

Abstract
BACKGROUND AND PURPOSE
Celecoxib is a selective cyclooxygenase-2 (COX-2) inhibitor used for the treatment of pain and inflammation. Emerging and accumulating evidence suggests that celecoxib can affect cellular targets other than COX, such as ion channels. In this study, we characterized the effects of celecoxib on Kv7 K+ channels and compared its effects with the well-established Kv7 channel opener retigabine.
EXPERIMENTAL APPROACH
A perforated whole-cell patch technique was used to record Kv7currents expressed in HEK 293 cells and M-type currents from rat superior cervical ganglion neurons.
KEY RESULTS
Celecoxib enhanced Kv7.2–7.4, Kv7.2/7.3 and Kv7.3/7.5 currents but inhibited Kv7.1 and Kv7.1/KCNE1 currents and these effects were concentration dependent. The IC50 value for inhibition of Kv7.1 channels was approximately 4 µM and the EC50 values for activation of Kv7.2–7.4, Kv7.2/Kv7.3 and Kv7.3/Kv7.5 channels were approximately 2–5 µM. The effects of celecoxib were manifested by increasing current amplitudes, shifting the voltage-dependent activation curve in a more negative direction and slowing the deactivation of Kv7 currents. 2,5-Dimethyl-celecoxib, a celecoxib analogue devoid of COX inhibition activity, has similar but greater effects on Kv7currents. Kv7.2(A235T) and Kv7.2(W236L) mutant channels, which have greatly attenuated responses to retigabine, showed a reversed response to celecoxib, from activation to inhibition.
CONCLUSIONS AND IMPLICATIONS
These results suggest that Kv7 channels are targets of celecoxib action and provide new mechanistic evidence for understanding the effects of celecoxib. They also provide a new approach to developing Kv7 modulators and for studying the structure–function relationship of Kv7 channels.
Keywords: activation of potassium channels, celecoxib, HEK 293 cell, Kv7, NSAIDs, retigabine, SCG
Introduction
The introduction of selective cyclooxygenase-2 (COX-2) inhibitors into the market was considered to be a major improvement on traditional non-steroidal anti-inflammatory drugs (NSAIDs) for the treatment of pain and inflammation. These COX-2 inhibitors were expected to be safer due to the lack of gastrointestinal and other NSAID side effects associated with inhibition of COX-1 (Flower, 2003). However, the safety of selective COX-2 inhibitors was soon in question, as rofecoxib, but not celecoxib, both selective COX-2 inhibitors, was shown to significantly increase the risk of cardiovascular events (McGettigan and Henry, 2006; White et al., 2007).
The factors that distinguish celecoxib from rofecoxib and other selective COX-2 inhibitors are still a matter for debate. One possible explanation is that some beneficial effects of celecoxib may antagonize the deleterious effects inherent in COX-2 inhibitors. Indeed, patients receiving celecoxib have a significantly lower risk of developing hypertension compared with those receiving rofecoxib (Cho et al., 2003; Aw et al., 2005). Emerging and accumulating evidence suggests that celecoxib can affect cellular targets other than COX. Ion channels are prominent among these non-COX targets as new candidates for the molecular basis of some of the actions of celecoxib. In this regard, recent work by Brueggemann and colleagues proposes the interesting and attractive hypothesis that the beneficial effects of celecoxib on the cardiovascular system may result from its actions on two types of ion channels, the Kv7 potassium and L-type calcium channels (Brueggemann et al., 2009, see also Shapiro, 2009; channel nomenclature follows Alexander et al., 2009). Celecoxib, but not rofecoxib or diclofenac, enhanced Kv7 (possibly Kv7.5) and suppressed L-type voltage-sensitive calcium currents in A7r5 rat aortic smooth muscle cells and freshly isolated rat mesenteric artery myocytes (Brueggemann et al., 2009). This study also showed that celecoxib, but not rofecoxib, inhibited calcium responses to vasopressin in A7r5 cells and dilated intact and endothelium-denuded rat mesenteric arteries. The authors concluded that these effects were independent of COX-2 inhibition and may explain the differential risk of cardiovascular events in patients taking different selective COX-2 inhibitors (Brueggemann et al., 2009).
KCNQ genes encode K+ channel subunits of the Kv7 family. There are five members of this family: Kv7.1 to Kv7.5 (corresponding to KCNQ1–KCNQ5). Of these members, four (Kv7.2– Kv7.5) are expressed in the nervous system (Jentsch, 2000; Robbins, 2001). Kv7.2 and Kv7.3 are the principal subunit components of the slow, voltage-gated M-channel, which widely regulates neuronal excitability, although other subunits may contribute to M-type currents in some locations (Brown and Passmore, 2009). In vascular smooth muscle cells, the discovery of several Kv7 subtypes raises the possibility of controlling vascular tone by M-channel activity (Mackie and Byron, 2008). Indeed, Kv7.1, Kv7.4 and Kv7.5 channels have all been identified in vascular smooth muscle cells (Yeung et al., 2007; Zhong et al., 2010), and Kv7 channel activity seems instrumental to the vasoconstrictive response to the hormone arginine vasopressin (Mackie et al., 2008; Brueggemann et al., 2009) or to phenylephrine (Yeung et al., 2007). The Kv7.1 channel is mainly expressed in cardiac myocytes and the Kv7.1/KCNE1 channel complex that underlies the delayed rectifier potassium current, IKs, is key for controlling the duration of the action potential of the human heart (Barhanin et al., 1996; Sanguinetti et al., 1996). The importance of Kv7 channels can best be assessed by the mutations in the genes for four of these subunits (Kv7.1– Kv7.4) that give rise to genetic disorders in humans (Jentsch, 2000).
Although the Kv7.5 subunit has been suggested as a target of celecoxib activation (Brueggemann et al., 2009), a systematic study on celecoxib modulation of Kv7 channels is lacking. This question is worthy of investigation, considering the physiological importance of Kv7 channels, the wide prescription of celecoxib as a NSAID and its other potential clinical uses. In this study, we have characterized the effects of celecoxib on all members of the Kv7 channel family and compared its effects with the well-established Kv7 channel opener, retigabine, and the non-COX-inhibitor analogue, 2,5-dimethyl-celecoxib (DM-celecoxib) (Figure S1). The mechanism of action for celecoxib was also studied.
Methods
cDNA constructs
Plasmids encoding human Kv7.1, human Kv7.2, rat Kv7.3, human Kv7.4 and human Kv7.5 channels (GenBank accession numbers: NM000218, AF110020, AF091247, AF105202 and AF249278 respectively) were kindly provided by Diomedes E. Logothetis (Virginia Commonwealth University, Richmond, VA, USA) and subcloned into pcDNA3. Kv7.2(A235T) and Kv7.2(W236L) mutants were kindly provided by Min Li (Johns Hopkins University, Baltimore, MD, USA). Kv7.1(T265A) and Kv7.1(L266W) mutants were produced by Pfu DNA polymerase with a QuickChange kit (Stratagene, La Jolla, CA, USA). The structure of the mutants was confirmed with DNA sequencing.
HEK293 cell culture and transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics in a humidified incubator at 37°C (5% CO2). The cells were seeded on glass coverslips in a 24-multiwell plate and transfected when 60–70% confluence was reached. For transfection of six wells of cells, a mixture of 3 µg Kv7 in pcDNA3 (1.5 µg cDNA for each channel subunit when two channel subunits were co-expressed), pEGFP-N1 cDNAs and 3 µL Lipofectamine 2000 reagent (Invitrogen) were prepared in 1.2 mL of DMEM and incubated for 20 min according to the manufacturer's instructions. The mixture was then applied to the cell culture wells and incubated for 4–6 h. Recordings were made 24 h after cell transfection, and the cells were used within 48 h.
Rat superior cervical ganglion neuron culture
All animal care and experimental procedures were approved by the Animal Care and Ethical Committee of Hebei Medical University (Shijiazhuang, China) under policies adhering to IASP guidelines for use of animals. Superior cervical ganglia (SCGs) were isolated from 10- to 17-day-old Sprague Dawley rats, cut into pieces, transferred into a collagenase solution (1 mg·mL−1) and incubated for 30 min at 37°C. The ganglia were then placed into trypsin solution (2.5 mg·mL−1) for 30 min at 37°C. The digested fragments were then rinsed with 2 mL DMEM plus 10% fetal bovine serum three times, centrifuged and dissociated by trituration. The isolated cells were plated onto glass coverslips pre-coated with poly-D-lysine and incubated at 37°C. After the neurons had attached to the coverslips, the cell culture medium was changed to Neurobasal plus B27 supplement (Invitrogen). The neurons were cultured for 1 day and used within 24 h.
Electrophysiology
For current measurements in the SCG neurons and HEK293 cells, recordings were performed using the perforated (amphotericin B, 250 µg·mL−1, Sigma) whole-cell configuration of the patch-clamp technique. The signals were amplified using an Axon 700B patch-clamp amplifier (Axon Instruments) and filtered at 2 kHz. Patch electrodes were pulled with a Flaming/Brown micropipette puller (Sutter Instruments) and fire-polished to a final resistance of 1–2 MΩ when filled with internal solution. To reduce the errors of the voltage clamping arising from the series resistance, the Kv7 channels were expressed in a low level so that the maximum current amplitudes were at most cases less than 3 nA; we also used series resistance compensation which normally reached 60–80%. The access resistance in our experiments was measured to be around 7–12 Mohms. Thus, the maximum voltage errors were less than 10 mV (around a few mV in most cases). Data acquisition was achieved using the pClamp 10 software. The internal solution for the HEK293 cell and rat SCG neuron recording was as follows (in mM): 150 KCl, 5 MgCl2 and 10 HEPES, adjusted to pH 7.4 with KOH. The external solution for the HEK293 cells and SCG neurons contained the following (in mM): 160 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES and 10 glucose, adjusted to pH 7.4 with NaOH. The perfusion system was a homemade 100 µL perfusion chamber through which solution flowed continuously at 1–2 mL·min−1. Drugs were applied to the cells by gravity via a BPS-8 valve control system (Scientific Instruments). All recordings were carried out at room temperature.
Homology modelling and docking
The method used for the homology modelling and docking is similar to the method described by Wuttke et al. (2005). The MthK channel structure (1LNQ) and the KcsA channel structure (1BL8) were downloaded from the Protein Data Bank (http://www.pdb.org). The three-dimensional structures of the S5 to S6 domains of Kv7.2 subunitswere constructed based on the solved crystal structures of the corresponding domains of MthK/KscA. The Kv7.2 structures were generated using SWISS-MODEL (Arnold et al., 2006; Kiefer et al., 2009) and were energy optimized using NAMD (Phillips et al., 2005) in default settings. Manual docking of celecoxib and DM-celecoxib molecules which were drawn with Gaussian viewer was performed with Auto dock 4.0.
Data analysis and statistics
Currents were analysed and fitted using the Clampfit 10 (Axon Instrument) and Origin 7.5 (Originlab Corporation) software. The current amplitudes were measured without leak subtraction and the details were given in the Figure legends. The current activation curves were generated by plotting the normalized tail current amplitudes against the step potentials and were fitted with a Boltzmann function: y = A/{1 + exp[(Vh− Vm)/k]}, where A is the amplitude of relationship, Vh is the voltage for half-maximal activation, Vm is the test potential and k is the slope. The kinetics of activation and deactivation were fitted with an exponential function (Clampfit 10): 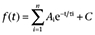 , from which the time constants τ were obtained. Results are expressed as the mean ± SEM. Differences between group means was analysed with one-way anova, followed by the Bonferroni post hoc test. The differences were considered significant if P < 0.05.
, from which the time constants τ were obtained. Results are expressed as the mean ± SEM. Differences between group means was analysed with one-way anova, followed by the Bonferroni post hoc test. The differences were considered significant if P < 0.05.
Materials
Celecoxib, DM-celecoxib and retigabine were synthesized in the Department of New Drug Development, School of Pharmacy, Hebei Medical University. Celecoxib was also purchased from Matrix Scientific (Columbia, SC, USA). The other chemicals were all purchased from Sigma (St. Louis, MO, USA). The stock solutions for celecoxib (100 mM), DM-celecoxib (100 mM) and RTG (100 mM) were made in dimethyl sulphoxide and were stored at −20°C. The celecoxib and RTG solutions were freshly prepared from stock solutions before each experiment and protected from light.
Results
Celecoxib inhibits Kv7.1 currents and enhances Kv7.2–7.4 and Kv7.3/ Kv7.5 currents
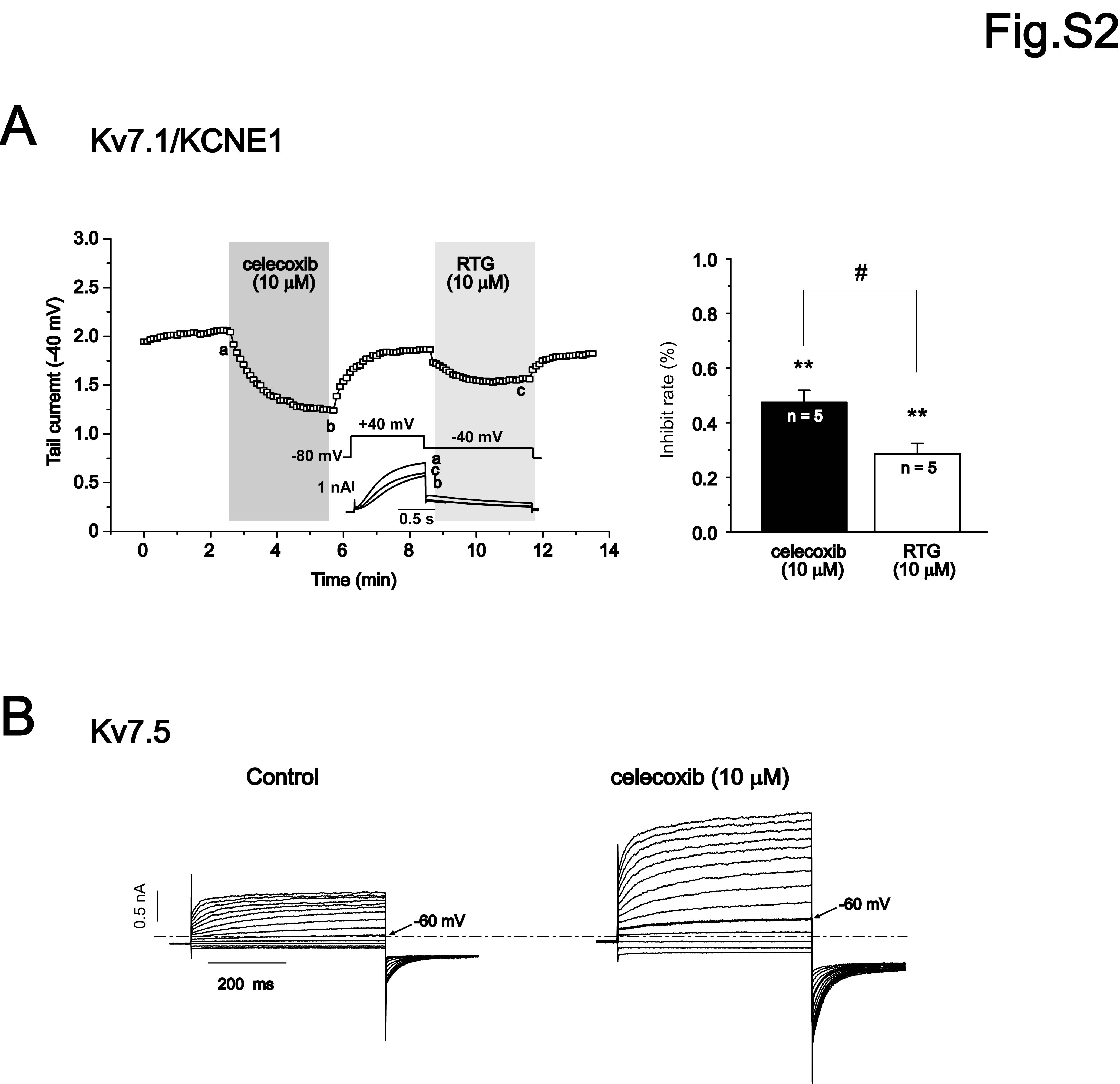
We started our experiments by studying the effects of celecoxib on homomeric Kv7.1–7.4 and heteromeric Kv7.3/ Kv7.5 channels expressed in HEK293 cells. The effects of celecoxib were compared with those of retigabine, a well established Kv7 channel opener (Figure S1) (Tatulian et al., 2001). Figure 1 shows the current traces of the Kv7 channels recorded using the voltage protocol shown at the top of the Figure. Celecoxib affected the Kv7 currents in a manner qualitatively similar to retigabine; thus, both drugs inhibited Kv7.1 currents while enhancing Kv7.2– Kv7.4 and Kv7.3/ Kv7.5 currents (Figure 1). The effects of celecoxib and retigabine were quantified by measuring the changes in the current amplitude at −20 mV induced by the drugs (indicated by the arrows in Figure 1A–E), and the results are summarized in Figure 1F. Both celecoxib and retigabine induced a significant inhibition in Kv7.1 currents, but the effect of celecoxib was more substantial (the maximum current at −20 mV was inhibited 50 ± 4% and 35 ± 8% by celecoxib and retigabine respectively (P < 0.05). Celecoxib 10 µM also inhibited the Kv7.1/KCNE1 currents (currents were reduced by 47 ± 4%, n = 5). Similarly, retigabine at 10 µM inhibited Kv7.1/KCNE1 currents (Figure S2A), although less than celecoxib (currents were reduced by 29 ± 4%, n = 5).
Figure 1.
The effects of celecoxib and retigabine on Kv7 currents. The whole-cell currents were recorded from HEK293 cells expressing Kv7 channels by using the voltage protocol shown at the top of the Figure. The current traces of homomeric Kv7.1–7.4 (A–D) and heteromeric Kv7.3/Kv7.5 channels (E) induced by the multiple depolarization steps were shown. The perforated whole-cell patch clamp technique was used. The cells were held at −80 mV. The arrows indicate the currents induced by the −20 mV step potential. For each cell, the effects of 10 µM celecoxib and 10 µM retigabine (RTG) on Kv7 currents were tested. Either celecoxib or retigabine was applied first and the second drug was always applied after the first drug had been washed out (3–5 min). Celecoxib and retigabine were applied until the effects were stabilized, normally 1–3 min. (F) Normalized current amplitudes at −20 mV. The stead state current amplitudes were measured at the end of the depolarizing steps. *P < 0.05, **P < 0.01, compared with the current amplitudes in the absence of celecoxib, which were taken as 1. n = 5–8.
Celecoxib significantly increased the currents of Kv7.2 and Kv7.4 at −20 mV by 95 ± 18%, and 144 ± 32% respectively (Figure 1F). However, the currents of Kv7.3 at −20 mV was not significantly affected (Figure 1F). Although we could sometimes detect the currents of Kv7.5 expressed in HEK293 cells and the currents were also enhanced by celecoxib (Figure S2B), the lack of consistency in measurable Kv7.5 currents prevented a systematic study on celecoxib modulation of homomeric Kv7.5 currents. We encountered the same problem in our previous study (Liu et al., 2008). Because we studied the effects of celecoxib in close comparison with retigabine and because most of the information concerning the effects of retigabine is from heteromeric Kv7.3/Kv7.5 currents (Wickenden et al., 2000; Wickenden et al., 2001), we characterized the effects of celecoxib on heteromeric Kv7.3/Kv7.5 currents instead. As shown in Figure 1E and F, celecoxib increased Kv7.3/ Kv7.5 currents measured at −20 mV by 117 ± 24%.
I-V curves for voltage-dependent activation of Kv7 currents were established from the tail currents at −140 mV, and the half-activation potential (V1/2) was obtained from the fitting functions described in Methods. The V1/2 changes (ΔV1/2) induced by celecoxib and retigabine are summarized in Table 1. Celecoxib and retigabine did not affect the V1/2 of Kv7.1 currents (Table 1). By contrast, celecoxib and retigabine significantly shifted the V1/2 of Kv7.2–7.4 and Kv7.3/Kv7.5 currents to more negative potentials (Table 1). However, it is clear from the data shown in Table 1 that retigabine is more effective at negatively shifting the voltage-dependent activation of Kv7 currents.
Table 1.
The effects of celecoxib and retigabine (RTG) on the voltage-dependent activation and activation and deactivation kinetics of Kv7 currents
| Kv7.1 | Kv7.2 | Kv7.3 | Kv7.4 | Kv7.3/Kv7.5 | Kv7.2/Kv7.3 | Kv7.2(A235T) | Kv7.2(W236L) | ||
|---|---|---|---|---|---|---|---|---|---|
| ΔV1/2 (mV) | Celecoxib (10 µM) | −5.7 ± 1.6 | −13.8 ± 0.9** | −7.8 ± 0.9** | −20.4 ± 4.2** | −7.8 ± 2.9** | −11.5 ± 2.1** | +32.3 ± 4.9** | −4.0 ± 3.2 |
| NS | NS | ||||||||
| RTG (10 µM) | −2.1 ± 1.0 | −40.0 ± 1.0** | −37.9 ± 2.1** | −27.3 ± 3.1** | −8.7 ± 2.5** | −35.6 ± 2.8** | −7.6 ± 5.1* | −3.0 ± 8.7 | |
| NS | NS | ||||||||
| τ (activation) (ms) | Control | 255 ± 18 | 119 ± 6 | 77 ± 5 | 98 ± 17 | 121 ± 13 | 60 ± 5 | ||
| Celecoxib (10 µM) | 318 ± 26** | 103 ± 6* | 58 ± 5** | 93 ± 15 | 105 ± 11** | 49 ± 3** | |||
| NS | |||||||||
| τ (deactivation) (ms) | Control | 208 ± 15 | 48 ± 2 | 124 ± 5 | 60 ± 4 | 120 ± 16 | 40 ± 2 | ||
| Celecoxib (10 µM) | 311 ± 16** | 73 ± 5** | 159 ± 9** | 90 ± 6** | 166 ± 21** | 64 ± 4** |
P < 0.05
P < 0.01 compared with the control
NS, not significant compared with the control.ΔV1/2– the difference of the half-activation potential before and after treatment with celecoxib or retigabine, ΔV1/2 = V1/2 (after) − V1/2 (before).
n = 4–6 for ΔV1/2 measurements; n = 5–6 for τ measurements.
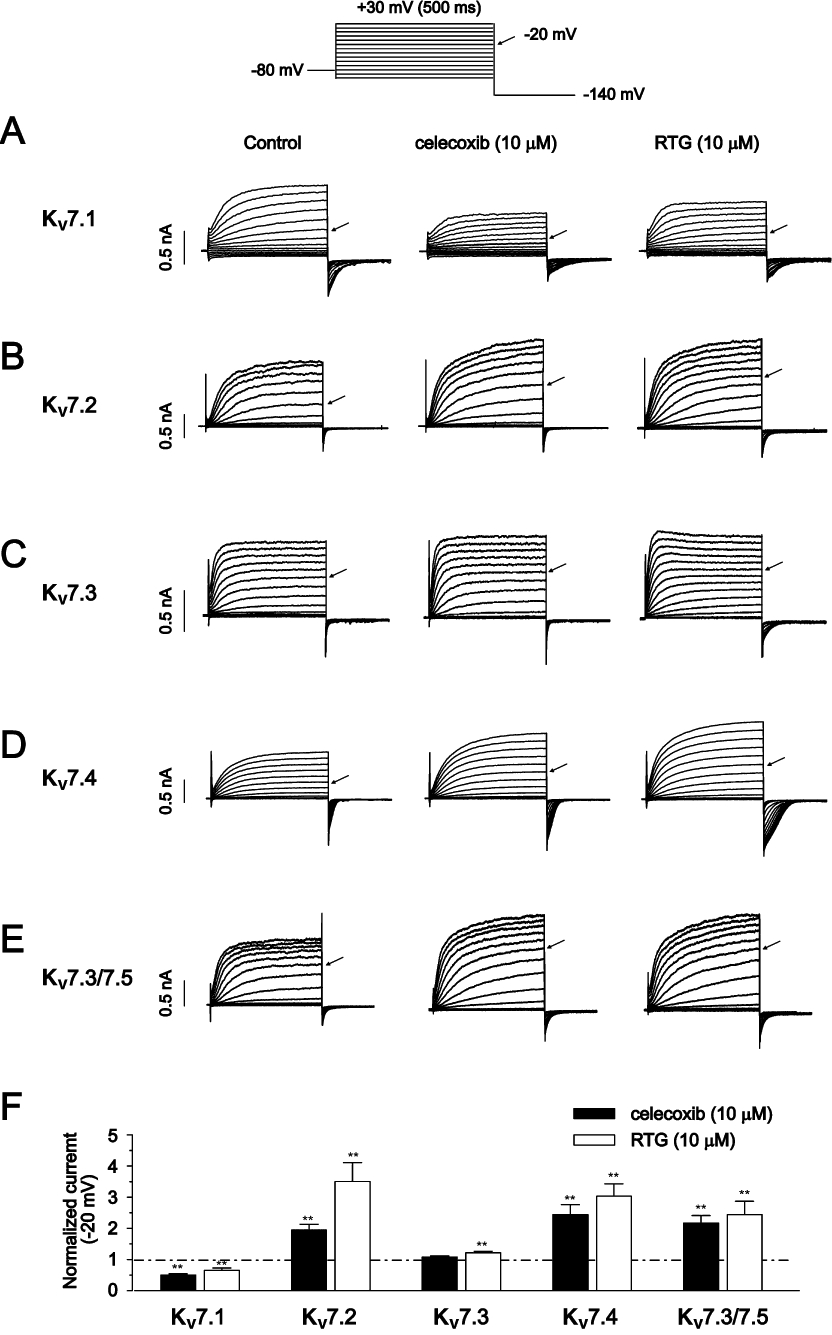
The effects of celecoxib on the activation and deactivation kinetics of Kv7 channels were also tested, and the results are shown in Figure 2. The current activation traces induced by the depolarization step to −20 mV and the tail current deactivation traces at −60 mV before and after application of celecoxib are shown. The activation and the deactivation current traces were fitted with exponential functions. Celecoxib slowed the activation of Kv7.1 currents (Figure 2A) while enhancing the activation of Kv7.2 (Figure 2B), Kv7.3 (Figure 2C) and Kv7.3/Kv7.5 (Figure 2E); the activation kinetics of Kv7.4 channels were not significantly affected (Figure 2D). Celecoxib also slowed the deactivation processes of all Kv7 currents (Figure 2A–E, Table 1).
Figure 2.
The effects of celecoxib on the activation and deactivation kinetics of Kv7 currents. The activation and deactivation of Kv7 currents were recorded by the depolarization step of −20 mV and the repolarization step of −60 mV, respectively, and the protocol was shown at the top of the figure. The amplitudes of the control current traces were either scaled down (for Kv7.1 currents in A) or scaled up (for Kv7.2–7.5 currents in B–E) to normalize to the amplitudes of Kv7 currents in the presence of 10 µM celecoxib. Both activation and deactivation of Kv7 currents were fitted with the exponential function described. The time constants from these fittings were shown in the right panel. n = 5–6. *P < 0.05, **P < 0.01 compared with the currents in the absence of celecoxib.
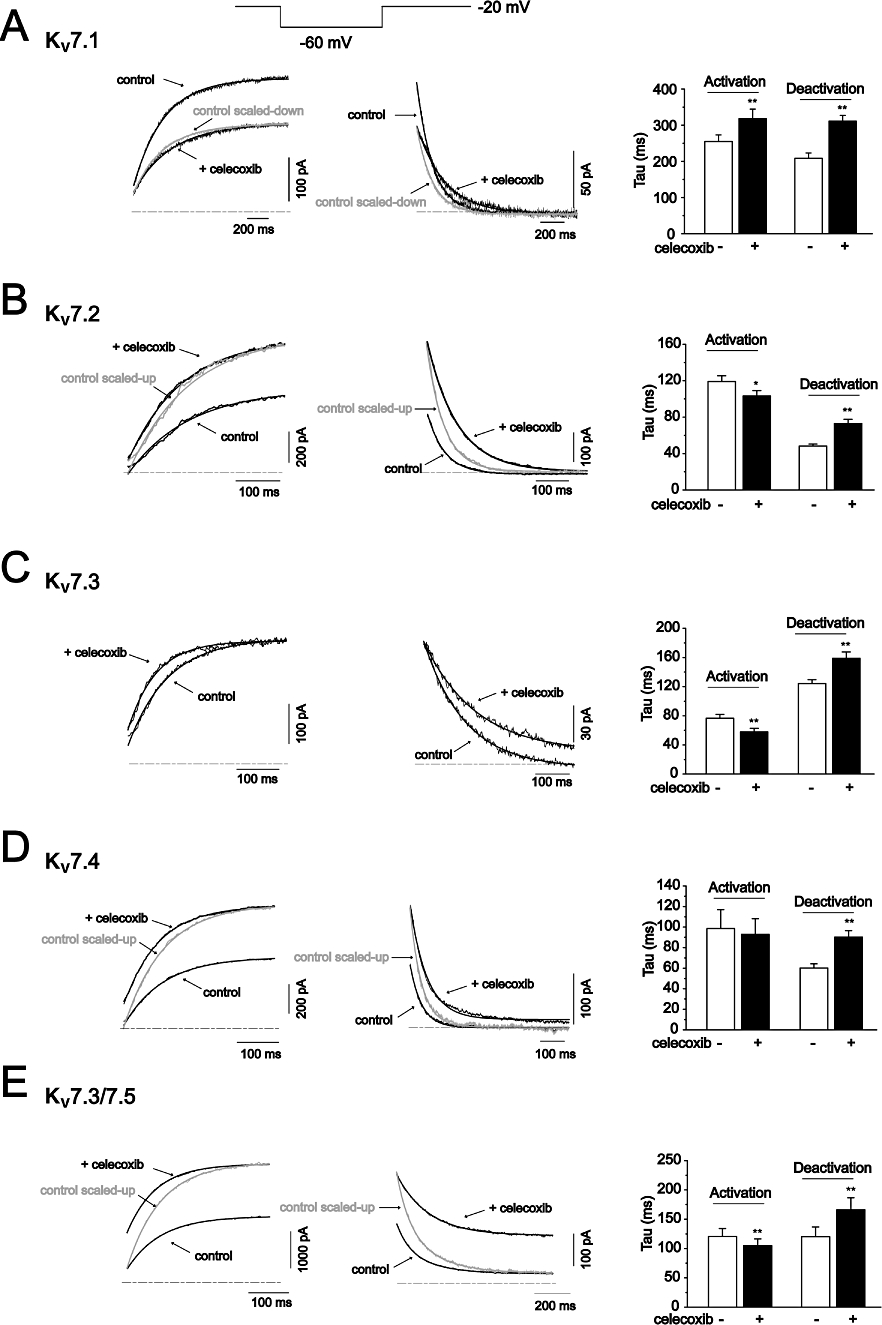
The concentration–response relationship of celecoxib was then established for each of the expressed Kv7 channels (Figure 3). For this investigation, Kv7 currents activated at −20 mV and −60 mV were measured at different concentrations of celecoxib. The concentration–response curves were constructed from the normalized currents over the control measured at −20 mV. Celecoxib concentration-dependently inhibited Kv7.1 currents and enhanced Kv7.2, Kv7.4 and Kv7.3/Kv7.5 currents at −20 mV. Celecoxib affected Kv7 currents with similar potency and efficacy. Thus, celecoxib began affecting Kv7 currents at approximately 1 µM and reached a maximal effect at approximately 10–30 µM (Figure 3). At the maximal effect, celecoxib inhibited Kv7.1 currents by ∼60% and increased Kv7.2, Kv7.4 and Kv7.3/ Kv7.5 currents by ∼100%. The IC50 of celecoxib for Kv7.1 was 4.00 ± 0.13 µM; and the EC50s of celecoxib for Kv7.2, Kv7.4 and Kv7.3/Kv7.5 were 3.09 ± 0.27 µM, 3.37 ± 0.26 µM and 2.27 ± 0.4 µM respectively. Celecoxib did not significantly affect Kv7.3 currents when measured at −20 mV (see also Figure 1F), but did concentration-dependently increase the current measured at −60 mV (Figure 3C). This result was consistent with celecoxib not increasing the maximum current amplitude but negatively shifting the voltage-dependent activation of Kv7.3 currents, as described above in Table 1.
Figure 3.
The concentration-dependent effects of celecoxib on Kv7 currents. The concentration–response relationships of celecoxib on homomeric Kv7.1–7.4 currents (A–D) and heteromeric Kv7.3/ Kv7.5 (E) were shown. The left panel shows the time courses of concentration-dependent modulation of the Kv7 currents recorded at −20 mV (upper line) and −60 mV (lower line). The currents were recorded using the protocol shown in Figure 3. The current amplitudes were measured every 1 s. The right panel shows the fitted curves for the concentration-dependent effects of celecoxib on Kv7 currents recorded at −20 mV. The dotted line indicates the control current level before celecoxib application. The concentration–response relationships were fitted with the logistic function. The IC50 is 4.00 ± 0.13 µM (Kv7.1). The EC50 values are 3.09 ± 0.27 µM (Kv7.2), 3.37 ± 0.26 µM (Kv7.4) and 2.27 ± 0.40 µM (Kv7.3/ Kv7.5), n = 2–14.
Celecoxib modulates Kv7 currents with a mechanism similar to that of retigabine
Both celecoxib and retigabine inhibited Kv7.1 and activate Kv7.2– Kv7.4 and Kv7.3/Kv7.5 currents, and they both negatively shifted the voltage-dependent activation and slowed the deactivation of Kv7.2– Kv7.4 and Kv7.3/Kv7.5 channels. These data suggest that celecoxib may share a mechanism with retigabine for modulating Kv7 channel functions. It has been suggested that retigabine opens Kv7.2 channels by binding to the channel activation gate (Wuttke et al., 2005). A tryptophan residue at the cytoplasmic end of S5, Trp236, has been suggested as critical for this binding and for activation of the channel. Trp236 is conserved in Kv7.2–7.5 but is replaced by a leucine in the case of Kv7.1 channels. Mutation of Trp236 to leucine (Kv7.2(W236L)) abolished the effects of retigabine on Kv7.2 channels (Wuttke et al., 2005). A residue adjacent to Trp236, Ala235, is also conserved in Kv7.2–7.5 channels but is replaced by a threonine in the case of Kv7.1 channels. This Ala235 seems to play a minor role in the activation of Kv7.2 channels by retigabine (Wuttke et al., 2005). We tested the effects of celecoxib on the mutant channels, Kv7.2(W236L) and Kv7.2(A235T), compared with those of retigabine (Figure 4). For Kv7.2(A235T) channels, the stimulatory effect of celecoxib on Kv7.2 currents was reversed to become a potent inhibitory effect, whereas the stimulatory effect of retigabine remained (Figure 4A and B). For Kv7.2(W236L) channels, the stimulatory effects of both celecoxib and retigabine disappeared, and celecoxib became inhibitory (Figure 4A and C). The effects of celecoxib and retigabine on these two Kv7.2 channel mutants were further quantified, as shown in Figure 4B–E. Celecoxib inhibited Kv7.2(A235T) and Kv7.2(W236L) currents, concentration-dependently (Figure 4B and C). By comparison, celecoxib at 10 µM increased Kv7.2 currents by 81 ± 6% and inhibited Kv7.2(A235T) currents by 82 ± 3% and Kv7.2(W236L) currents by 31 ± 3% (Figure 4E). By contrast, retigabine at 10 µM increased Kv7.2 currents by 309 ± 59% and increased Kv7.2(A235T) currents by only 49 ± 4% and did not affect Kv7.2(W236L) currents (Figure 4E). Thus, as is the case for retigabine, Trp236 in Kv7.2 channels is critical for celecoxib-induced activation of the channels.
Figure 4.
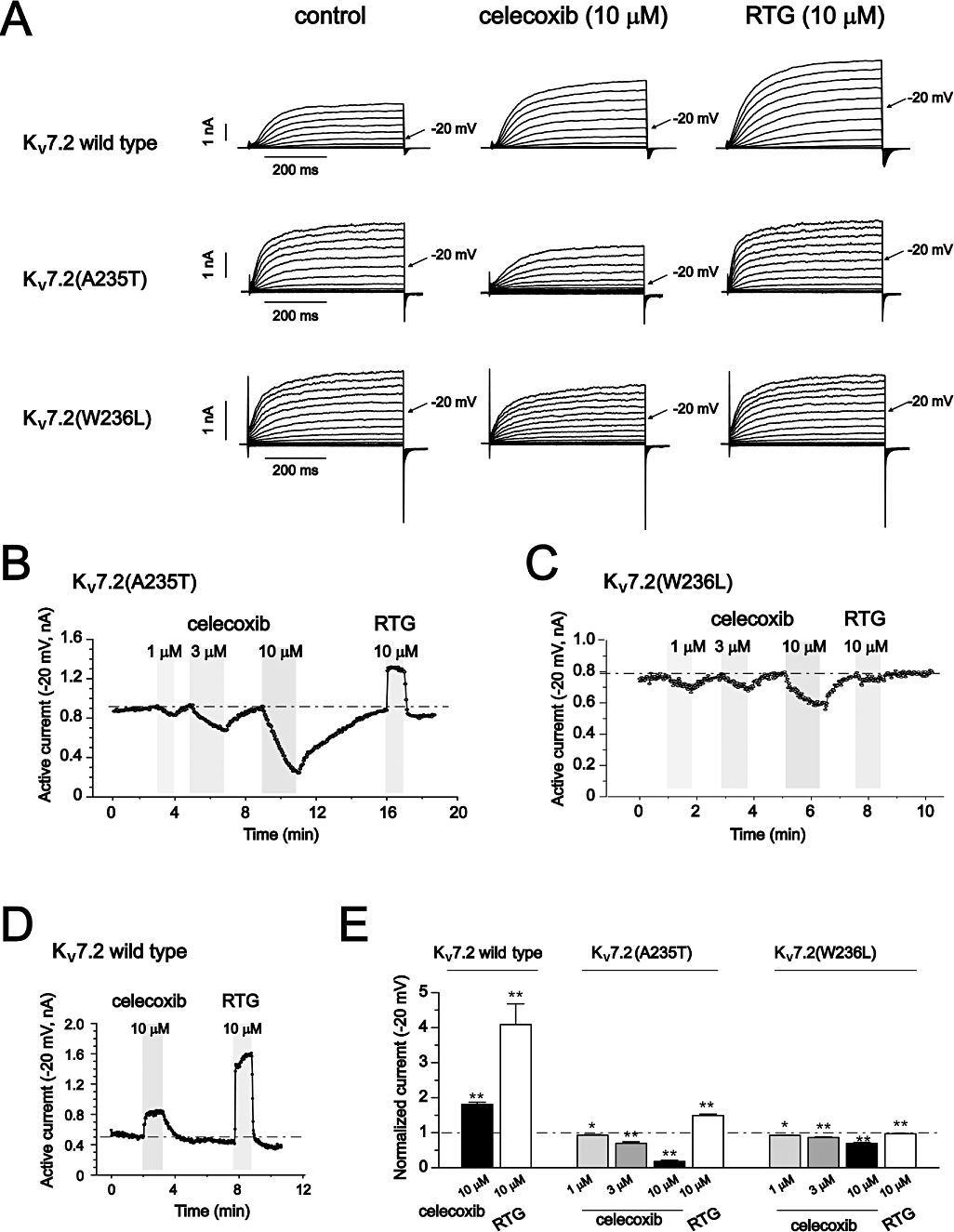
The effects of celecoxib and retigabine on Kv7.2(A235T) and Kv7.2(W236L) currents. (A) The current traces of Kv7.2, Kv7.2(A235T) and Kv7.2(W236L) recorded using the voltage protocol shown in Figure 1. The effects of 10 µM celecoxib and 10 µM retigabine (RTG) are shown. (B–D) The time courses for the effects of celecoxib and retigabine on Kv7.2(A235T) (B), Kv7.2(W236L) (C) and Kv7.2 (D) currents recorded at −20 mV. (E) The normalized currents recorded at −20 mV. The control current amplitudes before celecoxib and retigabine application were taken as 1. *P < 0.05, **P < 0.01 compared with the control currents in the absence of celecoxib or retigabine, n = 3–8.
Unlike wild-type Kv7.2, the voltage-dependent activation of Kv7.2(W236L) currents was no longer shifted by celecoxib or retigabine (Table 1). Furthermore, the voltage-dependent activation of Kv7.2(A235T) currents was positively shifted by celecoxib but was modestly shifted by retigabine towards a more negative potential (Table 1).
To further evaluate the importance of Ala235 and Trp236 in the effects of celecoxib described above, these residues were introduced into the corresponding sites in Kv7.1 channels and the effects of celecoxib and retigabine tested. For Kv7.1(L266W) channels, celecoxib and retigabine, both at 10 µM, inhibited the current amplitude recorded at −20 mV by 45 ± 2% (n = 5) and 29 ± 6% (n = 3) respectively, which are similar to celecoxib- and retigabine-induced inhibition of wild-type Kv7.1 channels (50 ± 4% and 35 ± 8%, Figure 1F). For the mutant Kv7.1(T265A) channels, celecoxib inhibited the current amplitude recorded at 0 mV by 39 ± 3% (n = 4), whereas retigabine did not inhibit the currents (Figure S3). We measured Kv7.1(T265A) currents at 0 mV because it has lower current density than the wild type Kv7.1 channels. For comparison, celecoxib and retigabine inhibited wild type Kv7.1 currents recorded at 0 mV by 40 ± 7% (n = 4) and 23 ± 4% (n = 4) respectively (Figure S3).
The effects of celecoxib on Kv7 channels do not depend on its inhibition of COX
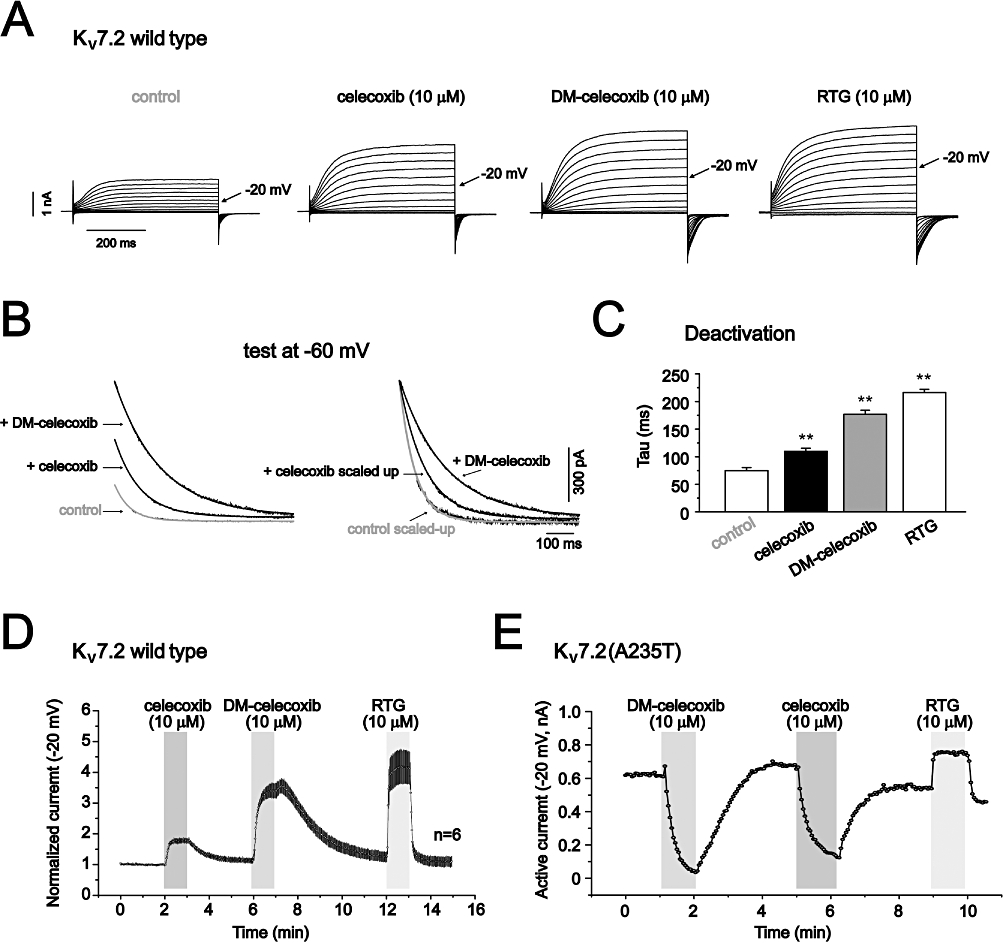
Previous work has shown that DM-celecoxib, a celecoxib analogue that does not inhibit COX-2 (Schonthal et al., 2008), mimics the effects of celecoxib on activation of K+ currents and inhibition of Ca2+ currents (Brueggemann et al., 2009). We tested whether DM-celecoxib could also mimic the effects of celecoxib on Kv7.2, Kv7.2(A235T) and Kv7.2(W236L) currents. Indeed, as shown in Figure 5, DM-celecoxib had similar effects to celecoxib on currents in both Kv7.2 channels and its mutants. At the same concentration, in fact, DM-celecoxib affected Kv7.2 currents to a greater extent than did celecoxib (Figure 5A). The voltage-dependent activation was further shifted to more negative potentials (−22.5 ± 1.9 mV, n = 3), and the deactivation kinetics were further slowed (Figure 5B and C). Actually, the effects of DM-celecoxib were closer to those of retigabine than those of celecoxib, with respect to the increase of the current amplitude (Figure 5A, D and G), the shifting of voltage-dependent activation and the slowing of the deactivation processes (Figure 5B and C).
Figure 5.
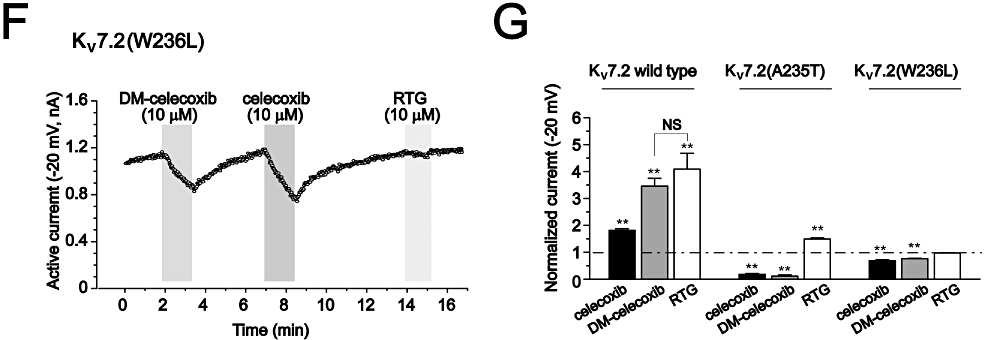
The effects of DM-celecoxib on Kv7 currents. (A) The currents traces of Kv7.2 recorded using the voltage protocol shown in Figure 1. The effects of 10 µM celecoxib, 10 µM DM-celecoxib and 10 µM retigabine (RTG) were shown. (B) The effects of celecoxib and DM-celecoxib on the deactivation kinetics of Kv7.2 currents recorded at −60 mV (the currents were first activated at −20 mV). The control and the celecoxib-activated currents were normalized based on the currents activated by DM-celecoxib. (C) Summarized data of the deactivation time constants. (D–F) The time courses for the effects of celecoxib, DM-celecoxib and retigabine on Kv7.2 (D), Kv7.2(A235T) (E) and Kv7.2(W236L) (F) currents recorded at −20 mV. Note that the Kv7.2 (D) currents were from normalized currents of six recordings. (G) Normalized currents recorded at −20 mV. The control current amplitude before celecoxib, DM-celecoxib and retigabine were taken as 1. *P < 0.05, **P < 0.01 compared with the control currents in the absence of celecoxib, DM-celecoxib or retigabine, n = 6–8.
Importantly, DM-celecoxib affected Kv7.2(A235T) and Kv7.2(W236L) currents in a manner similar to celecoxib (Figure 5E–G). Also, in both Kv7.2(A235T) and Kv7.2(W236L) channels, the effects of DM-celecoxib were reversed from activation to inhibition (Figure 5E and F). DM-celecoxib and celecoxib inhibited Kv7.2(A235T) and Kv7.2(W236L) currents to a similar extent (Figure 5G).
The effects of celecoxib on Kv7/M-type currents
We then examined whether celecoxib could also modulate native Kv7 currents. The Kv7 currents in neuronal cells are best manifested by the M-type currents found in these cells. M-type currents from sympathetic neurons (Brown and Adams, 1980) have been used extensively for the study of M/Kv7 channel modulations. M-channels are most likely composed of Kv7.2 and Kv7.3 subunits (Wang et al., 1998; Hadley et al., 2000; Shapiro et al., 2000). Thus, we first tested the effects of celecoxib on heteromeric Kv7.2/Kv7.3 currents expressed in HEK293 cells. Celecoxib at 10 µM slightly increased the maximal amplitude of Kv7.2/Kv7.3 currents (Figure 6A), but significantly shifted the voltage-dependent activation of Kv7.2/Kv7.3 currents (Table 1). Similar to other Kv7 currents, the deactivation of Kv7.2/Kv7.3 currents was also significantly slowed, and the activation was mildly enhanced (Figure 6B and C, Table 1). Celecoxib increased Kv7.2/Kv7.3 currents in a concentration-dependent manner (Figure 6D) with an EC50 of 4.95 ± 1.16 µM (Figure 6E).
Figure 6.
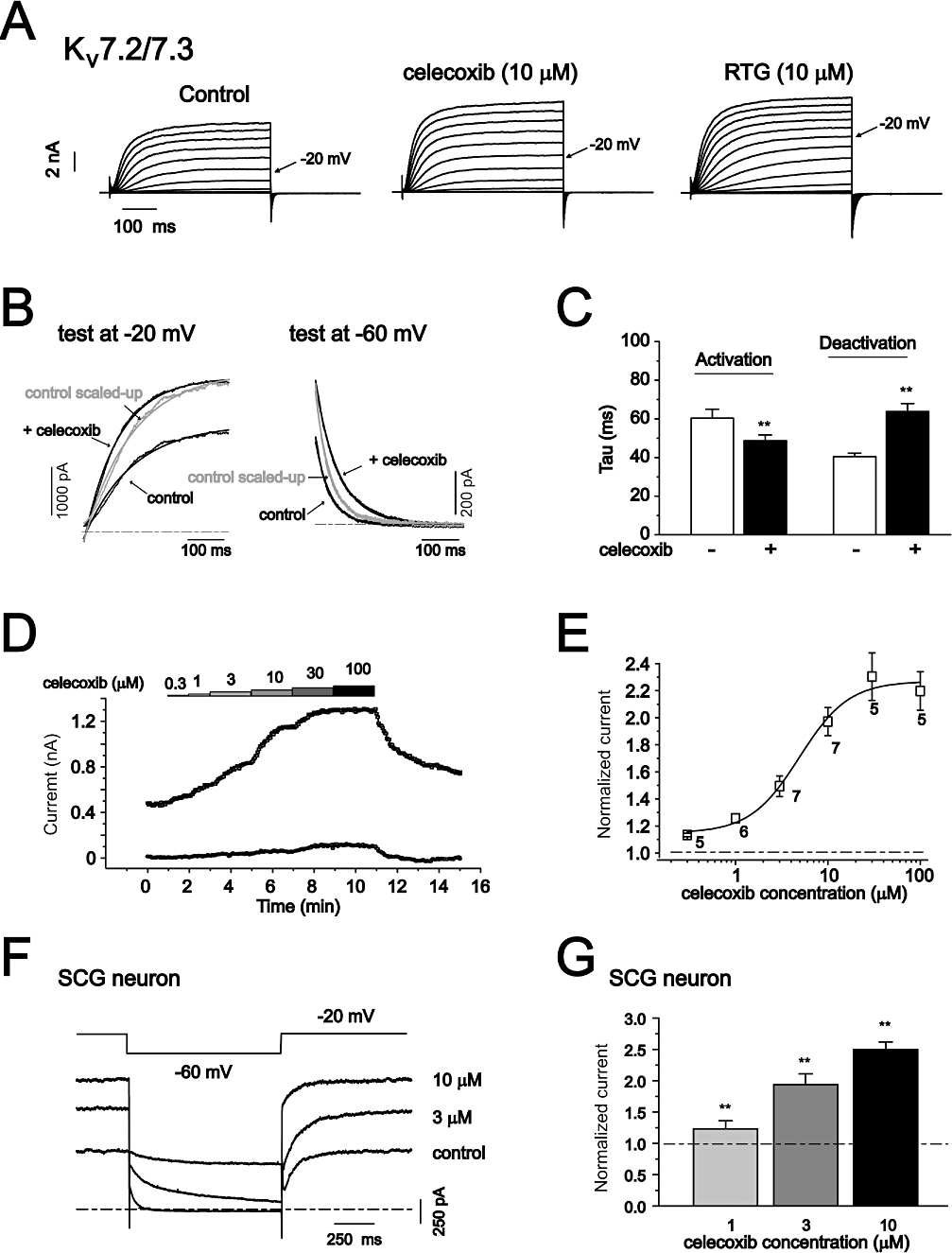
The effects of celecoxib on Kv7.2 Kv7.3 currents and M-type currents from rat SCG neurons. (A) The currents traces of Kv7.2/ Kv7.3 recorded using the voltage protocol shown in Figure 1. The effects of 10 µM celecoxib and 10 µM retigabine (RTG) are shown. (B) The effects of celecoxib on the activation and deactivation kinetics of Kv7.2/ Kv7.3 currents. (C) Summarized data of the activation and deactivation time constants. (D) The time course of concentration-dependent effects of celecoxib on Kv7.2/ Kv7.3 currents recorded using the protocol shown at the right panel. The current traces at −20 mV (upper line) and −60 mV (lower line) were shown. (E) The fitted curve for the concentration–response effect of celecoxib. The EC50 is 4.95 ± 1.16 µM, n = 5–7. (F) M-type currents from rat SCG neurons were recorded using the voltage shown and the effects of different concentration of celecoxib were shown. The M-type currents were measured at −20 mV and the deactivation of the currents were measured at −60 mV. (G) Concentration-dependent activation of M-type currents by celecoxib from SCG neurons. **P < 0.01 compared with the currents in the absence of celecoxib. n = 5.
The effects of celecoxib on the native M-type currents were tested on neurons from rat SCG using the protocol shown at the top of Figure 6F (Adams et al., 1982). Celecoxib enhanced M-type currents in a concentration-dependent manner (Figure 6F and G). At 10 µM, celecoxib increased M-type currents (at −20 mV) by 154 ± 24% (Figure 6G). As for the expressed Kv7.2/Kv7.3 currents, celecoxib also slowed the deactivation of M-type currents. The time constants of current deactivation at −60 mV were 61.2 ± 13.1 ms (n = 4) and 217.2 ± 74.5 ms (n = 4, P < 0.01 compared with the control) for control and celecoxib 10 µM respectively.
Discussion and conclusions
In the present study, we have characterized the effects of celecoxib on the homomeric Kv7.2–7.4 and heteromeric Kv7.2/ Kv7.3, Kv7.3/ Kv7.5 currents of Kv7 channels and on the native neuronal M/ Kv7 currents. The results demonstrate that all Kv7 channels are targets for modulation by celecoxib. Importantly, celecoxib shares a similar mechanism of action with retigabine but also utilizes a distinct mechanism of its own.
In a previous report, Brueggemann and colleagues suggested that celecoxib can activate Kv7 currents (possibly Kv7.5) in smooth muscle cells (Brueggemann et al., 2009). However, it was not clear whether celecoxib modulates all Kv7 channels or affects Kv7 channels in an isoform-specific manner. It was also not clear how the Kv7 channels are modulated by celecoxib. We planned our experiments to use retigabine in parallel with celecoxib throughout the investigations to better understand the effects of celecoxib. The effects of retigabine on expressed Kv7 currents and native M-type currents have been well characterized (Wickenden et al., 2000; Tatulian et al., 2001; Wickenden et al., 2001; Wuttke et al., 2005). Our results with retigabine are generally in agreement with published data. The outstanding features of retigabine's effects on Kv7 currents (with the exception of Kv7.1) include negatively shifting voltage-dependent activation and slowing deactivation of Kv7 currents (Tatulian et al., 2001). In this regard, the effects of celecoxib are similar to those of retigabine. Celecoxib also produced a negative shifting of voltage-dependent activation and a slowing of current deactivation for Kv7.2–7.4, Kv7.2/ Kv7.3 and Kv7.3/ Kv7.5 (Table 1). As has been previously reported for retigabine (Tatulian et al., 2001), neither retigabine nor celecoxib altered the voltage-dependent activation of Kv7.1 currents (Table 1). We also found that both celecoxib and retigabine inhibited Kv7.1 current amplitudes (Figure 1). Thus, in general, the effects of celecoxib on Kv7 currents were qualitatively similar to those of retigabine.
Apart from the effects on the voltage-dependent activation and the deactivation kinetics, we also found that both retigabine and celecoxib enhanced the maximal current amplitudes of Kv7.2, Kv7.4 and Kv7.5, but not Kv7.3 channels. These results were different from those of a previous report, which showed retigabine enhancing the maximal current amplitude from Kv7.3 and Kv7.4, but not Kv7.2 channels (Tatulian et al., 2001). It is not clear what caused this discrepancy, but the different expression systems used (CHO vs. HEK cells) could be one possibility. Nevertheless, celecoxib and retigabine still shared a similar pattern of effects on the maximal currents amplitudes in the present study.
Our results provide the first insight into the mechanism of celecoxib modulation of Kv7 channels. It is almost certain that Trp236 in Kv7.2, and very likely in all of Kv7.2–7.5 channels (as Trp236 is conserved in all these channel proteins), serves as the binding site for retigabine and celecoxib because the mutant channel Kv7.2(W236L) was not activated by retigabine and celecoxib and was in fact modestly inhibited by celecoxib (Figure 4, Table 1). It has been suggested that retigabine can bind to the cytoplasmic sites of the S5 and S6 segments of Kv7.2 channels and that these bindings involve Trp236 in S5 and the gating hinge Gly301 in S6 (Wuttke et al., 2005). These binding sites would favour Kv7.2 channels being in the open conformation, as manifested by the negatively shifted voltage-dependent activation, the slowing of the deactivation and the increased current amplitude, which are shared effects of both retigabine and celecoxib. Our docking simulation results shown in Figure 7 further support that celecoxib and retigabine share a similar mechanism for activation of Kv7 channels. The docking modelling suggests that although the binding energies for both celecoxib and DM-celecoxib to closed and open conformations of Kv7.2 channels are not very different, celecoxib or DM-celecoxib can form hydrogen bonds with open Kv7.2 channels more easily than with closed Kv7.2 channels. Thus, at open state, residues Trp236, Gly239 and Phe304 of Kv7.2 channels form five hydrogen bonds with celecoxib and seven hydrogen bonds with DM-celecoxib. By contrast, at closed state, only the Phe305 of Kv7.2 channels forms one hydrogen bond with DM-celecoxib (Figure 7). Thus, as suggested previously for retigabine, Trp236 is indeed also a binding site for celecoxib and DM-celecoxib at the open state of Kv7.2 channels. Our docking results do not suggest a direct interaction between Gly301 of Kv7.2 channels with celecoxib. However, Phe304 of the Kv7.2 channel, which has been proposed as important for the sensitivity of Kv7.2 channels to retigabine (Wuttke et al., 2005), does interact with celecoxib in our simulation. Thus, as suggested by Wuttke et al. (2005) for retigabine, celecoxib (and DM-celecoxib) are likely to form a interaction complex with Kv7.2 channels in the open conformation, involving some residues around Trp236 of S5 and around the gating hinge (S301) of S6. Introducing Trp236 into Kv7.1 channels did not allow these channels to be activated by celecoxib; instead, Kv7.1(L266W) currents were still inhibited by celecoxib, with to the same extent as the wild type Kv7.1 currents These results are consistent with the previous work (Wuttke et al., 2005). The overall pore structure of Kv7.1 channels might be quite different from Kv7.2 channels, and introduction of a single amino acid may not be sufficient to induce celecoxib activation of Kv7.1 channels, as observed and suggested by Wuttke et al. (2005) for retigabine.
Figure 7.
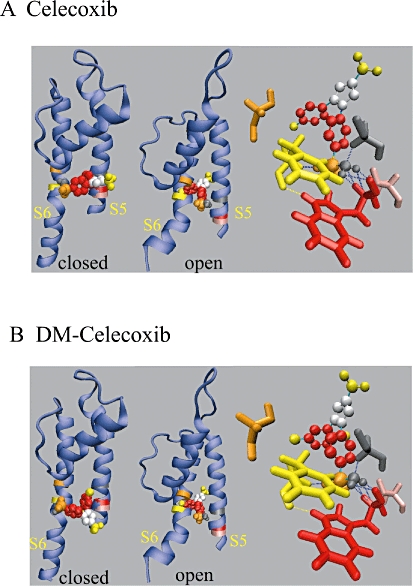
Docking results for the interaction of celecoxib or DM-celecoxib with residues within S5 and S6 of the Kv7.2 channel. Docking results for celecoxib (A) and DM-celecoxib (B) binding to Kv7.2 channels. The closed and the open conformations of S5 and S6 segments with the pore loop were shown. The right panel shows the hydrogen bonds (blue dotted lines) between celecoxib (A), DM-celecoxib (B) and W236, G239 and F304 in open channel state. Residues A235, W236, G239, G301, F304 are shown in pink, red, gray, orange and yellow colour respectively. For the structure of celecoxib and DM-celecoxib (see Figure S1), -CF3, benzene, -SO2, -NH2 are shown as yellow, red, orange and gray spheres respectively. The five-carbon ring is shown as silver spheres. Methyls in the benzene are shown as yellow spheres.
However, it should be noted that there are some distinct differences between celecoxib and retigabine. It is clear that celecoxib has much stronger inhibitory effects on Kv7 1 (and Kv7.1/KCNE1) currents than retigabine, while it has weaker stimulatory effects on Kv7.2–7.5 currents than retigabine. Furthermore, the experiments with Kv7.2(A235T) further suggest a difference between celecoxib and retigabine, as retigabine still enhanced Kv7.2(A235T) currents and negatively shifted voltage-dependent activation, whereas celecoxib profoundly inhibited Kv7.2(A235T) currents and positively shifted the voltage-dependent activation. Because celecoxib inhibited potently both Kv7.1 and Kv7.2(A235T) currents, we thought that the Thr265 residue in Kv7.1 channels might be the crucial amino acid for celecoxib-induced inhibition. However, celecoxib still inhibited Kv7.1(T265A) currents, as well as it did the wild type Kv7.1 currents (Figure S3). Interestingly, contrary to wild-type Kv7.1, the mutant Kv7.1(T265A) channels were no longer inhibited by retigabine (Figure S3). These results suggest that Thr265 did play some role in Kv7.1 channel inhibition (at least for retigabine), and the results also indicate there are some other sites which are also involved in celecoxib-mediated inhibition of Kv7.1 channels.
It is interesting to note that the celecoxib analogue DM-celecoxib, which has no COX-inhibition activity, has similar activity in regulating activity of Kv7 channels. If there is any difference between DM-celecoxib and celecoxib, it is that the former is a stronger activator of Kv7 channel functions (Figure 5). It is clear that DM-celecoxib utilizes the same sites for modulating Kv7 channels as described above for celecoxib (Figures 5 and 7). Thus, it is likely that the difference in potency between DM-celecoxib and celecoxib in their modulation of Kv7 channels arises from their different abilities to interact with the Kv7 channel structures, at sites such as Ala235 and Trp236 in the Kv7.2 subunit, which could be the result of the subtle structural differences between DM-celecoxib and celecoxib (Figure S1). However, our docking modelling work did not give a clear explanation for the differences between celecoxib and DM-celecoxib. The methyls in the benzene ring (the numbers and the positions are different between celecoxib and DM-celecoxib, Figure S1) are not involved in the direct interaction with residues of Kv7.2 channels (Figure 7) and the orientation of two small molecules within Kv7.2 channels is similar (Figure 7). Anyway, the quantitative functional difference between celecoxib and DM-celecoxib may be too small to be resolved by the docking modelling in this present study.
These results provide a new template for developing new modulators for Kv7 channels and provide a new dimension to the study of COX inhibitors and the structure–function relationship of Kv7 and other ion channels. Celecoxib and DM-celecoxib are being investigated for their therapeutic potentials as anticancer agents (Schonthal et al., 2008). Intriguingly, the two pharmacological effects, inhibition of COX-2 and suppression of tumour growth, were found to reside in different structural aspects of the celecoxib molecule; therefore, they could be separated. It will also be interesting to see whether the modulatory effects of celecoxib and its analogue on ion channels contribute to their anticancer activity (Brueggemann et al., 2010).
Finally, what are the physiological implications of the present study? The concentrations at which celecoxib modulates Kv7 channel functions could be clinically relevant. At a therapeutic dose of 200 mg, the peak plasma concentration of celecoxib in healthy adults is around 2 µM (Davies et al., 2000). Clearly, celecoxib at this concentration significantly affected Kv7 channels (Figure 3). Thus, the selective modulation of the Kv7 channels expressed in the vascular system (Kv7.4, Kv7.5) (Yeung et al., 2007; Zhong et al., 2010) by celecoxib, but not by rofecoxib, may underlie their different cardiovascular side effects (Shapiro, 2009; Brueggemann et al., 2010). Kv7.2– Kv7.5 channels are expressed in the nervous system (Jentsch, 2000; Robbins, 2001) and contribute differentially to neuronal M-type currents, depending on the location (Brown and Passmore, 2009). Recently, M/Kv7 currents were found to be important for controlling the excitability of sensory neurons related to pain transduction and opening M/Kv7 channels can antagonize nociceptive behaviour (Passmore et al., 2003; Linley et al., 2008; Liu et al., 2010). It will be interesting to know whether any analgesic effects of celecoxib could result from the activation of M/Kv7channels. Along the same lines, DM-celecoxib, which lacks COX-2 inhibition but has greater Kv7 channel activation activity, would be expected to exert analgesic and anti-epileptic effects, similar to those of retigabine, because retigabine has been developed as a new class of anti-epileptic drug. Another intriguing issue raised recently by Zhou et al. (2010) is that persistent opening of M/Kv7 channels in hippocampal neurons can induce apoptosis of these neurons. In this case, could celecoxib cause neuronal cell death through activation of M/Kv7 channels? Considering that celecoxib is already a widely prescribed COX-2 inhibitor and the new potential clinical indications, such as cancer, are being assessed for celecoxib, these issues are worthy of further investigation.
Acknowledgments
This work was supported by National Natural Science Foundation of China (30730031) to H. Z., (30500112) X. D. and by the 973 Program (2007CB512100) to H. Z. H. Z. was a recipient of National Science Fund for Distinguished Young Scholars of China (30325038). Bingcai Guan read the manuscript and gave valuable suggestions.
Glossary
Abbreviations
- NSAIDs
non-steroidal anti-inflammatory drugs
- SCG
superior cervical ganglion
Conflicts of interest
There are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 The structures of celecoxib, DM-celecoxib and RTG.
{kind=link}
Figure S2 The effects of celecoxib on Kv7.1/KCNEE1 and Kv7.5 currents expressed in HEK 293 cells. (A) The whole-cell currents were recorded from HEK293 cells expressing Kv7.1/E1 with the protocol shown in the inset. The time course of celecoxib and retigabine effects on Kv7.1/E1 tail currents recorded at −40 mV was shown. The percentage inhibition of Kv7.1/E1 currents induced by celecoxib and retigabine was shown in the right panel. (B) The effects of celecoxib on currents of KV7.5 expressed in HEK293. The whole-cell currents were recorded using the voltage protocol shown in Figure 1.
{kind=link}
Figure S3 The effects of celecoxib and retigabine on Kv7.1(T265A) currents. (A) The current traces of Kv7.1, Kv7.1(T265A) recorded using the voltage protocol shown in Figure 1. The effects of 10 μM celecoxib and 10 μM retigabine were shown. (B) The summarized data for the percentage inhibition of Kv7.1 and Kv7.1(T265A) currents recorded at 0 mV. **P < 0.01 compared with the control currents in the absence of celecoxib or RTG, n = 3–5.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams PR, Brown DA, Constanti A. M-currents and other potassium currents in bullfrog sympathetic neurones. J Physiol. 1982;330:537–572. doi: 10.1113/jphysiol.1982.sp014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Aw TJ, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165:490–496. doi: 10.1001/archinte.165.5.IOI50013. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Mani BK, Cribbs LL, Byron KL. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol. 2009;76:1053–1061. doi: 10.1124/mol.109.057844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mani BK, Mackie AR, Cribbs LL, Byron KL. Novel actions of nonsteroidal anti-inflammatory drugs on vascular ion channels: accounting for cardiovascular side effects and identifying new therapeutic applications. Mol Cell Pharmacol. 2010;2:15–19. [PMC free article] [PubMed] [Google Scholar]
- Cho J, Cooke CE, Proveaux W. A retrospective review of the effect of COX-2 inhibitors on blood pressure change. Am J Ther. 2003;10:311–317. doi: 10.1097/00045391-200309000-00002. [DOI] [PubMed] [Google Scholar]
- Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet. 2000;38:225–242. doi: 10.2165/00003088-200038030-00003. [DOI] [PubMed] [Google Scholar]
- Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov. 2003;2:179–191. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci. 2008;28:11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zhang X, Wang C, Zhang G, Zhang H. Antihistamine mepyramine directly inhibits KCNQ/M channel and depolarizes rat superior cervical ganglion neurons. Neuropharmacology. 2008;54:629–639. doi: 10.1016/j.neuropharm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, et al. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J Clin Invest. 2010;120:1240–1252. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie AR, Byron KL. Cardiovascular KCNQ (Kv7) potassium channels: physiological regulators and new targets for therapeutic intervention. Mol Pharmacol. 2008;74:1171–1179. doi: 10.1124/mol.108.049825. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schonthal AH, Chen TC, Hofman FM, Louie SG, Petasis NA. Celecoxib analogs that lack COX-2 inhibitory function: preclinical development of novel anticancer drugs. Expert Opin Investig Drugs. 2008;17:197–208. doi: 10.1517/13543784.17.2.197. [DOI] [PubMed] [Google Scholar]
- Shapiro MS. An ion channel hypothesis to explain divergent cardiovascular safety of cyclooxygenase-2 inhibitors: the answer to a hotly debated puzzle? Mol Pharmacol. 2009;76:942–945. doi: 10.1124/mol.109.059683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- White WB, West CR, Borer JS, Gorelick PB, Lavange L, Pan SX, et al. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol. 2007;99:91–98. doi: 10.1016/j.amjcard.2006.07.069. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Zou A, Wagoner PK, Jegla T. Characterization of KCNQ5/Q3 potassium channels expressed in mammalian cells. Br J Pharmacol. 2001;132:381–384. doi: 10.1038/sj.bjp.0703861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, et al. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wei J, Song M, Francis K, Yu SP. Novel role of KCNQ2/3 channels in regulating neuronal cell viability. Cell Death Differ. 2010;18:493–505. doi: 10.1038/cdd.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.