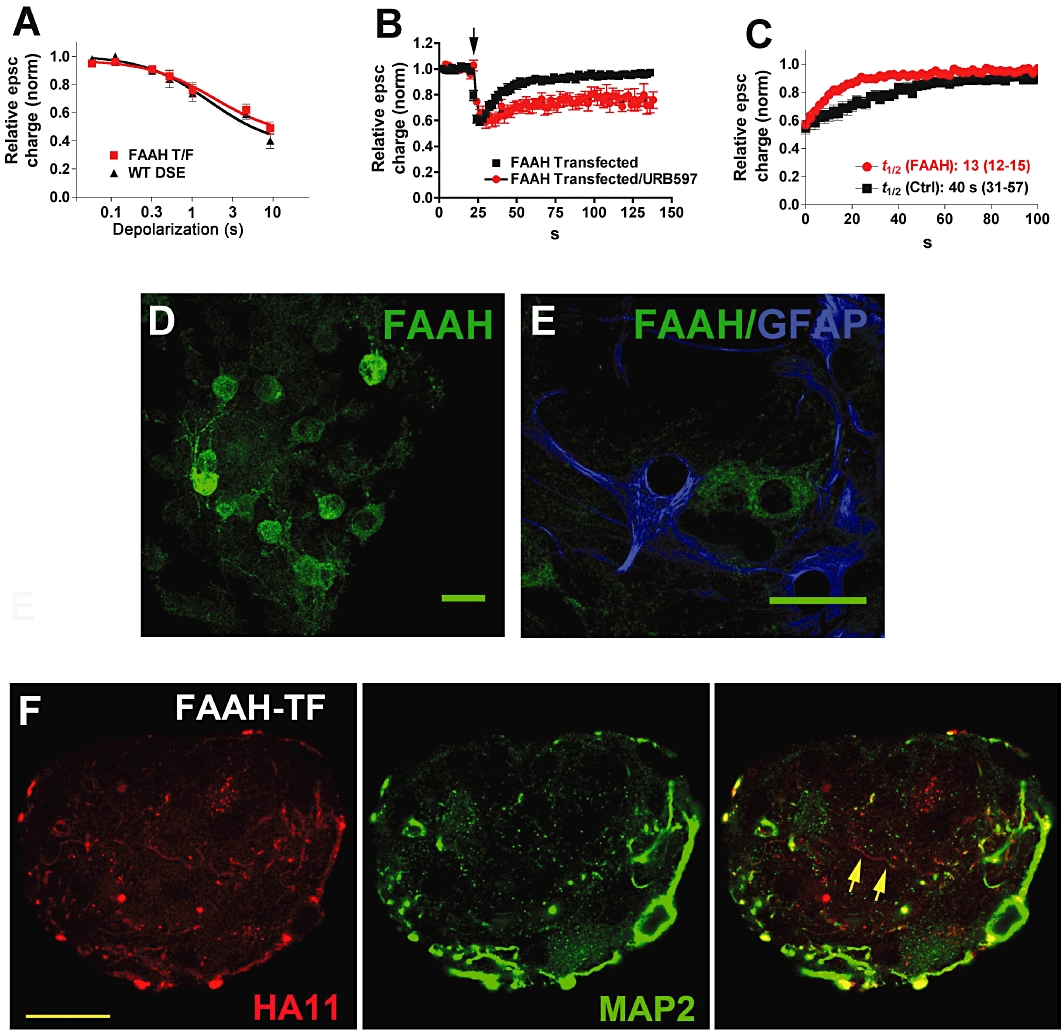
Overexpression of FAAH with endogenous MGL also shortens DSE duration. (A) ‘Dose’–response for DSE of FAAH-transfected neurons (FAAH T/F) using a range of depolarizations from 50 ms to 10 s. The wild-type DSE dose–response is shown for comparison. (B) Averaged DSE time courses for neurons transfected with FAAH and FAAH transfected neurons subsequently treated with URB597 (250 nM). (C) DSE recovery time courses for FAAH-transfected and control cells. t1/2-values with 95% CIs are listed below the curves. (D) Micrograph shows FAAH staining in autaptic hippocampal cultures. Scale bar = 25 µm. (E) Micrograph shows FAAH staining does not colocalize with the astrocyte cell marker GFAP. Scale bar = 25 µm. (F) Micrograph shows that FAAH is expressed in neurons transfected with FAAH, and that it is expressed in processes that are labelled and unlabelled with dendritic marker MAP2. Left panel shows HA11 staining against the HA-tagged FAAH protein. Centre panel shows dendritic marker MAP2. Right panel shows the composite of the left and centre panels, with overlap in yellow. Scale bar = 15 µm.
Although pharmacological studies have not uncovered any role for FAAH in the modulation of neurotransmission in cultured hippocampal neurons, FAAH is prominently expressed in the hippocampal pyramidal cells (Egertova et al., 1998; Tsou et al., 1998). We therefore examined FAAH expression in autaptic hippocampal cultures using a previously characterized FAAH antibody (Bracey et al., 2002; Hu et al., 2010). We found that FAAH protein is endogenously expressed in these cultured neurons, but that this expression is primarily detectable in somata and proximal dendrites (Figure 2D). The somatic enrichment of FAAH is consistent with an absence of acute modulation by FAAH of synaptic transmission. FAAH expression does not overlap with that of the astrocyte marker GFAP (Figure 2E), suggesting that FAAH expression is restricted to neurons in these cultures. We separately examined where FAAH was trafficked when overexpressed, using an HA11 antibody against an HA tag included in the transfected FAAH (Figure 2F). We found that while FAAH staining coincided with MAP2 staining, indicating dendritic trafficking, MAP2-negative, HA11-positive fibres were also seen, suggesting that overexpressed FAAH is also trafficked to axons [HA11-positive, MAP2-negative fibres in Figure 2F (arrows)].
ABHD6 and ABHD12 have little effect on the induction or duration of DSE
MGL is expressed presynaptically in autaptic hippocampal neurons, leaving it well placed to break down 2-AG after activation of CB1 (Straiker et al., 2009). In contrast, ABHD6 is found post-synaptically (Straiker et al., 2009). We first showed that overexpression of ABHD6 did not alter the sensitivity of the synapses to DSE (Figure 3A). Transfection with ABHD6 resulted in a DSE recovery rate that closely mimicked that in wild-type cells, suggesting that it is not able to play an active role in breaking down 2-AG after DSE (Figures 3B,C and 4; t1/2 ABHD6 = 32 s, 95% CI, 25–47, n = 6, overlapping CIs). We also examined the trafficking of ABHD6 after overexpression using an antibody against an HA tag on the expressed protein (Figure 3G), and found that ABHD6 is expressed in both dendritic, MAP2 labelled, and presumed axonal MAP2-unlabelled processes.
Figure 3.
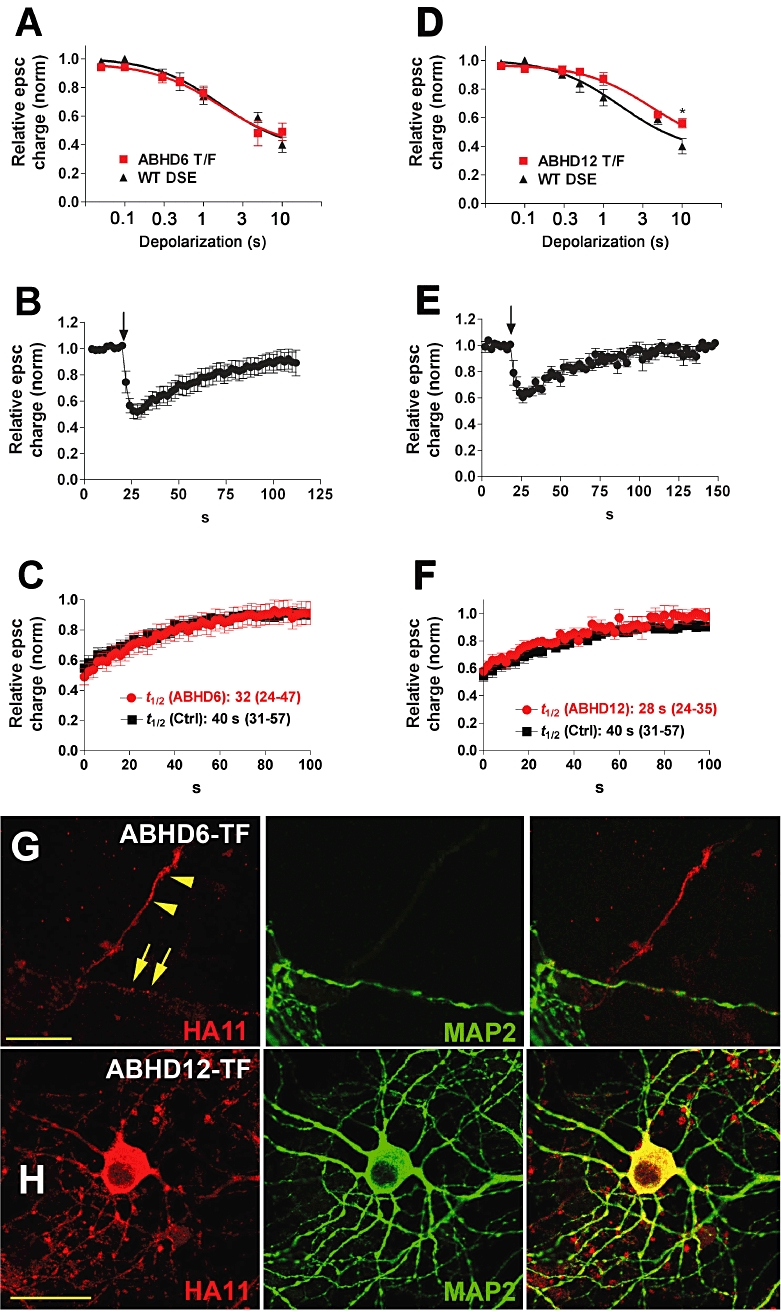
ABHD6 and ABHD12 do not speed up DSE recovery. (A) ‘Dose’–response for DSE in ABHD6-transfected neurons (ABHD6 T/F) using a range of depolarizations from 50 ms to 10 s. The wild-type DSE dose–response is shown for comparison. (B) Averaged DSE time courses for neurons transfected with ABHD6. Arrow indicates the time point at which the neuron is depolarized for 3 s. (C) Recovery time courses for ABHD6-transfected and control cells. t1/2-values with 95% CIs are listed as inset. (D) ‘Dose’–response for DSE in ABHD12-transfected neurons (ABHD12 T/F) using a range of depolarizations from 50 ms to 10 s. The wild-type DSE dose–response is shown for comparison. ABHD12 decreases the maximum inhibition for 10 s depolarization. (E) Averaged DSE time courses for neurons transfected with ABHD12. Arrow indicates the point at which the neuron is depolarized for 3 s. (F) Recovery time courses for ABHD12-transfected and control neurons. t1/2-values with 95% CIs are listed as insets. (G) Micrograph shows that ABHD6 is expressed in neurons transfected with ABHD6 in processes that are labelled and unlabelled with the dendritic marker MAP2. Left panel shows HA11 staining of the HA-tagged ABHD6 protein (red). Centre panel shows the dendritic marker MAP2. Right panel shows the composite. Scale bar = 15 µm. (H) Transfected ABHD12 is also widely expressed in neurons. Left panel shows ABHD12-HA11 staining. Centre panel shows MAP2 staining. Right panel is the composite. Scale bar = 25 µm.
Figure 4.
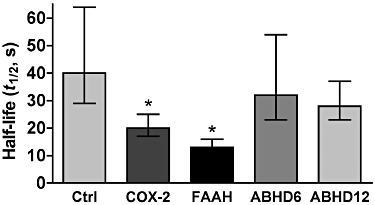
Summary of changes to DSE t1/2 after overexpression of four endocannabinoid-metabolizing enzymes. Bar graph shows t1/2-values of DSE (s) for untransfected neurons (control) versus neurons transfected with COX-2, FAAH, ABHD6 or ABHD12. Error bars represent 95% CIs with a Bonferroni correction for multiple comparisons (see Methods).
ABHD12 is also capable of breaking down 2-AG. However, we found that ABHD12 – like ABHD6 – did not alter the recovery of DSE (Figures 3E, F and 4; t1/2 ABHD12 = 28 s, 95% CI, 24–35, n = 6, overlapping 95% CIs). This suggests that ABHD12 is not able to play an active role in breaking down 2-AG after DSE. ABHD12 was found to alter DSE signalling – albeit modestly – doing so by decreasing the maximum inhibition for 10 s depolarizations (Figure 3D, *P < 0.05, two-way anova with Bonferroni post hoc test; P < 0.05 for dose × transfection interaction). The successful overexpression of ABHD12 was verified using an antibody against an HA tag on the protein (Figure 3H); double staining with dendritic marker MAP2 indicated that ABHD12 traffics throughout a given neuron. Figure 4 summarizes the comparison of the half-life values of untransfected neurons and those transfected with COX-2, FAAH, ABHD6 or ABHD12 (Figure 4).
Robust DSE requires an optimal level of MGL
We have previously presented pharmacological and immunocytochemical evidence that MGL regulates the recovery time course of DSE and DSI in cultured autaptic neurons (Straiker and Mackie, 2009; Straiker et al., 2009). In these next experiments, we tested DSE in hippocampal neurons cultured from MGL−/− mice (Schlosburg et al., 2010). We found that neurons lacking MGL had a markedly slowed time course of DSE recovery, as predicted from our previous results (Figure 5A and B). In a finding that is consistent with a very recent report describing altered cannabinoid-mediated synaptic plasticity in MGL−/− neurons (Schlosburg et al., 2010), we observed that in many neurons DSE was absent, and that average DSE was much diminished from that expected in wild-type neurons [Figure 5C and D, relative epsc charge after 3 s depolarization in MGL KO: 0.75 ± 0.04, n = 23; in WT (MGL+/+): 0.52 ± 0.05, n = 5; P < 0.05 unpaired t-test vs. WT DSE responses]. These results suggest that functional MGL is an essential component of rapid and robust DSE. In MGL−/− neurons, the inhibition observed following the addition of exogenous 2-AG was also diminished [Figure 5D: relative epsc charge after 5 µM 2-AG in MGL KO: 0.64 ± 0.05, n = 13; in WT (C57/Bl6): 0.48 ± 0.04, n = 13; P < 0.05 unpaired t-test vs. WT 2-AG responses], suggesting impaired CB1-mediated responses, consistent with CB1 desensitization (Straiker and Mackie, 2005; Schlosburg et al., 2010). Responses of the synthetic agonist WIN-55212–2 (WIN) were similarly diminished in MGL KO vs. wild-type responses [Figure 5E, EC50: 550nM (95% CI, 400–766) for WIN in MGL KO; 7.3nM (95% CI, 6.7–7.9) for WIN in WT]. As noted previously (Straiker and Mackie, 2005), we did not observe a strain difference in WIN sensitivity between neurons cultured from C57Bl6 and CD1 strains (e.g. Figure 5E).
Figure 5.
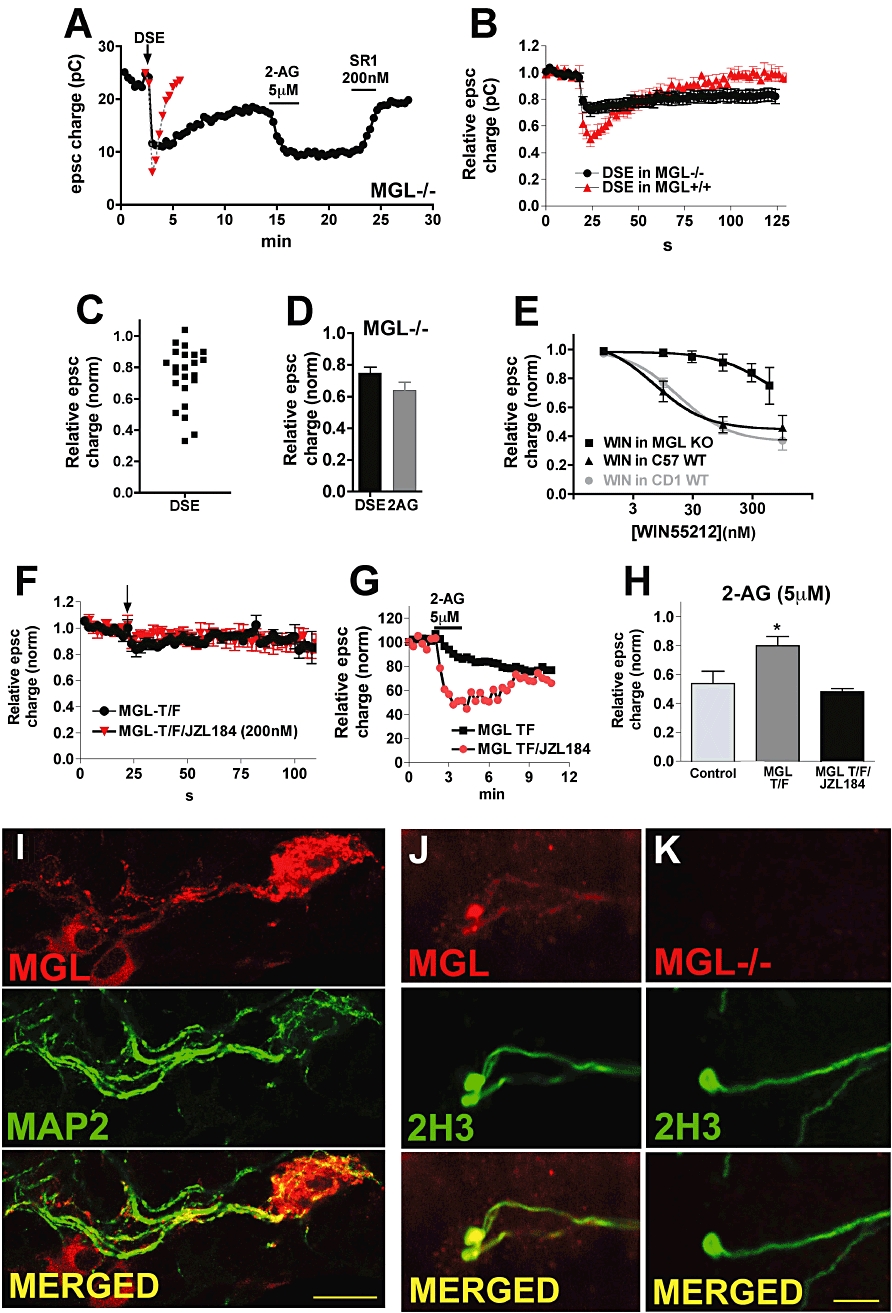
Robust DSE requires an optimal level of MGL. (A) Sample time course shows epscs in MGL−/− neuron after DSE stimulation (arrow), 2-AG treatment (5 µM) and reversal by CB1 antagonist SR141716 (200 nM). Example of WT DSE time course is included for comparison. (B) Averaged DSE time courses from MGL−/− and MGL+/+ cultures. (C) Scatter plot of inhibition values after the DSE stimulus in MGL−/− neurons shows that DSE is diminished in most neurons. (D) Bar graph shows epsc inhibition in response to DSE or 2-AG (5 µM) in MGL−/− neurons. (E) WIN55212-2 responses are desensitized in MGL KO neurons. Concentration–response curves for WIN55212-2 in MGL KO and wild-type (C57) neurons. The CD1 wild-type WIN55212-2 concentration–response is shown for comparison (adapted from Straiker and Mackie, 2005). (F) Averaged DSE time course for wild-type neurons transfected with MGL under control conditions or after treatment overnight with MGL blocker JZL184 (200 nM). Arrow indicates the point at which the cell was depolarized for 3 s. (G) Sample time courses in response to 2-AG (5 µM) in MGL-transfected neuron (the incomplete recovery is probably due to 2-AG-induced LTD; Kellogg et al., 2009) and in MGL-transfected neurons treated overnight with JZL 184 (200 nM). (H) Bar graph shows average 5 µM 2-AG responses for untransfected neurons, MGL-transfected neurons and MGL-transfected neurons treated overnight with JZL184 (200nM). (I) Micrograph shows MGL-transfected neuron stained for HA11 and MAP2, a dendritic marker. Bottom panel shows overlay of the two channels (yellow represents overlap, scale bar = 25 µm). (J) Micrographs show MGL antibody (top panel) staining for the axonal marker, 2H3 (centre panel) and overlap (bottom panel, overlap in yellow) in a WT untransfected neuron. (K) Same layout as (J), but in MGL−/− neuronal cultures. Panels (J–K) show that the MGL antibody stains axons. Scale bar = 20 µm.
If COX-2 and FAAH can co-operatively increase the rate of 2-AG breakdown and shorten the duration of DSE, then it is possible that overexpression of MGL could also shorten DSE. Unexpectedly, however, transfection of MGL in wild-type neurons did not shorten the duration of DSE but rather markedly reduced DSE [Figure 5F, relative epsc charge after 3 s depolarization: 0.86 ± 0.04, n = 9; after 10 s depolarization (data not shown): 0.85 ± 0.12, n = 5]. In these neurons 2-AG signalling was also diminished relative to controls (Figure 5H, relative epsc charge after 5 µM 2-AG: 0.80 ± 0.06, n = 8; 2-AG response in untransfected neurons: 0.54 ± 0.09, n = 6. P < 0.05 unpaired t-test vs. 2-AG responses in untransfected neurons). Overnight treatment of MGL-transfected neurons with the MGL blocker JZL184 (200 nM) did not rescue the DSE response [Figure 5F, relative epsc charge after 3 s depolarization in MGL-transfected neurons treated with JZL184 (200 nM, overnight): 0.96 ± 0.03, n = 8]. In contrast, overnight treatment with 200 nM JZL184 did restore the 2-AG response back to wild-type levels. A representative experiment is shown in Figure 5G, and summary data are shown in Figure 5H (relative epsc charge in MGL-transfected and JZL184-treated neurons after 5 µM 2-AG: 0.48 ± 0.02, n = 3). Taken together, these results suggest that overexpression of MGL impairs both CB1 signalling and eCB production, with the former recovering partially after acute MGL inhibition. We also tested whether MGL transfection into MGL−/− cultures could rescue DSE or CB1 signalling, and found that in the case of DSE it did not, but that part of the 2-AG response was recovered [data not shown; relative epsc charge after 3 s depolarization in MGL-transfected MGL−/− neurons: 0.87 ± 0.06, n = 11, P < 0.05 unpaired t-test vs. WT (C57/Bl6) control DSE; relative epsc charge after 5 µM 2-AG: 0.71 ± 0.09, n = 5; P > 0.05 unpaired t-test vs. WT control 2-AG].
We have previously reported that MGL is expressed in presynaptic terminals in autaptic hippocampal neurons (Straiker et al., 2009). We monitored trafficking of transfected MGL via HA staining, and found that it travelled throughout the neuron, including dendrites, as determined by colocalization with the dendritic marker MAP2 (Figure 5I). Trafficking of MGL to dendrites, where it might limit effective eCB release, offers a potential mechanism for MGL interference with DSE. We also tested MGL staining in MGL−/− neurons, confirming that MGL is indeed absent in neurons cultured from MGL−/− animals (Figure 5J) (Schlosburg et al., 2010).
Discussion
Our most significant finding is that expression of COX-2 or FAAH in excitatory neurons modulates endocannabinoid-mediated signalling. (We use the term ‘endocannabinoid’ here to denote a molecule that is metabolized by FAAH or COX-2 and activates CB1 receptors, realizing that it could be any number of bioactive lipids.) Co-expression of either protein, together with the MGL constitutively expressed in excitatory autaptic neurons, results in a briefer DSE compared to that seen under control conditions. Peak DSE and the sensitivity of DSE induction to the duration of depolarization are unaltered by expression of either enzyme. Thus, the net effect of up-regulation of FAAH or COX expression is a diminishment of eCB signalling.
A role for COX-2 in DSE is consistent with our recent findings that in two populations of inhibitory hippocampal neurons distinguished by the duration of DSI, COX-2 is functionally co-expressed with MGL only in the subpopulation with brief DSI. In this population, blockade of COX-2 modestly prolongs DSI, while blockade of both COX-2 and MGL results in a near complete failure to recover after DSI. A COX-2 role in modulation of excitatory transmission is also consistent with the findings of Slanina and Schweitzer (2005), who reported that COX-2 blockade enhanced excitatory neurotransmission in a CB1-dependent manner but independent of GABAergic signalling. COX-2 is expressed in numerous cell types, but it is of considerable interest that COX-2 is induced in neurons in response to neuronal stimulation (Adams et al., 1996) as well as a variety of pathological states including transient brain ischaemia (Planas et al., 1995), inflammation (Patrignani et al., 2005), Parkinson's disease (Teismann et al., 2003) and kainate-induced seizure (Tocco et al., 1997), as well as in response to insults such as glutamate toxicity (Tocco et al., 1997) and glucocorticoid excess (Yamagata et al., 1993). Our results suggest that when COX-2 is induced under pathological conditions such as those described above, eCB inhibition of synaptic transmission will be suppressed. Depending on the neurons most affected, COX-2 induction could have far-reaching consequences on downstream pathways (e.g. either promote or suppress excitotoxicity). In keeping with this, a recent report by Telleria-Diaz et al. (2010)) has implicated COX-2 modulation of eCBs in spinal hyperalgesia. In addition, it is important to recall that the COX-2 metabolite of 2-AG, PGE2-G, has been shown to potentiate synaptic transmission in excitatory hippocampal neurons and to modulate inflammation and pain (Sang et al., 2006; Hu et al., 2008). Depending on the configuration of enzymes and receptors, one might observe a co-operative activation of synaptic transmission by a diminishment of cannabinoid inhibition of excitatory transmission combined with an activation of an as yet uncharacterized PGE2-G-based signalling pathway.
A somewhat unexpected finding was that FAAH played a similar role to COX-2 when co-expressed with endogenous MGL, implying that FAAH and MGL can co-operatively regulate endocannabinoid signalling. To our knowledge, this is the first example to show that FAAH can enhance neurotransmission by speeding up 2-AG breakdown. Naturally, the significance of this finding will depend on the degree to which such an effect of FAAH is found in vivo (Maccarrone et al., 2003), but the present results suggest that FAAH is clearly capable of interacting co-operatively with MGL to limit the duration of 2-AG-mediated signalling.
A recent study of cannabinoid function in MGL−/− mice found dramatic desensitization of brain CB1 receptors (Schlosburg et al., 2010). We found that eCB signalling in neurons cultured from MGL−/− mice was bimodal: consistent with Schlosburg (Schlosburg et al., 2010), many neurons no longer exhibited DSE, presumably due to CB1 receptor desensitization, which is prominent in autaptic cultures (Straiker and Mackie, 2005), with the remaining neurons expressing a slowly recovering DSE consistent with the predicted role of MGL in regulating the time course of DSE. Transfection of MGL into MGL−/− neurons did not rescue DSE, as might have been expected given our results for COX-2 and FAAH. Instead, DSE was greatly diminished, as were responses to exogenous 2-AG. Our results suggest that MGL provides a window for optimal DSE: too little MGL results in an excess of 2-AG with consequent CB1 receptor desensitization or down-regulation, while an excess of MGL, or at least improperly distributed MGL, eliminates DSE, possibly by perturbing phospholipid pools. The distribution of MGL is an important consideration given that the distribution of MGL is normally restricted to axonal terminals in autaptic neurons (Straiker et al., 2009), but that overexpressed MGL is also trafficked to dendrites (Figure 5H). Interference with 2-AG production by dendritically localized MGL might account for the recovery of 2-AG inhibition but not DSE in JZL184-treated MGL overexpressing neurons. The differential wild-type trafficking of MGL and FAAH – axon terminal localization for MGL and somatic localization of FAAH – is likely to explain a somewhat surprising result: transfected MGL and FAAH both alter autaptic cannabinoid signalling but only MGL knockout animals exhibit an altered cannabinoid signalling profile. In contrast to MGL, FAAH in autaptic neurons is excluded from presynaptic terminals, leaving FAAH unable to degrade presynaptic 2-AG. When overexpressed both FAAH and MGL traffic indiscriminately to axonal and dendritic compartments, maximizing endocannabinoid degradation.
ABHD6 and ABHD12 have only recently been shown to be involved in 2-AG metabolism, but the observation that they account for ∼15% of 2-AG breakdown in brain homogenates has generated considerable interest (MGL accounted for 80%, FAAH accounted for ∼2%, and the contribution of COX-2 was not assessed) (Blankman et al., 2007) and mutations of ABHD12 in humans result in profound tissue-specific effects (Fiskerstrand et al., 2010). We previously reported that ABHD6 is expressed in dendrites and therefore not as well positioned (compared with MGL) to break down 2-AG close to presynaptic CB1 receptors (Straiker et al., 2009). We also previously found that the ABHD6 blocker WWL70 did not alter the time course of DSE or DSI (Straiker and Mackie, 2009; Straiker et al., 2009). In the current study, transfected ABHD6 had no effect on cannabinoid signalling on either the DSE time course or the DSE ‘dose–response’. It remains entirely possible that ABHD6 has a role in neurotransmission under other conditions such as the induction of long-term depression (LTD) when considerable quantities of 2-AG, spilling beyond the synapse, may be generated during prolonged DGL activation. A recent report examining LTD in the cortex supports this hypothesis. In that study, inhibition of ABHD6 decreased the stimulus needed to produce LTD (Marrs et al., 2010). The same may be true for ABHD12. Interestingly, ABHD12 was the only enzyme that altered the extent of DSE, albeit only at the most prolonged depolarizations. This result may indicate a role for ABHD12 in clearing 2-AG when 2-AG levels are very high or that ABHD12 overexpression limits the amount of 2-AG formed by decreasing PIP2 levels.
In summary, increasing the expression of COX-2 and FAAH restricts the temporal window for endogenous cannabinoid signalling at excitatory synapses; this modulation takes the form of reducing the duration of DSE, which will serve to potentiate glutamatergic synaptic transmission. Thus, conditions that induce COX-2 expression in hippocampal pyramidal cells (e.g. Alzheimer's disease, stroke, neurotoxic damage and seizures) may enhance glutamatergic signalling. As the oxidative metabolite of 2-AG, PGE2-G, is pro-excitatory, it, in combination with increased glutamatergic signalling may further contribute to excitotoxicity following COX-2 induction. Surprisingly, FAAH is able to act in a similar manner to COX-2 to modulate endocannabinoid signalling, an unexpected finding for an enzyme thought to act chiefly on anandamide and other acyl amides, which don't seem to be involved in autaptic DSE (Straiker and Mackie, 2005). It is important to note that the inducible expression of FAAH or COX-2 may regulate other forms of cannabinoid-mediated signalling such as metabotropic suppression of excitation/inhibition (Maejima et al., 2001; Kim et al., 2002) and LTD (Marsicano et al., 2002; Robbe et al., 2002). Ultimately, our results underscore the centrality of MGL to DSE while emphasizing the notion that regulated expression of multiple eCB-metabolizing enzymes can serve to shape the duration of eCB-mediated synaptic plasticity.
Acknowledgments
Excellent technical assistance was provided by Ben Cornett and Alhasan Elghouche. This work was supported by National Institutes of Health (grants DA11322, DA021696, DA024122, DA009158); the Indiana METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc; and by the Indiana University Light Microscopy Imaging Center.
Glossary
Abbreviations
- 2-AG
2-arachidonylglycerol
- ABHD6
α/β hydrolase domain 6
- ABHD12
α/β hydrolase domain 12
- AEA
arachidonyl ethanolamide
- DSE
depolarization-induced suppression of excitation
- DSI
depolarization-induced suppression of inhibition
- eCB
endocannabinoid
- FAAH
fatty acid amide hydrolase
- MGL
monoacylglycerol lipase
Conflicts of interest
None.
References
- Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. 2009;158(Suppl 1):254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N, et al. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- Burian M, Geisslinger G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol Ther. 2005;107:139–154. doi: 10.1016/j.pharmthera.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Gheusi G, Cremona S, Laye S, Parnet P, et al. Molecular basis of sickness behavior. Ann NY Acad Sci. 1998;856:132–138. doi: 10.1111/j.1749-6632.1998.tb08321.x. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, Sugiura T, Melck D, De Petrocellis L. The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem J. 1998;331(Pt 1):15–19. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, et al. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005;569:751–760. doi: 10.1113/jphysiol.2005.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertova M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc R Soc Lond B Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskerstrand T, H'Mida-Ben Brahim D, Johansson S, M'Zahem A, Haukanes BI, Drouot N, et al. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am J Hum Genet. 2010;87:410–417. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci USA. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86:1646–1647. [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci. 2007;27:1211–1219. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer's disease. J Neurosci Res. 1999;57:295–303. doi: 10.1002/(SICI)1097-4547(19990801)57:3<295::AID-JNR1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Arnold A, Hutchens JM, Radicke J, Cravatt BF, Wager-Miller J, et al. Architecture of cannabinoid signaling in mouse retina. J Comp Neurol. 2010;518:3848–3866. doi: 10.1002/cne.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol. 2007;152:624–632. doi: 10.1038/sj.bjp.0707433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Deng L, Chen G. High Ca(2+)-phosphate transfection efficiency enables single neuron gene analysis. Gene Ther. 2004;11:1303–1311. doi: 10.1038/sj.gt.3302305. [DOI] [PubMed] [Google Scholar]
- Kellogg R, Mackie K, Straiker A. Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol. 2009;102:1160–1171. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JJ, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Curr Pharm Des. 2004;10:659–667. doi: 10.2174/1381612043453081. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Levison SW, McCarthy KD. Characterization and partial purification of AIM: a plasma protein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. J Neurochem. 1991;57:782–794. doi: 10.1111/j.1471-4159.1991.tb08220.x. [DOI] [PubMed] [Google Scholar]
- McAdam BF, Mardini IA, Habib A, Burke A, Lawson JA, Kapoor S, et al. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J Clin Invest. 2000;105:1473–1482. doi: 10.1172/JCI9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Cecconi S, Rossi G, Battista N, Pauselli R, Finazzi-Agro A. Anandamide activity and degradation are regulated by early postnatal aging and follicle-stimulating hormone in mouse Sertoli cells. Endocrinology. 2003;144:20–28. doi: 10.1210/en.2002-220544. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Patrignani P, Tacconelli S, Sciulli MG, Capone ML. New insights into COX-2 biology and inhibition. Brain Res Brain Res Rev. 2005;48:352–359. doi: 10.1016/j.brainresrev.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Rodriguez-Farre E, Ferrer I. Induction of cyclooxygenase-2 mRNA and protein following transient focal ischemia in the rat brain. Neurosci Lett. 1995;200:187–190. doi: 10.1016/0304-3940(95)12108-g. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Chen C. PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol. 2006;572(Pt 3):735–745. doi: 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long J, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slanina KA, Schweitzer P. Inhibition of cyclooxygenase-2 elicits a CB1-mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacology. 2005;49:653–659. doi: 10.1016/j.neuropharm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neuroscience. 2009;163:11. doi: 10.1016/j.neuroscience.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, et al. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, et al. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci USA. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telleria-Diaz A, Schmidt M, Kreusch S, Neubert AK, Schache F, Vazquez E, et al. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: involvement of prostaglandins and endocannabinoids. Pain. 2010;148:26–35. doi: 10.1016/j.pain.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM. Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: implications for Alzheimer's disease. Exp Neurol. 1997;144:339–349. doi: 10.1006/exnr.1997.6429. [DOI] [PubMed] [Google Scholar]
- Trettel J, Levine ES. Endocannabinoids mediate rapid retrograde signaling at interneuron right-arrow pyramidal neuron synapses of the neocortex. J Neurophysiol. 2003;89:2334–2338. doi: 10.1152/jn.01037.2002. [DOI] [PubMed] [Google Scholar]
- Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, et al. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, et al. COX-2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119:287–294. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waleh NS, Cravatt BF, Apte-Deshpande A, Terao A, Kilduff TS. Transcriptional regulation of the mouse fatty acid amide hydrolase gene. Gene. 2002;291:203–210. doi: 10.1016/s0378-1119(02)00598-x. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Yanovsky Y, Mades S, Misgeld U. Retrograde signaling changes the venue of postsynaptic inhibition in rat substantia nigra. Neuroscience. 2003;122:317–328. doi: 10.1016/s0306-4522(03)00607-9. [DOI] [PubMed] [Google Scholar]