

Abstract
BACKGROUND AND PURPOSE
Reduced NO availability has been described as a key mechanism responsible for endothelial dysfunction in atherosclerosis. We previously reported that neuronal NOS (nNOS)-derived H2O2 is an important endothelium-derived relaxant factor in the mouse aorta. The role of H2O2 and nNOS in endothelial dysfunction in atherosclerosis remains undetermined. We hypothesized that a decrease in nNOS-derived H2O2 contributes to the impaired vasodilatation in apolipoprotein E-deficient mice (ApoE−/−).
EXPERIMENTAL APPROACH
Changes in isometric tension were recorded on a myograph; simultaneously, NO and H2O2 were measured using carbon microsensors. Antisense oligodeoxynucleotides were used to knockdown eNOS and nNOS in vivo. Western blot and confocal microscopy were used to analyse the expression and localization of NOS isoforms.
KEY RESULTS
Aortas from ApoE−/− mice showed impaired vasodilatation paralleled by decreased NO and H2O2 production. Inhibition of nNOS with L-ArgNO2-L-Dbu, knockdown of nNOS and catalase, which decomposes H2O2 into oxygen and water, decreased ACh-induced relaxation by half, produced a small diminution of NO production and abolished H2O2 in wild-type animals, but had no effect in ApoE−/− mice. Confocal microscopy showed increased nNOS immunostaining in endothelial cells of ApoE−/− mice. However, ACh stimulation of vessels resulted in less phosphorylation on Ser852 in ApoE−/− mice.
CONCLUSIONS AND IMPLICATIONS
Our data show that endothelial nNOS-derived H2O2 production is impaired and contributes to endothelial dysfunction in ApoE−/− aorta. The present study provides a new mechanism for endothelial dysfunction in atherosclerosis and may represent a novel target to elaborate the therapeutic strategy for vascular atherosclerosis.
Keywords: nNOS, H2O2, endothelial dysfunction, atherosclerosis
Introduction
Atherosclerosis is the major cause of cardiovascular disease, which still has the leading position in morbidity and mortality in the Western world. Endothelial dysfunction is considered an earlier marker for atherosclerosis, preceding angiographic or ultrasonic evidence of atherosclerotic plaques (Busse and Fleming, 1996; Luscher and Barton, 1997; Ross, 1999; Cai and Harrison, 2000; Higashi et al., 2009a).
As the major regulator of vascular homeostasis, the endothelium not only modulates the tone of the underling vascular smooth muscle but also inhibits several pro-atherogenic processes, including smooth muscle cell proliferation and migration, platelet aggregation, oxidation of low-density lipoproteins (LDL), monocyte and platelet adhesion and synthesis of inflammatory cytokines, thus exhibiting important anti-atherogenic effects (Vanhoutte, 1986; Kubes et al., 1991; Freedman et al., 1999; Shimokawa, 1999; Leopold and Loscalzo, 2009; Sima et al., 2009). Many of these effects are largely mediated by NO.
NO is produced by NOS enzymes classified as endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS) (Alderton et al., 2001). Although, eNOS is predominantly expressed in endothelial cells, and nNOS in neurons, many tissues express more than one isoform. The vasculature has the potential to express nNOS and eNOS (Rosenblum and Murata, 1996; Boulanger et al., 1998; Toda and Okamura, 2003; Bachetti et al., 2004; Capettini et al., 2008). A physiologically relevant role for nNOS has been attributed in the modulation of myogenic tone (Fleming, 2003), systemic arterial pressure (Kurihara et al., 1998) and cerebral blood flow (Hagioka et al., 2005).
Recently, we have shown that nNOS is constitutively expressed in the endothelium of the mouse aorta and, besides NO, also produces hydrogen peroxide (H2O2) (Capettini et al., 2008; 2010;). H2O2 has been considered an endothelium-derived hyperpolarizing factor (EDHF) in mesenteric (Matoba et al., 2000; 2002;), coronary (Miura et al., 2003; Yada et al., 2003) and cerebral arteries (Sobey et al., 1997; Iida and Katusic, 2000). In the mouse aorta, nNOS-derived H2O2 equally contributes with eNOS-derived NO to endothelium-dependent vascular relaxation (Capettini et al., 2010).
nNOS has been recently proposed as a new anti-atherogenic factor (Tsutsui, 2004; Kuhlencordt et al., 2006; Schodel et al., 2009) preventing neointima formation in carotid artery ligation model (Morishita et al., 2002). An increase in nNOS expression was found in conductance vessels with atherosclerotic plaque in human and murine models (Wilcox et al., 1997). In addition, gene deletion of nNOS in apolipoprotein E-deficient mice (ApoE−/−) accelerates atherosclerotic plaque formation in the aortic root and descending thoracic aorta (Kuhlencordt et al., 2006; Schodel et al., 2009).
Murine models of atherosclerosis have impaired endothelium-dependent relaxation (Busse and Fleming, 1996; Bouloumie et al., 1997; Luscher and Barton, 1997; Deckert et al., 1999; Rabelo et al., 2003; Sima et al., 2009). Chemical inactivation and reduced biosynthesis of NO have been described as key mechanisms responsible for endothelial dysfunction in aortas from atherosclerotic animals (Bouloumie et al., 1997; Laursen et al., 2001; Higashi et al., 2009b). The role of H2O2 and nNOS to endothelial dysfunction in atherosclerosis remains so far unknown. The aim of this study was to investigate the contribution of nNOS-derived H2O2 to the impaired endothelium-dependent relaxation in a murine model of atherosclerosis. We hypothesize that an impairment of the nNOS/H2O2 axis might contribute to endothelium dysfunction in ApoE−/− mice.
Methods
Animals
All animal care and experimental procedures complied with guidelines for the humane use of laboratory animals and were approved by the animal ethics committee of the Federal University of Minas Gerais (protocol # 155/10). We used 12 week-old male homozygous ApoE−/− (29.7 ± 0.4 g; n = 22) mice and age-matched wild-type C57BL/6J (28.0 ± 2.6 g; n = 25) mice. ApoE−/− mice were originally obtained from Jackson Laboratories (Bar Harbor, ME, USA) and breed in animal facilities of Federal University of Minas Gerais. C57BL/6J mice were obtained from CEBIO/ICB (UFMG, Brazil). All animals were fed a non-atherogenic diet. For metabolic characterization of the animals and morphological and histological characterization of the aorta, see online supporting information and Figure S1.
Simultaneous measurements of NO, H2O2 and vascular function
Simultaneous measurements of vasodilatation, NO and H2O2 production, induced by ACh were performed as previously described (Capettini et al., 2010). In brief, rings from the thoracic aorta were obtained, mounted in organ bath system, washed in Krebs–Henseleit solution (in mmol·L−1: 110.8 NaCl, 5.9 KCl, 25.0 NaHCO3, 1.07 MgSO4, 2.49 CaCl2, 2.33 NaH2PO4 and 11.51 glucose, pH 7.4) and stabilized for 60 min. Concentration–response curves to ACh were constructed in vessels precontracted to the same tension level (approximately 2.5 mN.mm) with submaximal concentrations of phenylephrine (0.03–0.1 µmol·L−1). Measurements of isometric tension were recorded by a force transducer (World Precision Instruments, Inc., Sarasota, FL, USA) and were fed to an amplifier-recorder (TBM-4 model; World Precision Instruments, Inc.) and to a personal computer equipped with an analogue-to-digital converter board (AD16JR; World Precision Instruments Inc.). Changes in isometric tension were analysed using WinDaq Data Acquisition software (Dataq® Instruments, Akron, OH, USA). Carbon microsensors with a NO and H2O2 permeable membrane (ISO-NOPF100 and ISO-HPO100, respectively; World Precision Instruments Inc.) were placed next to the lumen of vessels before ACh (0.001–300 µmol·L−1) stimulus and currents (nA) were measured. NO and H2O2 concentrations were determined by calibrations curves of known concentrations of S-nitroso-n-acetylpeniciline (SNAP, 0.2 to 500 nmol·L−1; World Precision Instruments, Inc.) or H2O2 (0.001 to 10 µmol·L−1; Merck, Darmstadt, Germany) freshly prepared.
Antisense oligonucleotides
Antisense oligodeoxynucleotides (AS-ODN) were used to knockdown in vivo eNOS (eNOS-KD) and nNOS (nNOS-KD) in wild-type and ApoE−/− mice (Capettini et al., 2008; 2010;). The 19-base phosphorothioated AS-ODN were constructed based on the mouse sequence. We used the following specific sequences: 5′-CTCTTCAAGTTGCCCATGT-3′ for eNOS and 5′-AACGTGTGCTCTTCCATGG-3′ for nNOS (GenBank accession numbers NM 008713 and NM 008712) purchased from Eurogentech North America Inc. (San Diego, CA, USA). The phosphorothioated mismatch ODN (MM-ODN) sequence with the base composition, 5′-GTCTTGAACTTCCCGATCT-3′, was used as control ODN.
In vivo treatment with AS-ODN to nNOS and eNOS
The mice received 2 nmol AS-ODN or MM-ODN i.v., 24 and 48 h before the experiments (Capettini et al., 2008; 2010;). The AS-ODN and MM-ODN were dissolved in a total volume of 200 µL saline and injected with a 26-gauge needle in the penile vein. The efficiency of the AS-ODN to block expression of nNOS and eNOS was evaluated by Western blot analysis and by functional assay of ACh-induced vasorelaxation.
Western blot analysis
Western blot was performed as previously described with some modifications (Capettini et al., 2008). Aortic rings were dissected and stabilized in Krebs–Henseleit solution for 15 min. ACh (100 µmol·L−1) was then applied, and the aortas were collected 8 min after and immediately frozen at −80°C. Non-stimulated aortas were used for basal condition assays. The frozen aortas were homogenized in lyses buffer (in mmol·L−1): 150 NaCl, 50 Tris–HCl, 5 EDTA.2Na, and 1 MgCl2 containing 1% Triton X-100 and 0.5% SDS plus a cocktail of protease inhibitors (SigmaFAST®, Sigma, St. Louis, MO, USA) and phosphatase inhibitors (20 mmol·L−1 NaF; 0.1 mmol·L−1 Na3VO4); 50 µg of protein were denatured and separated in denaturing SDS/7.5% polyacrylamide gel. Proteins were transferred onto a polyvinylidene fluoride membrane (Immobilon P; Millipore, MA, USA). Blots were blocked at room temperature with 2.5% non-fat dry milk in PBS plus 0.1% Tween 20 before incubation with rabbit polyclonal anti-nNOS (1:1000), mouse monoclonal anti-nNOS Ser852 (1:1000), rabbit polyclonal anti-eNOS (1:1000), goat polyclonal anti-eNOS Ser1177 (1:1000), goat polyclonal anti-eNOS Thr495 (1:1000) or rabbit polyclonal anti-β-actin (1:3000), at room temperature. Immunoreactive bands were detected using the Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA). Rabbit polyclonal anti-eNOS was purchased from Sigma. The other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Confocal microscopy
Thirty percent sucrose (in PBS)-fixed cryosections (6 µm) of the thoracic aorta from wild-type and ApoE−/− mice were rinsed in wash buffer (3% BSA + 0.3% Triton X-100, in PBS). Following appropriate blocking procedures (3% BSA in PBS; 30 min), cross-reactivity of secondary antibodies with the alternating primary antibodies was removed. Slides were incubated with mouse monoclonal anti-GAPDH (1:100; Santa Cruz Biotechnology) and rabbit anti-nNOS (1:50; Santa Cruz Biotechnology) or rabbit anti-eNOS (1:50; Sigma) overnight at 4°C followed by incubation with goat anti-rabbit secondary antibody conjugated with Alexa Fluor 633 (1:500; Invitrogen, Carlsbad, CA, USA) and goat anti-mouse secondary antibody conjugated with Alexa Fluor 488 (1:500; Invitrogen) for 1 h. The sections were examined with a Zeiss LSM 510 confocal microscope (Thornwood, NY, USA) with excitation at 488/633 nm and emission at 505–530/650 nm. The fluorescence (arbitraries unities) intensity was measured using ImageJ® software 1.42q (Wayne Rasband, NIH). Ten fields per slide of endothelial and media layers were measured. The mean of fluorescence from each slide was plotted and analysed using GraphPad Prism 4 (Graphpad Software Inc., La Jolla, CA, USA). Fluorescence intensity in ApoE−/− aorta was expressed as fold increase compared with wild-type animals.
Statistical analysis
Results are expressed as mean ± SEM. Two-way anova with Bonferroni's multiple comparisons post-test was used to compare concentration–response curves. Student's t-test was used in the other experiments. All statistical analyses were considered significant when P < 0.05.
Materials
ACh, catalase, Nω-nitro-l-arginine methyl ester hydrochloride, L-ArgNO2-l-Dbu-NH2 2TFA and phenylephrine were purchased from Sigma.
Results
Role of nNOS on ACh-induced vasodilatation, and NO and H2O2 production in the ApoE−/− mice aorta
Aortic rings from ApoE−/− mice showed a reduced vasodilatation in response to ACh (Emax = 54.0 ± 4.1%; P < 0.001), compared with wild-type animals (Emax = 95.04 ± 1.55%; Figure 1A,B). The impaired vasodilator response in ApoE−/− arteries was accompanied by a severe impairment in NO (Figure 1A) and H2O2 production (Figure 1B). Non-selective inhibition of NOS with l-NAME (300 µmol·L−1) abolished the vasodilator response (Figure 2A) and NO production (Figure 2B) in wild-type and ApoE−/− mice. H2O2 production was almost completely abolished by l-NAME in wild-type vessels. However, in ApoE−/− animals, the production of H2O2 was very low and not affected by l-NAME (Figure 2C). Interestingly, selective inhibition of nNOS with 1 µmol·L−1l-ArgNO2-l-Dbu, a concentration that inhibits nNOS without affecting eNOS (Huang et al., 1999), reduced ACh-induced relaxation in wild-type but not in ApoE−/− aorta (Figure 2D), suggesting a reduction in nNOS function and or expression in ApoE−/− vessels. In wild-type animals, nNOS inhibition modestly decreased NO (Figure 2E) but abolished H2O2 production (Figure 2F). In ApoE−/− animals, NO and H2O2 production were not affected by selective nNOS inhibition. Catalase (2400 U·mL−1), which specifically decomposes H2O2 into oxygen and water, reduced vasodilatation in wild-type animals in the same proportion as that obtained with nNOS inhibition. However, catalase had no effect in ApoE−/− aortas (Figure S2). Vasorelaxation in response to H2O2 was not different between the strains (Figure S3).
Figure 1.
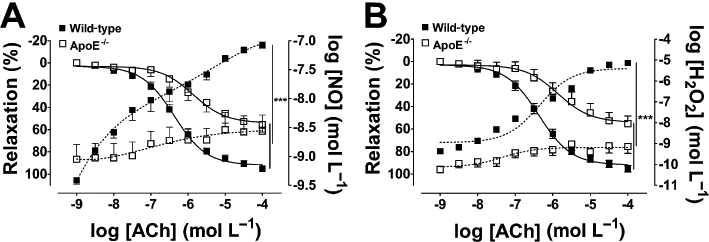
Simultaneous measurements of vasodilatation (A,B), and NO (A) and H2O2 (B) production stimulated by ACh in the aortas of wild-type and ApoE−/− mice. Continuous lines represent vasodilatation (left axis); dotted lines: NO (A) and H2O2 (B) measurements (right axis). The data are shown as mean ± SEM of at least five experiments. ***P < 0.001.
Figure 2.
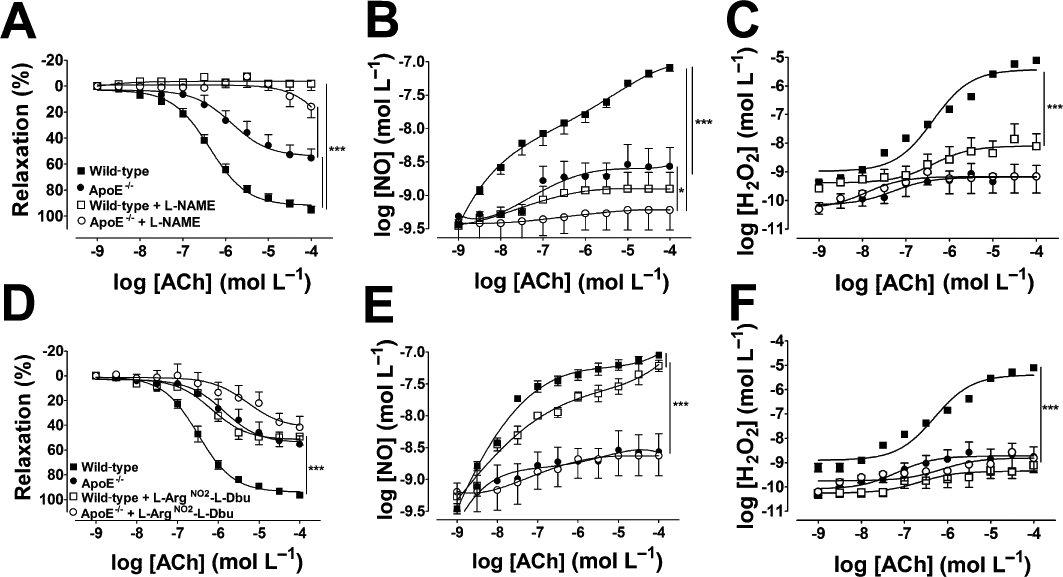
(A–C) Effect of l-NAME (300 µmol·L−1) on ACh-induced vasodilatation (A), NO (B) and H2O2 (C) production in the aortas of wild-type and ApoE−/− mice. (D–F) Selective pharmacological inhibition of nNOS with l-ArgNO2-l-Dbu (1 µmol·L−1) reduced vasodilatation in wild-type but not in ApoE−/− mice (D). In wild-type mice, l-ArgNO2-l-Dbu produced a small reduction in NO production (E) and abolished H2O2 (F). NO (E) and H2O2 (F) were not modified by l-ArgNO2-l-Dbu in ApoE−/− mice. The data represent mean ± SEM of at least five experiments. ***P < 0.001.
Expression and localization of eNOS and nNOS
Western blot analysis showed that expression of eNOS and nNOS were increased in the aortas from ApoE−/− mice (Figure 3A,D). Confocal experiments to immunolocalize eNOS and nNOS showed that in the wild-type animals, both enzymes were present in the endothelial cells but were absent in the media layer (Figures 4A and 5A). In ApoE−/− arteries, eNOS (Figure 4C) and nNOS (Figure 5C) immunostaining were increased in the endothelium as compared with wild-type animals. Moreover, in ApoE−/− vessels, both enzymes were also shown to be present in the smooth muscle layer of the aorta (Figures 4A,B and 5A,B).
Figure 3.
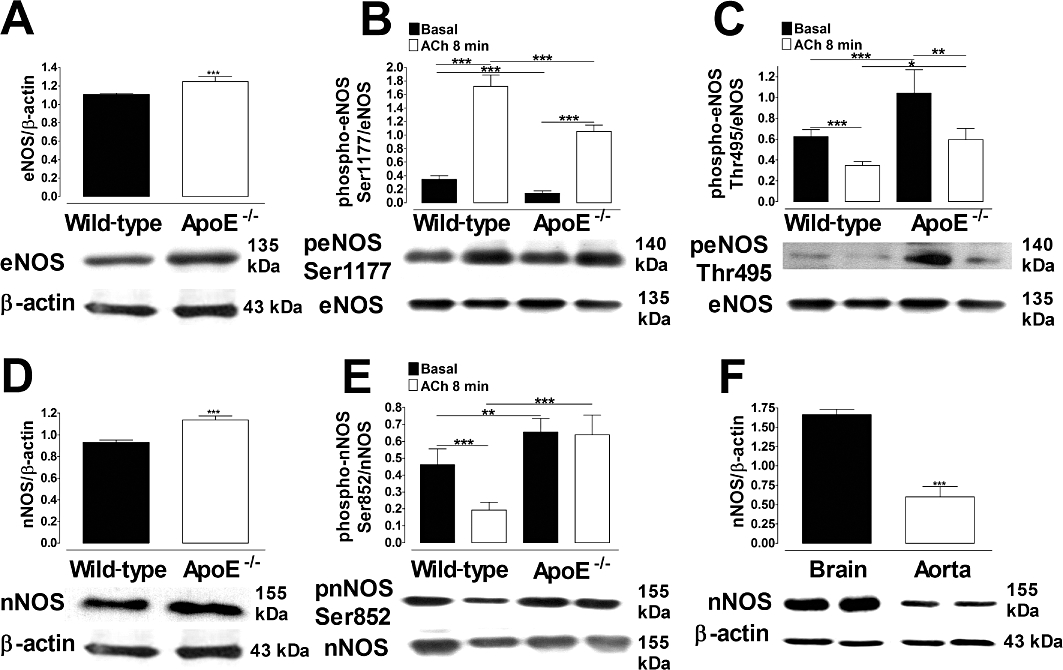
eNOS (A) and nNOS (B) expression in aortas from wild-type and ApoE−/− mice. ACh-induced changes in the phosphorylation of eNOS-Ser1177 (B), eNOS-Thr495 (C) and nNOS-Ser852 (E). (F) Positive control for nNOS in brain. The vessels were stimulated with 100 µmol·L−1 ACh. The histograms represent mean ± SEM of four experiments. ***P < 0.001; **P < 0.01. Images are representative blots from four separate experiments.
Figure 4.
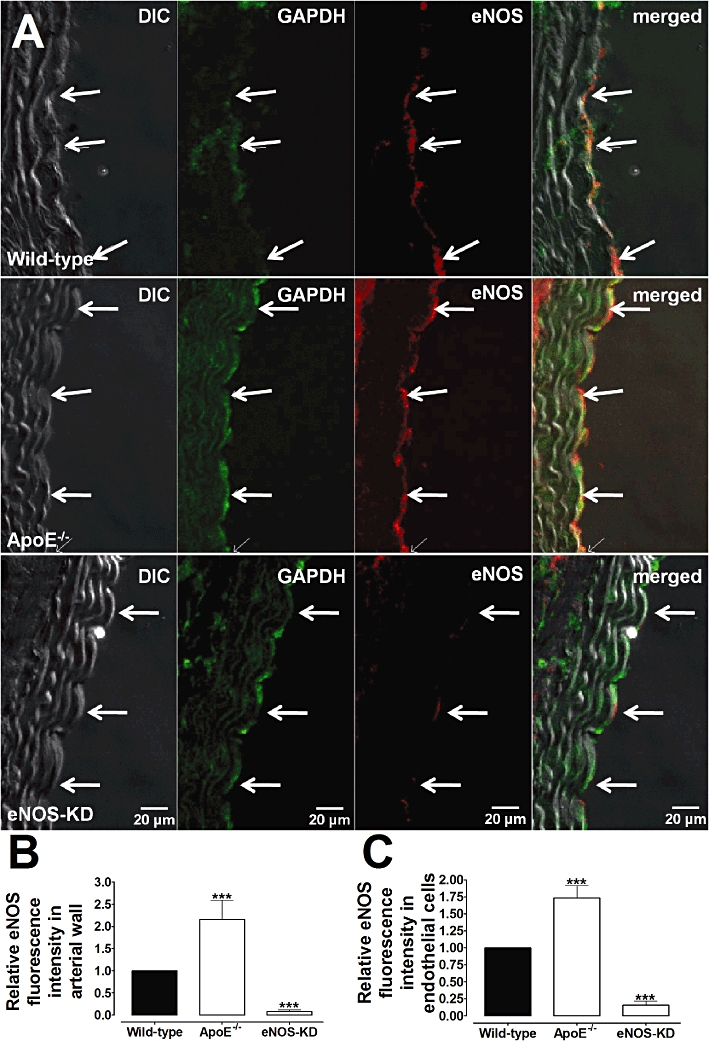
Immunofluorescence detection of eNOS in aortic rings from wild-type (A, higher panel), ApoE−/− (A, middle panel) and eNOS-KD (A, lower panel) mice. Immunostaining for eNOS was present in endothelial cells (arrows) in wild-type and increased in ApoE−/− vessels. Note the strong decrease in eNOS immunostaining in eNOS KD (knockdown) animals. (B,C) The graphical representation of the relative eNOS fluorescence in the arterial wall (B) and in endothelial cells (C) from wild-type, ApoE−/− and eNOS-KD arteries. GAPDH was used for control loading purposes. Images are representative of five animals for each group. ***P < 0.001 compared with wild-type vessels.
Figure 5.
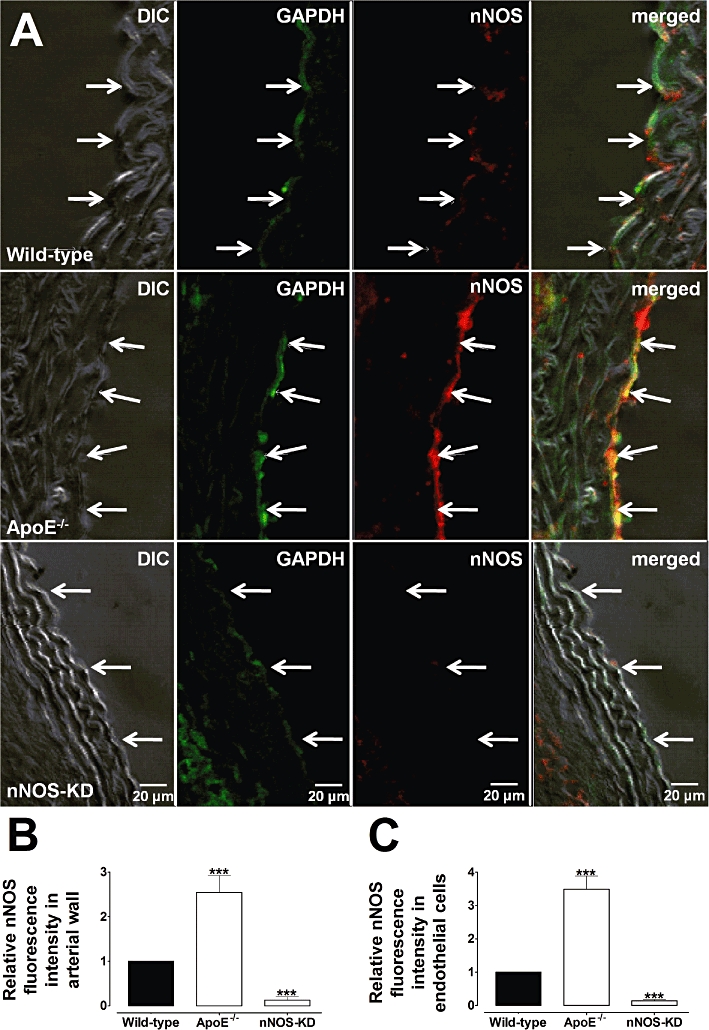
Immunofluorescence detection of nNOS in aortic rings from wild-type (A, higher panel), ApoE−/− (A, middle panel) and nNOS-KD (A, lower panel) mice. Immunostaining for nNOS was increased in endothelial cells (arrows) from ApoE−/− compared with wild-type vessels. Staining in nNOS-KD (knockdown) animals was strongly diminished. (B,C) Graphical representation of the relative nNOS fluorescence in the arterial wall (B) and in endothelial cells (C) from wild-type, ApoE−/− and nNOS-KD arteries. GAPDH was used for control loading purposes. Images are representative of five animals for each group. ***P < 0.001 compared with wild-type vessels.
Determination of eNOS and nNOS phosphorylation by Western blot
We analysed the phosphorylation state on serine (Ser) and threonine (Thr) sites of eNOS and nNOS by Western blot. In wild-type animals, ACh produced an increase in phosphorylation on Ser1177, the activation site of eNOS (Figure 3B); conversely, the phosphorylation state of the inactivation site of the enzyme on Thr495 was decreased (Figure 3C). In ApoE−/− mice, the increase in phosphorylation on eNOS- Ser1177 and dephosphorylation on eNOS-Thr495 induced by ACh was smaller compared with wild-type vessels (Figure 3B,C). Note that in basal conditions, ApoE−/− aortas also showed a less significant phosphorylation level on eNOS-Ser1177 and a higher level on eNOS-Thr495, compared with wild-type animals.
The inactivation site for nNOS on Ser852 showed higher phosphorylation levels on aortas from ApoE−/− mouse compared with control animals in the basal conditions. After stimulation with ACh, the phosphorylation level on the inactivation site of the enzyme was decreased in wild-type aorta but unchanged in ApoE−/− vessels (Figure 3E).
Vasodilator response, NO and H2O2 production in eNOS and nNOS knockdown animals
The individual contribution of eNOS and nNOS to the vascular responses, and NO and H2O2 production in ApoE−/− mice, was evaluated by the use of in vivo AS-ODN knockdown of eNOS and nNOS expression. Wild-type eNOS-KD mice showed a reduced ACh-induced vasodilatation (Figure 6A) that was accompanied by a strong decrease in NO production (Figure 6B), without changes in H2O2 (Figure 6C). ACh-induced relaxation and NO production were attenuated in ApoE−/− eNOS-KD mice (Figure 6D,E). H2O2 production was already impaired in ApoE−/− aortas and was not modified in eNOS-KD animals (Figure 6F). Aortas from wild-type nNOS-KD mice showed reduced vascular responses to ACh (Figure 7A) in the same proportion as those seen with in vitro pharmacological inhibition of nNOS and with catalase, accompanied by a small reduction in NO production (Figure 7B). However, H2O2 synthesis was strongly decreased in these animals (Figure 7C). In aortas from ApoE−/− animals, nNOS knockdown did not change the relaxant response to ACh (Figure 7D), or the production of NO and H2O2 (Figure 7E,F). The ability of AS-ODN treatment to reduce eNOS and nNOS expression was confirmed by Western blot analysis as shown in Figure S4.
Figure 6.

Effect of in vivo antisense oligodeoxynucleotide eNOS knockdown (eNOS KD) on vasodilatation (A,D), NO (B,E) and H2O2 (C,F) production in aortas from wild-type (A–C) and ApoE−/− (D–F) mice. The data represent mean ± SEM of at least five experiments. ***P < 0.001.
Figure 7.

Effect of in vivo antisense oligodeoxynucleotide nNOS knockdown (nNOS KD) on vasodilatation (A,D), NO (B,E) and H2O2 (C,F) production in aortas from wild-type (A–C) and ApoE−/− (D–F) mice. The data represent mean ± SEM of at least five experiments. ***P < 0.001.
Discussion and conclusions
The major finding of this work is that the function of endothelial nNOS in ApoE−/− mouse aorta is decreased, leading to a deficiency in H2O2 production and this contributes to the endothelial dysfunction in ApoE−/− mouse.
It is well known that in the cardiovascular system, eNOS-derived NO plays an important role in the regulation of vascular tone (Garland et al., 1995; Urakami-Harasawa et al., 1997). However, there is increasing evidence attributing a physiologically relevant role for nNOS in the control of vascular homeostasis. The expression of nNOS in vascular smooth muscle and endothelial cells has been commonly associated with the control of brain blood flow (Wei et al., 1999; Atochin et al., 2003; Hagioka et al., 2005; Kitaura et al., 2007). In addition, in eNOS−/− mice, nNOS plays a major role in the control of the coronary circulation (Huang et al., 2002; Talukder et al., 2004; Chlopicki et al., 2005). Recently, we have shown that nNOS is constitutively expressed in the endothelium of mouse aorta and contributes to the endothelium-derived vasodilatation induced by ACh (Capettini et al., 2008). These findings are consistent with the reduced vasodilatation found in the aorta from nNOS−/− mice (Nangle et al., 2004). Interestingly, we have shown that besides NO, nNOS also produces H2O2 in physiological conditions. Using pharmacological inhibitors and antisense nNOS knockdown, and simultaneous measurement of NO, H2O2 and vascular function, we demonstrated that nNOS-derived H2O2 is a major endothelium-dependent relaxing factor in the mouse aorta and importantly contributes to endothelial-dependent vasodilatation in the mouse aorta (Rabelo et al., 2003; Capettini et al., 2008; 2010).
It is well-established that endothelium-dependent vasodilatation is attenuated by hyperlipidaemia and atherosclerosis in animals (Busse and Fleming, 1996; Nangle et al., 2003; Rabelo et al., 2003) and human (Higashi et al., 2009a; Toma et al., 2009). Endothelial dysfunction in atherosclerosis has been commonly associated with a reduction in eNOS-derived NO bioavailability; the involvement of nNOS and H2O2 in endothelial dysfunction in atherosclerosis had not been clarified. In this work, we provide consistent evidence that an impairment in nNOS-derived H2O2 also contributes to endothelial dysfunction in the aorta from ApoE−/− mouse. In line with this proposal, selective pharmacological inhibition of nNOS reduced ACh-induced relaxation in control but not in ApoE−/− mouse. These results were further corroborated by specific antisense nNOS knockdown that showed similar results. These data point to a decreased function and/or expression of nNOS in ApoE−/− animals. However, the level of nNOS protein was increased in ApoE−/− mouse aortas, as assessed by Western blot analysis. Increased expression of nNOS in the media layer from ApoE−/− mice aortas has been reported previously (Schodel et al., 2009).
Our confocal data showed the presence of nNOS immunostaining in the endothelial cell layer in control animals, consistent with previous reports using different assays (Loesch and Burnstock, 1998; Capettini et al., 2008). Interestingly, we found that nNOS immunoreactivity was increased in the smooth muscle cell layer as well as in the endothelial cells from ApoE−/− mice aorta. Nonetheless, an important finding from the present work was the difference in the phosphorylation state of nNOS-Ser852, between the strains. In wild-type aortas ACh produced a decrease in phosphorylation of the inactivation site of the enzyme, while in ApoE−/− vessels nNOS-Ser852 remained unchanged by ACh stimulation. Together, these data indicate a decreased functioning of endothelial nNOS and suggest a role for this enzyme in the impaired vasorelaxation in the aorta from ApoE−/− mouse.
Oxidative stress has been implicated in impaired endothelium-dependent relaxations in atherosclerosis. An increased level of reactive oxygen species (ROS) in the vessel wall leads to modifications in calcium handling, expression of voltage-dependent l-type Ca2+ channels, reduction in tetrahydrobiopterin availability and depletion of l-arginine in endothelial cells (Laursen et al., 2001; Katusic and d'Uscio, 2004; Fransen et al., 2008; Erdely et al., 2010; Fu et al., 2010). Therefore, we speculated that oxidative stress may well contribute to the reduced functionality of nNOS leading to impaired H2O2-induced relaxations.
The expression of eNOS was also reported to be increased in aortas from ApoE−/− mice in despite of the decreased NO production (Bouloumie et al., 1997; Loesch and Burnstock, 1998; Laursen et al., 2001; Kuhlencordt et al., 2004). The decrease in NO production has been associated with changes in the phosphorylation state of the eNOS (Fernández-Hernando et al., 2007; Wang et al., 2010; Yamashiro et al., 2010). Consistent with these data, we found an increased expression of eNOS in aortas from ApoE−/− mice and this was accompanied by a decreased vasodilator response, a diminution of eNOS function and a severe impairment in NO production.
In summary, our data show that vasodilatation in wild-type animals was decreased by half with catalase, nNOS knockdown or pharmacological nNOS inhibition. Endothelial dysfunction in ApoE−/− vessels was accompanied by abolishment of H2O2 production and vasorelaxation was not affected by catalase, nNOS knockdown or selective pharmacological nNOS inhibition. Conversely, eNOS knockdown abolished the vasorelaxation in ApoE−/− mice and reduced vasodilatation by half in wild-type animals. Vasodilatation in response to exogenous H2O2 was not different between strains. Together, these results show that (i) H2O2 production is suppressed in aortas from ApoE−/− animals and might contribute to the endothelial dysfunction in atherosclerosis; (ii) although severely impaired, NO accounts for the residual vasorelaxation in ApoE−/− mice.
In conclusion, our data show that endothelial nNOS activity is decreased in ApoE−/− mouse aorta. The reduced nNOS activity leads to an impairment in H2O2 production that contributes to the attenuated endothelium-dependent vasodilator response. These results indicate a new mechanism for endothelial dysfunction showing a critical role for nNOS-derived H2O2 in the impaired vasodilator response in atherosclerosis. nNOS may represent a novel target to elaborate the therapeutic strategy for vascular atherosclerosis.
Acknowledgments
This study was supported by FAPEMIG (Fundação de Apoio a Pesquisa do Estado de Minas Gerais), CNPq/Brazil (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).
Glossary
Abbreviations
- ApoE−/−
apolipoprotein E-deficient mice
- AS-ODN
antisense oligodeoxynucleotide
- EDRF
endothelium-derived relaxing factor
- eNOS
endothelial NOS
- iNOS
inducible NOS
- KD
knockdown
- LDL
low density lipoprotein
- MM-ODN
mismatch oligodeoxynucleotide
- nNOS
neuronal NOS
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Morphological analyses in the structure of the aortic arch (A) and thoracic aorta (B) from wild-type and ApoE−/− mice. Representative H&E-stained sections from 5 experiments. Scale bar: 10 µm.
Figure S2 Effect of catalase (2400 U·mL−1) on vasodilation induced by ACh on wild-type and ApoE−/− mice aortas. The data represent mean ± SEM of five experiments. ***P < 0.001.
Figure S3 Vasodilator effect of exogenous H2O2 in aortas from wild type and ApoE−/− mice. The experiments were performed in the presence of 50 mmol·L−1 aminotriazole (Sigma) to inhibit endogenous peroxidises. The data represent mean ± SEM of five experiments.
Figure S4 Western-blot analysis of the effect of in vivo antisense eNOS (eNOS-KD; A) and nNOS (n-NOS-KD; B) knockdown in wild-type and ApoE−/− mice aorta. (C) Control experiments for nNOS in eNOS-KD animals and (D) controls Western blot for eNOS in nNOS-KD aortas. Images are representative of four experiments. Bar graphs represent mean ± SEM of four experiments. ***P < 0.001.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atochin DN, Demchenko IT, Astern J, Boso AE, Piantadosi CA, Huang PL. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J Cereb Blood Flow Metab. 2003;23:1219–1226. doi: 10.1097/01.WCB.0000089601.87125.E4. [DOI] [PubMed] [Google Scholar]
- Bachetti T, Comini L, Curello S, Bastianon D, Palmieri M, Bresciani G, et al. Co-expression and modulation of neuronal and endothelial nitric oxide synthase in human endothelial cells. J Mol Cell Cardiol. 2004;37:939–945. doi: 10.1016/j.yjmcc.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Boulanger CM, Heymes C, Benessiano J, Geske RS, Levy BI, Vanhoutte PM. Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension. Circ Res. 1998;83:1271–1278. doi: 10.1161/01.res.83.12.1271. [DOI] [PubMed] [Google Scholar]
- Bouloumie A, Bauersachs J, Linz W, Scholkens BA, Wiemer G, Fleming I, et al. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension. 1997;30:934–941. doi: 10.1161/01.hyp.30.4.934. [DOI] [PubMed] [Google Scholar]
- Busse R, Fleming I. Endothelial dysfunction in atherosclerosis. J Vasc Res. 1996;33:181–194. doi: 10.1159/000159147. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Capettini LS, Cortes SF, Gomes MA, Silva GA, Pesquero JL, Lopes MJ, et al. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circ Physiol. 2008;295:H2503–H2511. doi: 10.1152/ajpheart.00731.2008. [DOI] [PubMed] [Google Scholar]
- Capettini LS, Cortes SF, Lemos VS. Relative contribution of eNOS and nNOS to endothelium-dependent vasodilation in the mouse aorta. Eur J Pharmacol. 2010;643:260–266. doi: 10.1016/j.ejphar.2010.06.066. [DOI] [PubMed] [Google Scholar]
- Chlopicki S, Kozlovski VI, Lorkowska B, Drelicharz L, Gebska A. Compensation of endothelium-dependent responses in coronary circulation of eNOS-deficient mice. J Cardiovasc Pharmacol. 2005;46:115–123. doi: 10.1097/01.fjc.0000164093.88821.00. [DOI] [PubMed] [Google Scholar]
- Deckert V, Lizard G, Duverger N, Athias A, Palleau V, Emmanuel F, et al. Impairment of endothelium-dependent arterial relaxation by high-fat feeding in ApoE-deficient mice: toward normalization by human ApoA-I expression. Circulation. 1999;100:1230–1235. doi: 10.1161/01.cir.100.11.1230. [DOI] [PubMed] [Google Scholar]
- Erdely A, Kepka-Lenhart D, Salmen-Muniz R, Chapman R, Hulderman T, Kashon M, et al. Arginase activities and global arginine bioavailability in wild-type and ApoE-deficient mice: responses to high fat and high cholesterol diets. PLoS ONE. 2010;5:e15253. doi: 10.1371/journal.pone.0015253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Hernando C, Ackah E, Yu J, Suárez Y, Murata T, Iwakiri Y, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–457. doi: 10.1016/j.cmet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I. Brain in the brawn: the neuronal nitric oxide synthase as a regulator of myogenic tone. Circ Res. 2003;93:586–588. doi: 10.1161/01.RES.0000095380.06622.D8. [DOI] [PubMed] [Google Scholar]
- Fransen P, Van Assche T, Guns PJ, Van Hove CE, De Keuleaner GW, Herman AG, et al. Endothelial function in aorta segments of apolipoprotein E-deficient mice before development of atherosclerotic lesions. Pflugers Arch. 2008;455:811–815. doi: 10.1007/s00424-007-0337-9. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Sauter R, Battinelli EM, Ault K, Knowles C, Huang PL, et al. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ Res. 1999;84:1416–1421. doi: 10.1161/01.res.84.12.1416. [DOI] [PubMed] [Google Scholar]
- Fu C, He J, Li C, Shyy JYJ, Zhu Y. Cholesterol increases adhesion of monocytes to endothelium by moving adhesion molecules out of caveolae. Biochim Biophys Acta. 2010;1801:702–710. doi: 10.1016/j.bbalip.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- Hagioka S, Takeda Y, Zhang S, Sato T, Morita K. Effects of 7-nitroindazole and N-nitro-L-arginine methyl ester on changes in cerebral blood flow and nitric oxide production preceding development of hyperbaric oxygen-induced seizures in rats. Neurosci Lett. 2005;382:206–210. doi: 10.1016/j.neulet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Maysuoka H, Umei H, Sugano R, Fujii Y, Soga J, et al. Endothelial function in subjects with isolated low HDL cholesterol: role of nitric oxide and circulating progenitor cells. Am J Physiol Endocrinol Metab. 2009a;298:e202–e209. doi: 10.1152/ajpendo.00394.2009. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009b;73:411–418. doi: 10.1253/circj.cj-08-1102. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Shesely EG, Levee EM, Koller A, Kaley G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2002;282:H429–H436. doi: 10.1152/ajpheart.00501.2001. [DOI] [PubMed] [Google Scholar]
- Huang H, Martasek P, Roman LJ, Masters BSS, Silverman RB. Nω-Nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- Iida Y, Katusic ZS. Mechanisms of cerebral arterial relaxations to hydrogen peroxide. Stroke. 2000;31:2224–2230. doi: 10.1161/01.str.31.9.2224. [DOI] [PubMed] [Google Scholar]
- Katusic ZS, d'Uscio LV. Tetrahydrobiopterin: mediator of endothelial protection. Arterioscler Thromb Vasc Biol. 2004;24:F552–F558. doi: 10.1161/01.ATV.0000121569.76931.0b. [DOI] [PubMed] [Google Scholar]
- Kitaura H, Uozumi N, Tohmi M, Yamazaki M, Sakimura K, Kudoh M, et al. Roles of nitric oxide as a vasodilator in neurovascular coupling of mouse somatosensory cortex. Neurosci Res. 2007;59:160–171. doi: 10.1016/j.neures.2007.06.1469. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Rosel E, Gerszten RE, Morales-Ruiz M, Dombkowski D, Atkinson WJ, et al. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. Am J Physiol Cell Physiol. 2004;286:C1195–C1202. doi: 10.1152/ajpcell.00546.2002. [DOI] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Hotten S, Schodel J, Rutzel S, Hu K, Widder J, et al. Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein-e knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:1539–1544. doi: 10.1161/01.ATV.0000223143.88128.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara N, Alfie ME, Sigmon DH, Rhaleb NE, Shesely EG, Carretero OA. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension. 1998;32:856–861. doi: 10.1161/01.hyp.32.5.856. [DOI] [PubMed] [Google Scholar]
- Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- Leopold JA, Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radic Biol Med. 2009;47:1673–1706. doi: 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch A, Burnstock G. Perivascular nerve fibres and endothelial cells of the rat basilar artery: immuno-gold labelling of antigenic sites for type I and type III nitric oxide synthase. J Neurocytol. 1998;27:197–204. doi: 10.1023/a:1026493425977. [DOI] [PubMed] [Google Scholar]
- Luscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20:II3–II10. [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290:909–913. doi: 10.1006/bbrc.2001.6278. [DOI] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- Morishita T, Tsutsui M, Shimokawa H, Horiuchi M, Tanimoto A, Suda O, et al. Vasculoprotective roles of neuronal nitric oxide synthase. FASEB J. 2002;16:1994–1996. doi: 10.1096/fj.02-0155fje. [DOI] [PubMed] [Google Scholar]
- Nangle MR, Cotter MA, Cameron NE. Effects of rosuvastatin on nitric oxide-dependent function in aorta and corpus cavernosum of diabetic mice: relationship to cholesterol biosynthesis pathway inhibition and lipid lowering. Diabetes. 2003;52:2396–2402. doi: 10.2337/diabetes.52.9.2396. [DOI] [PubMed] [Google Scholar]
- Nangle MR, Cotter MA, Cameron NE. An in vitro investigation of aorta and corpus cavernosum from eNOS and nNOS gene-deficient mice. Eur J Physiol. 2004;448:139–145. doi: 10.1007/s00424-003-1232-7. [DOI] [PubMed] [Google Scholar]
- Rabelo LA, Cortes SF, Alvarez-Leite JI, Lemos VS. Endothelium dysfunction in LDL receptor knockout mice: a role for H2O2. Br J Pharmacol. 2003;138:1215–1220. doi: 10.1038/sj.bjp.0705164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum WI, Murata S. Antisense evidence for two functionally active forms of nitric oxide synthase in brain microvascular endothelium. Biochem Biophys Res Commun. 1996;224:535–543. doi: 10.1006/bbrc.1996.1061. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138:S419–S420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- Schodel J, Padmapriya P, Marx A, Huang PL, Ertl G, Kuhlencordt PJ. Expression of neuronal nitric oxide synthase splice variants in atherosclerotic plaques of apoE knockout mice. Atherosclerosis. 2009;206:383–389. doi: 10.1016/j.atherosclerosis.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H. Primary endothelial dysfunction: atherosclerosis. J Mol Cell Cardiol. 1999;31:23–37. doi: 10.1006/jmcc.1998.0841. [DOI] [PubMed] [Google Scholar]
- Sima AV, Stancu CS, Simionescu M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009;335:191–203. doi: 10.1007/s00441-008-0678-5. [DOI] [PubMed] [Google Scholar]
- Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke. 1997;28:2290–2295. doi: 10.1161/01.str.28.11.2290. [DOI] [PubMed] [Google Scholar]
- Talukder MA, Fujiki T, Morikawa K, Motoishi M, Kubota H, Morishita T, et al. Up-regulated neuronal nitric oxide synthase compensates coronary flow response to bradykinin in endothelial nitric oxide synthase-deficient mice. J Cardiovasc Pharmacol. 2004;44:437–445. doi: 10.1097/01.fjc.0000139450.64337.cd. [DOI] [PubMed] [Google Scholar]
- Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271–324. doi: 10.1124/pr.55.2.3. [DOI] [PubMed] [Google Scholar]
- Toma L, Stancu CS, Botez GM, Sima AV, Simionescu M. Irreversibly glycated LDL induce oxidative and inflammatory state in human endothelial cells; added effect of high glucose. Biochem Biophys Res Commun. 2009;390:877–882. doi: 10.1016/j.bbrc.2009.10.066. [DOI] [PubMed] [Google Scholar]
- Tsutsui M. Neuronal nitric oxide synthase as a novel anti-atherogenic factor. J Atheroscler Thromb. 2004;11:41–48. doi: 10.5551/jat.11.41. [DOI] [PubMed] [Google Scholar]
- Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest. 1997;100:2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM. The endothelium and arterial reactivity. J Mal Vasc. 1986;11:213–221. [PubMed] [Google Scholar]
- Wang F, Okamoto Y, Inoki I, Yoshioka K, Du W, Qi X, et al. Sphingosine-1-phosphate receptor-2 deficiency leads to inhibition of macrophage proinflammatory activities and atherosclerosis in apoE-deficient mice. J Clin Invest. 2010;120:3979–3995. doi: 10.1172/JCI42315. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wei G, Dawson VL, Zweier JL. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim Biophys Acta. 1999;1455:23–34. doi: 10.1016/s0925-4439(99)00051-4. [DOI] [PubMed] [Google Scholar]
- Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17:2479–2488. doi: 10.1161/01.atv.17.11.2479. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, et al. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107:1040–1045. doi: 10.1161/01.cir.0000050145.25589.65. [DOI] [PubMed] [Google Scholar]
- Yamashiro K, Milsom AB, Duchene J, Panayiotou C, Urabe T, Hattori N, et al. Alterations in nitric oxide and endothelin-1 bioactivity underlie cerebrovascular dysfunction in ApoE-deficient mice. J Cereb Blood Flow Metab. 2010;30:1494–1503. doi: 10.1038/jcbfm.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.