

Abstract
The mechanisms underlying the muscle wasting that accompanies CKD are not well understood. Animal models suggest that impaired differentiation of muscle progenitor cells may contribute. Expression of the myogenesis-suppressing transcription factor Ying Yang-1 increases in muscle of animals with CKD, but the mechanism underlying this increased expression is unknown. Here, we examined a profile of microRNAs in muscles from mice with CKD and observed downregulation of both microRNA-29a (miR-29a) and miR-29b. Because miR-29 has a complementary sequence to the 3′-untranslated region of Ying Yang-1 mRNA, a decrease in miR-29 could increase Ying Yang-1. We used adenovirus-mediated gene transfer to express miR-29 in C2C12 myoblasts and measured its effect on both Ying Yang-1 and myoblast differentiation. An increase in miR-29 decreased the abundance of Ying Yang-1 and improved the differentiation of myoblasts into myotubes. Similarly, using myoblasts isolated from muscles of mice with CKD, an increase in miR-29 improved differentiation of muscle progenitor cells into myotubes. In conclusion, CKD suppresses miR-29 in muscle, which leads to higher expression of the transcription factor Ying Yang-1, thereby suppressing myogenesis. These data suggest a potential mechanism for the impaired muscle cell differentiation associated with CKD.
In chronic kidney disease (CKD), muscle atrophy is a serious complication because it is associated with excess morbidity and mortality.1 Although mechanisms underlying muscle wasting have been identified, there are few reliable treatment strategies that successfully overcome this complication. Understanding the mechanism causing muscle wasting is an initial step in conceiving of therapeutic options. In earlier studies of a rodent model of CKD, we found that the low muscle mass is due in part to increased protein degradation and suppressed protein synthesis.2,3 Recently, we identified another mechanism that contributes to the development of muscle atrophy associated with CKD, namely, there are defects in the function of muscle progenitor cells (MPCs or satellite cells) that reduce their regenerative capacity.4,5 This adverse response is relevant to muscle wasting because MPCs are required for muscle growth, the maintenance of muscle protein synthesis, and the repair of injured muscles.6
In mammalian skeletal muscle, muscle fibers are postmitotic and, hence, do not reenter the cell cycle. Consequently, MPCs in muscle are typically quiescent, but during muscle growth or in response to muscle trauma, they are activated to proliferate and then differentiate into myotubes that synthesize structural proteins such as embryonic myosin heavy chain (eMyHC) and α-actin. New myotubes can fuse to produce mature muscle fibers.7,8 The differentiation of MPCs can also be influenced by the transcription factor, Yin Yang 1 (YY1), an ubiquitously expressed protein that is capable of influencing biologic and pathologic processes. For example, in skeletal muscle, YY1 can inhibit muscle cell differentiation by inhibiting the synthesis of late-stage, differentiation genes including skeletal α-actin, muscle creatine kinase, and myosin heavy chain IIb.9–11 Because defects in the activity of MPCs can be detected in mice with CKD, we proposed that an increase in the expression of YY1 should contribute to CKD-induced defects in MPC function.4,12 This led to the following question: What influences the level of YY1?
MicroRNAs are relatively short (21 to 24 nucleotides), noncoding RNAs that are evolutionarily conserved. In general, they function as negative regulators of gene expression13 and are involved in a variety of biologic processes and diverse pathologic conditions.14 These microRNAs can influence gene expression in the following way: specific microRNAs bind to target sequences in the 3′-untranslated region (3′-UTR) of a complementary mRNA, and this binding results in decreased translation of this specific mRNA to its corresponding protein.15 In this formulation, a decrease in a specific microRNA would promote uninhibited translation of mRNA to protein. Notably, this sequence is not a one-to-one relationship between a specific microRNA and protein because several microRNAs can be involved in regulating the expression of one protein and individual microRNAs can influence the expression of a number of different proteins.15
On the basis of an array of microRNAs in muscle, CKD was associated with a lower level of microRNA-29 (miR-29), which contains a complementary sequence to the 3′-UTR of the YY1 mRNA in muscle.12 We found an increase in the muscle level of the transcription factor, YY1, under conditions of muscle wasting, and because YY1 can decrease myogenesis, we speculated that increased level of YY1 could be related to the lower level of a miR-29. The microarray data combined with the YY1 results suggested a new mechanism to explain how the differentiation of MPCs is impaired in CKD. That is, miR-29, by being reduced, will result in increased YY1 leading to decreased muscle myogenesis and CKD-induced muscle atrophy.3–5,16 Furthermore, it suggests a potential pathway that could be altered to prevent the CKD-induced muscle atrophy.
RESULTS
CKD Increases YY1 Protein Abundance in Muscle
A potential signal for the CKD-induced suppression of MPC myogenesis4,5 could be a higher level of the YY1 transcription factor because it reportedly inhibits myoblast differentiation. While exploring this possibility, we found a 1.9-fold (P < 0.05; Figure 1) increase in the level of YY1 protein in muscles of mice with CKD compared with the level present in muscles of sham-operated, pair-fed control mice.
Figure 1.
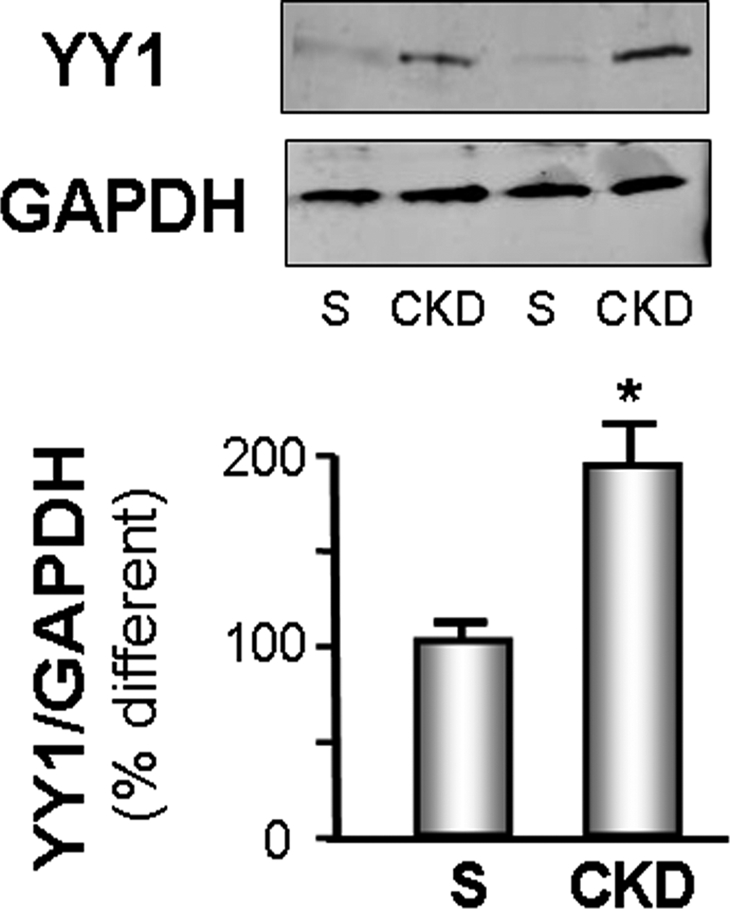
YY1 is increased in CKD muscle. The level of YY1 protein in gastrocnemius muscles of CKD and sham-operated, pair-fed, control mice (S) was measured by Western blot; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was the loading control. The bar graphs (mean ± SEM) represent the density of YY1 bands expressed as a percentage of control levels after correcting for GAPDH (n = 4 pairs; *P < 0.05 versus control [S]).
CKD Decreases the Expression of miR-29s in Muscle
To examine a potential mechanism for the increase in the YY1 transcription factor in muscles of CKD mice, we compared the levels of microRNAs in muscles of CKD and sham-operated, pair-fed control mice. Using a Rodent miRNA Taqman Low Density Array, we found that CKD was associated with significantly different levels of 12 microRNAs in muscle (Table 1). On the basis of an analysis of TargetScan MicroRNA targeting program (http://www.targetscan.org), only three microRNAs had complementary sequences to YY1's 3′-UTR mRNA: miR-181d, miR-29a, and miR-29b. We concentrated on studying miR-29 because the miR-181d level was increased in muscles of CKD mice. If it follows the general rule, an increase in miR-181d would inhibit the target mRNA (i.e., YY1) and suppress its translation leading to a decrease in the level of YY1 in muscles. Because YY1 was increased in muscle of CKD mice, we did not investigate miR-181d further.
Table 1.
MicroRNA array
| Detector | Array ID | Log10RQ (CKD-ctrl) | Fold Change | Inverted Fold When <1 | Significant (CKD versus Control) |
|---|---|---|---|---|---|
| hsa-miR-181d | 4373180 | 0.706 | 5.08 | Significant | |
| mmu-miR-434–5p | 4373359 | 0.821 | 6.624 | Significant | |
| mmu-miR-455–3p | 4386774 | 0.454 | 2.848 | Significant | |
| mmu-miR-496 | 4386771 | 0.564 | 3.669 | Significant | |
| mmu-miR-668 | 4386767 | 0.611 | 4.088 | Significant | |
| hsa-miR-29c | 4373289 | 0.057 | 1.058 | Nonsignificant | |
| hsa-miR-23a | 4373074 | −0.571 | 0.268 | −3.726 | Significant |
| hsa-miR-29a | 4373065 | −0.621 | 0.239 | −4.181 | Significant |
| hsa-miR-29b | 4373288 | −0.572 | 0.268 | −3.732 | Significant |
| mmu-miR-124a | 4373295 | −0.621 | 0.239 | −4.181 | Significant |
| mmu-miR-187 | 4373307 | −1.642 | 0.023 | −43.93 | Significant |
| mmu-miR-199b | 4373309 | −0.572 | 0.268 | −3.732 | Significant |
| hsa-miR-376a | 4378104 | −1.001 | 0.098 | −10.204 | Significant |
The remaining microRNAs that exhibit significantly decreased expression in muscles of CKD mice, miR-29a and miR-29b, have consensus sequences that could target the 3′-UTR of the YY1 in humans, mice, rats, and dogs (http://www.targetscan.org). Specifically, a 76% decrease in miR-29a (expressed as a 4.2-fold decrease) and a 73% decrease in miR-29b (expressed as a 3.7-fold decrease) in muscles of CKD mice could lead to an increase in YY1 expression. Therefore, a decrease in miR-29 offers a potential explanation for the increase in muscle YY1 expression in CKD mice.
To determine that CKD changes microRNA levels in muscle, we isolated MPCs from muscles of CKD and control mice. In these cells, miR-29a was decreased fivefold (P < 0.01) and miR-29b was fourfold lower (P < 0.01) compared with values in MPCs from muscles of sham-operated, control mice (Figure 2A). There also was a 2.5-fold decrease in the level of miR-29c (P < 0.05), but as noted, the microarray analysis did not detect this difference.
Figure 2.

MiR-29 is decreased in muscles of CKD. (A) miR-29 in MPCs isolated from hind-limb muscles of CKD or control (sham) mice were measured by real-time qPCR. The bar graph shows miR-29a, miR-29b, and miR-29c from CKD mice expressed as a percentage of levels in MPCs of control mice; results are normalized to U6 RNA as an internal control (n = 4 pairs; *P < 0.05 or **P < 0.01 versus sham). (B) RT-PCR was used to detect the pri-miR-29a and pre-miR-29a in MPCs isolated from hind-limb muscles of CKD or control (sham) mice. The bar graph shows the density of pri-miR-29a or pre-miR-29a bands expressed as a percentage change from control levels after normalization to the density of U6 RNA (n = 6 pairs; *P < 0.05 or **P < 0.01 versus sham).
To investigate why miR-29 isoforms are depressed in muscles of CKD mice, we examined different forms of microRNAs to determine if the low level of miR-29 is due to depressed production or increased destruction.17 MicroRNAs are initially transcribed from nuclear DNA producing a primary microRNA (pri-miR) that is subsequently cleaved to form a precursor microRNA (pre-miR), which can migrate into the cytosol where it is cleaved to produce a mature microRNA. In muscles of CKD compared with those of control mice, we found that the levels of both pri-miR-29a and pre-miR-29a were decreased (P < 0.05; Figure 2B). The findings suggest that the low levels of miR-29a result from decreased production rather than accelerated destruction.
miR-29 Combats Impaired Muscle Cell Differentiation Induced by YY1
We studied the C2C12 muscle cell line, which exhibits characteristics of satellite cells.18 Because miR-29a, miR-29b, and miR-29c differ by only a few nucleotides, we used the Ad-miR-29 adenovirus to express all three miRs, miR-29a, miR-29b, and miR-29c. When compared with C2C12 myoblasts treated with the control adenovirus (Ad-Ctrl), the miR-29–treated cells showed 50% lower YY1 (Figure 3). To determine whether this lower YY1 had an effect on MPC differentiation, we expressed both miR-29 and YY1, separately and together, in cultured C2C12 cells and determined the degree of differentiation. We initiated differentiation in the cells by treating with a differentiation medium. The degree of differentiation into myotubes was evaluated by calculating a fusion index on day 4 (methods).19 On the basis of an analysis of three randomly chosen fields, the fusion index was greater in C2C12 myotubes that had been treated with Ad-miR-29 compared with results from myotubes treated with the control virus. C2C12 myoblasts transfected to increase the expression of YY1 resulted in decreased differentiation of C2C12 myoblasts into myotubes. However, when myoblasts were also treated with Ad-miR-29, their differentiation increased (Figure 4, A and B).
Figure 3.

miR-29 decreases YY1 protein abundance in C2C12 cells. C2C12 cells were transduced with either Ad-miR-29 or control adenovirus (Ad-C) and their differentiations were assessed by changing the growth media to differentiation media. Proteins isolated from each group of cells were used to measure YY1 protein by Western blotting; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was the loading control. The bar graph shows the density of YY1 protein expressed as a percent of control value after being normalized to GAPDH (n = 6; *P < 0.05 versus Ad-C).
Figure 4.
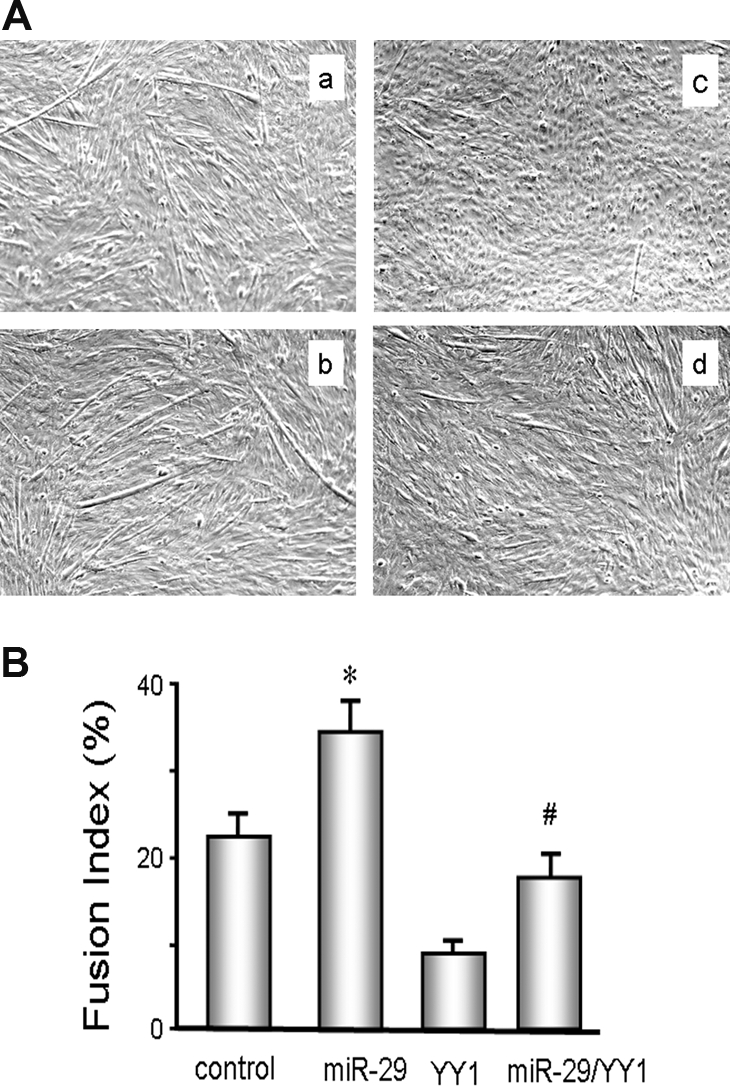
miR-29 in C2C12 muscle cells protects against YY1-induced suppression of their differentiation. (A) C2C12 myoblasts were expressed by (a) Ad-ctrl; (b) miR-29 (Ad-miR-29); (c) YY1 (pCMV-YY1); or (d) miR29 + YY1 (Ad-miR-29 and pCMV-YY1). After culturing in differentiation media, cell morphology was visualized using an Olympus 1 × 51 inverted microscope. Nuclei were stained with 4-diamidino-2-phenylindole. (B) The fusion index was calculated as the ratio of the number of nuclei within myotubes to the total number of nuclei in predefined fields of >450 nuclei. The experiment was repeated an additional three times, yielding the same conclusion (n = 4; *P < 0.05 versus control; #P < 0.05 versus YY1 treatment).
miR-29 Binds to the 3′-UTR of YY1, Evidence for Regulation of YY1 Expression
To examine whether miR-29 directly interacts with the YY1 mRNA, we used a firefly luciferase construct containing the 3′-UTR sequence of YY1 mRNA (pLuc-3URT-YY1) and measured luciferase activity and correcting for renilla luciferase activity. In some experiments, C2C12 myoblasts expressing the firefly luciferase construct (pLuc-3URT-YY1) were also treated with 107 to 109 TU of Ad-miR-29 to increase miR-29 expression. Luciferase activity was significantly suppressed in myoblasts that were transduced with 108 or 109 TU of Ad-miR-29 compared with cells treated with the control virus. These results indicate that miR-29 specifically interacts with the 3′-UTR of YY1 and decreases its ability to produce YY1. In contrast, in C2C12 cells that had been transfected with a luciferase construct in which the miR-29 binding site on YY1 mRNA was mutated to prohibit binding (pLuc-3URTmutant-YY1), there was an increase in luciferase activity in both control and Ad-miR-29–treated cells (Figure 5A). We believe that the luciferase activity in mutant YY1 was significant higher because the endogenous miR-29 that originally bound to the pLuc-3UTR-YY1 was unable to bind to the mutated pLuc-3UTRmutant-YY1 and hence unable to decrease luciferase activity. Similarly, the mutant provides no binding site for exogenously added miR-29, so luciferase activity was not inhibited. To confirm these results, we transfected C2C12 myoblasts with vectors to express pLuc-3UTR-YY1 with pmiR-Zip29a or/and the pmiRZip-29b (which provide antisense sequences to miR-29). The presence of the antisense produced a two- to threefold increase in YY1 luciferase activity (Figure 5B). These results demonstrate that an inhibition of endogenous miR-29 increases the luciferase activity of YY1.
Figure 5.
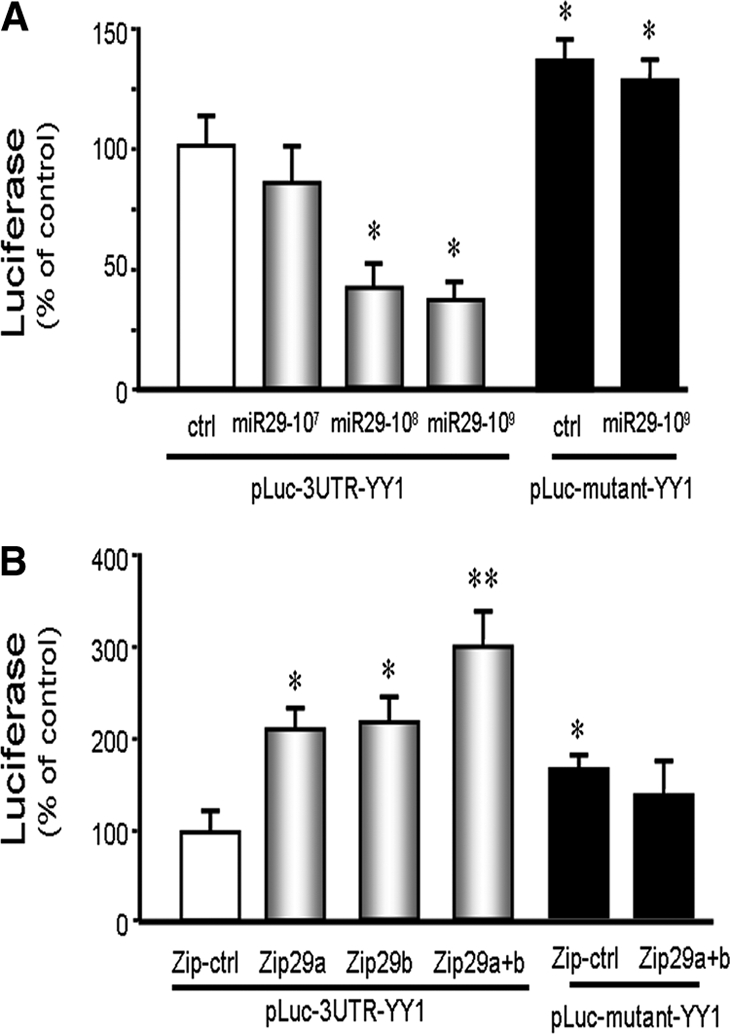
miR-29 binds to the 3′-UTR of YY1, suppressing YY1 luciferase activation in C2C12 muscle cells. (A) C2C12 myoblasts were transfected with pLuc-3URT-YY1 and treated with control virus or 107, 108, or 109 TU of Ad-miR-29. The control virus (ctrl) is represented by the white bar and expressed as 100%. The gray bars indicate that progressively higher levels of miR-29 interact with the 3′-UTR of YY1 to suppress luciferase activity. The black bars were cells transfected with pLuc-3URTmutant-YY1 (pLuc-mutant-YY1, lacking the miR-29 recognition sequence) in cells without (left) or with (right) exogenously added miR-29. Triplicate determinations were made for cells in each condition, and each experiment was repeated two additional times; the results were combined to calculate luciferase activity. The data represent the means ± SEM; n = 9; *P < 0.05 versus pLuc-3URT-YY1 plus ctrl (white bar). (B) To block miR-29 expression, vectors that express an antisense of miR-29a or miR-29b (pmiRZip29a or/and pmiRZip29b) were transfected into myoblasts with pLuc-3UTR-YY1. The control vector (zip-ctrl) is represented by the white bar and expressed as 100%. The black control bars were cells transfected with pLuc-3URTmutant-YY1 with pmiRZip-ctrl (left) or pmiRZip29a+b (right). The experiment was repeated three times with triplicate determinations for each plasmid. The data represent the means ± SEM; n = 9, *P < 0.05 or **P < 0.01 versus pLuc-3URT-YY1 plus zip-ctrl (white bar).
MiR-29 Improves MPC Differentiation
The relationships among an increase in YY1 protein, a decrease in miR-29, and the repression of MPC differentiation suggest that the CKD-induced defect in MPC differentiation could be improved if miR-29 expression were stimulated. To test this hypothesis, MPCs were isolated from muscles of CKD and control mice and incubated in primary growth media (see Concise Methods). When MPCs reached about 60% to 70% confluence, the culture media was changed to the differentiation media. After 4 to 5 days, multinucleated myotubes were detected in cultures of MPCs that had been obtained from control mice. These cells also expressed the eMyHC protein, indicating that the MPCs had differentiation and fusion. In contrast, MPCs isolated from muscles of CKD mice differentiated poorly; only a few myotubes were formed. However, when MPCs were isolated from muscles of CKD mice and then were transduced by Ad-miR-29, the formation of multinucleated myotubes increased (Figure 6).
Figure 6.

miR-29 protects against CKD-induced suppression of MPCs differentiation. MPCs isolated from hind-limb muscles of CKD or sham-operated, pair-fed mice (sham) were cultured in differentiation media on Entactin, Collagen IV, and Laminin matrix coated plates. Immunohistochemistry with an antibody against eMyHC was used to assess MPCs differentiation into myotubes. Nuclei (blue) were stained with 4-diamidino-2-phenylindole, and red identifies eMyHC.
DISCUSSION
Our results uncover a novel pathway that interferes with muscle metabolism in a mouse model of CKD. This pathway consists of increased expression of YY1 in muscles of CKD mice (Figure 1) because of a low level of the microRNA, miR-29. These relationships are relevant to CKD-induced muscle wasting because an increase in YY1 will interfere with the normal differentiation of MPCs, a process that is necessary for muscle growth, the maintenance of protein synthesis, and the repair of injuries in muscle.
Why pursue miR-29 as a key mediator of MPCs differentiation impaired by CKD? First, miR-29a, miR-29b, and miR-29c were significantly downregulated in muscles of CKD mice (Table 1 and Figure 2A). Second, the other five microRNAs (miR-23a, miR-124a, miR-187, miR199b, and miR-376a) that were downregulated in muscles of CKD mice do not have consensus sequences that can interact with the YY1 mRNA. Subsequently, we examined whether miR-29 will interact with the 3′-UTR of YY1 mRNA, yielding changes in YY1 function. The experiments were positive. When both miR-29 and a firefly luciferase reporter for YY1 (pLuc-3UTR-YY1) were expressed in myoblasts, miR-29 expressing cells exhibited decreased luciferase activity compared with non–miR-29 expressing cells. Luciferase activity was suppressed because miR-29 contains a consensus sequence that targets the YY1 mRNA. Therefore, this interaction would reduce the translation of YY1 ultimately, decreasing YY1 in muscle. These results demonstrate that miR-29 directly interacts with the 3′-UTR of the YY1 mRNA. As an additional control, we showed that luciferase activity increased when myoblasts expressing pLuc-3URTmutant-YY1, which contains a mutated miR-29 binding site in YY1 mRNA, were treated with or without exogenous miR-29 (Figure 5A). We believe this increase reflects the removal of the influence of endogenous miR-29 that would be part of the original response to pLuc-3UTR-YY1 with an intact binding site. In summary, the consequences of the decreased miR-29 were pursued because miR-29 can repress the production of YY1, and hence, a decrease in miR-29 would increase the YY1 transcription factor, which acts to suppress MPC differentiation.12 A similar relationship between miR-29 and YY1 was uncovered by Wang et al. in studies of rhabdomyosarcoma cells.20
The experiments were extended by determining that raising miR-29 not only increases its interaction with YY1 mRNA and decreases YY1 protein but also improves the differentiation of myoblasts. Using Ad-miR-29, we expressed miR-29 in C2C12 myoblasts and found an approximately 50% decrease in YY1 expression compared with results in myoblasts treated with the control adenovirus (Figure 3). There also was an improvement in the ability of myoblasts to differentiate (Figure 4, A and B). We also isolated MPCs from CKD and sham-operated, pair-fed control mice and found that MPCs from CKD mice developed fewer myotubes and exhibited reduced synthesis of eMyHC compared with control myoblasts. However, treatment of MPCs from CKD mice with Ad-miR-29 substantially improved their differentiation. The results indicate that a decrease in miR-29 in muscle induced by CKD leads to an increase in the level of YY1, and this response suppresses myoblast development into myotubes.4,5
A key factor in this pathway is the YY1 transcription factor that functions as a transcription repressor or activator.21 It is involved in several biologic processes such as embryogenesis, differentiation, replication, cellular proliferation, tumorigenesis, and apoptosis.21,22 Specifically, an increase in YY1 in muscle has been shown to interact with promoter regions of genes preventing the transcription of muscle-specific genes (skeletal α-actin, muscle creatine kinase, and myosin heavy chain IIb).9–11 In fact, Wang et al. proposed that interactions between miR-29 levels and YY1 production represent a negative feedback loop consisting of miR-29–induced blockade of YY1 translation leading to an increase in miR-29 transcription to regulate muscle cell growth.12 Additional evidence that YY1 can mediate defective muscle metabolism includes the reports that inflammatory cytokines, such as TNFα23 and IL-1β,24 stimulate YY1 protein expression. This is of interest because high circulating levels of inflammatory cytokines are associated with reduced protein stores or hypoalbuminemia, changes which have led to the speculation that inflammatory cytokines cause muscle wasting.25 There is a mechanism by which inflammation does cause muscle wasting. We showed that in muscle, IL-6 in synergy with the acute phase protein, serum amyloid A, stimulates activation of caspase-3 and the ubiquitin-proteasome system to increase protein degradation in muscle.26,27 In addition, a mechanism involving myostatin has revealed how inflammation contributes to muscle wasting.37 In that report, pharmacologic inhibition of myostatin not only reduced the high levels of circulating inflammatory cytokines including TNFα and IL-6 in CKD mice it also prevented muscle wasting and improved the differentiation of MPCs. Others have linked miR-29 to the regulation of muscle metabolism: Cacchiarelli et al. found decreased muscle levels of miR-29 in the X-linked recessive muscle disorder Duchenne's muscular dystrophy.28
Because increasing miR-29 expression can largely correct the impairment in MPC differentiation in muscle of CKD mice (Figure 6), we tentatively conclude that the CKD-induced decrease in miR-29 results in a cause-effect relationship that regulates muscle cell differentiation. We recognize, however, that proof of this conclusion would require additional experiments such as investigating the influence of CKD in genetically altered mice with knockout of YY1 or miR-29. There also should be gene transfer experiments to restore YY1 or miR-29 in muscles of the knockout mice to identify the roles of YY1 or miR-29. Unfortunately, we do not have these knockout mice.
A central issue is why is miR-29 depressed? As noted, microRNAs are transcribed from DNA forming pri-miR, which are processed in the nucleus yielding stem-loop structures of about 70 to 120 nucleotide pre-miR. We examined levels of these precursors and both were decreased in parallel with the level of the mature miR-29a. These results suggest that the lower abundance of miR-29 results from suppressed transcription of miR-29. There is precedence for such a mechanism as Wang et al. reported that an increase in NF-κB can inhibit the transcription of miR-29b/c and reduce differentiation of myoblasts.12 Mott et al. reported that c-Myc, Hedgehog, or NF-κB can suppress miR-29a/b promoter activity.17 Because CKD is associated with increased levels of inflammatory cytokines that could activate NF-κB, similar events could influence miR-29 gene transcription in CKD.25,29 After we submitted this manuscript, Winbanks et al. reported that TGF-β can regulate miR-29 and miR-206 in muscle cells, producing an improvement in the differentiation of C2C12 myoblasts.30 Instead of YY1, they found that the miR-29 influence was related to histone deacetylase 4, another factor that inhibits muscle differentiation. Our results based on miR-29 and YY1 when coupled with those of Winbanks et al. emphasize the complexity of the regulation of protein expression and demonstrate how a single microRNA can change cellular responses by regulating the expression of different proteins.
More than one microRNA could be involved in the regulation of muscle cell myogenesis. Because myogenesis or impaired myogenesis involves several different steps, there are other potential regulatory possibilities. For example, miR-1 and miR-206 have been reported to affect the differentiation of muscle cells,31 and it is highly likely that other participants in regulating muscle metabolism will be uncovered.
Our current results identify a new CKD-induced pathway that can influence the abnormal regulation of muscle mass. The pathway is initiated by complications of CKD (possibly inflammation) that suppress mature miR-29 production. The result is an increase in muscle levels of YY1 that impair myoblast differentiation and, hence, myogenesis (Figure 7). Strategies that increase miR-29, therefore, could stimulate myogenesis and overcome its contribution to the muscle atrophy of CKD and possibly other catabolic conditions.
Figure 7.
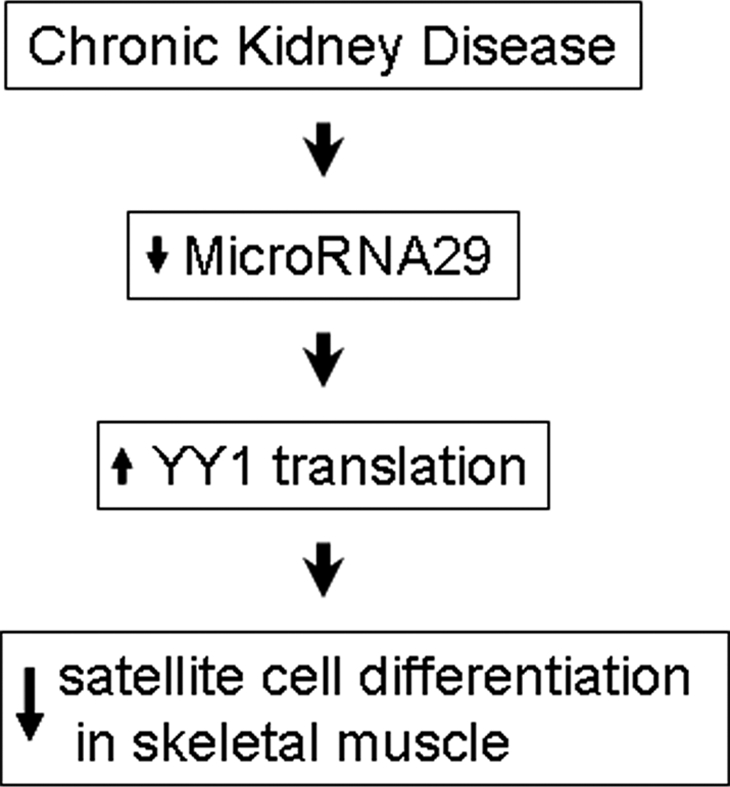
Decreased miR-29 causes increased YY1 to suppress satellite cell differentiation in mice with CKD.
CONCISE METHODS
Animals and CKD Model
The experiments were approved by the Emory University IACUC (protocol 141-2008). Mice (C57BL/6J) from Jackson Laboratories (Bar Harbor, ME) were kept in a 12-hour light/12-hour dark cycle. Under anesthesia (pentobarbital), CKD was produced by removing the right kidney and two poles of the left kidney; hemostasis was achieved by electric cautery and pressure.2 After surgery, CKD mice were fed 14% Protein Rodent Maintenance Diet Chow (Harlan Teklad, Madison, WI) ad libitum for 7 days before sham-operated mice were weight-matched with a CKD mouse and pair-fed a 40% protein diet for 2 weeks.2 CKD mice with blood urea nitrogen (BUN) >100 mg/dl (Reflotron Plus Diagnostic Device; Roche Diagnostics Corporation, Indianapolis) were studied.
Cell Culture and Myoblast Differentiation
C2C12 cells (ATCC, Manassas, VA) were cultured in growth media [Dulbecco modified Eagle medium plus 10% fetal bovine serum (FBS) and 10% cow serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine] and studied between passages 3 and 9. Cells (2 × 105) were cultured in a 6-well plate. To induce differentiation of C2C12 cells, the growth media was replaced by differentiation media (FBS and cow serum was replaced by 2% horse serum).32
Cell morphology was visualized using an Olympus 1 × 51 inverted microscope and photographed using a SIS-CC12 CLR camera. On day 4, pictures were used by a blinded observor to calculate a fusion index.19 The fusion index was calculated as the ratio of the number of nuclei present in myotubes to the total number of nuclei in three predefined, microscopic fields. Each contained >450 nuclei per field, and results from the three fields were averaged.
MPC Isolation and Differentiation
MPCs were isolated from hind-limb muscles of control and CKD mice.4 Muscles were minced into a coarse slurry and gently agitated for 1 hour at 37°C in Dulbecco-modified Eagle medium with 25 mM HEPES (pH 7.4) plus 0.1% pronase (Calbiochem, San Diego). The digest was passed through a 100-μm filter and then centrifuged through a Percoll gradient (70% overlaid with 40%).4 Cells from the gradient interface were collected and identified as MPC using anti–Pax-7 and anti-myoD antibodies (DSHB, University of Iowa). Cells collected were >90% Pax-7 and/or MyoD positive (data not shown).
For measurement of MPCs differentiation capacity, 4 × 105 MPCs were cultured in a Ham's F-10 medium contains 20% FBS, 5 ng/ml bFGF, 100 U/ml penicillin G, and 100 g/ml streptomycin (primary growth medium) in 6-well plates that had been coated with Entactin, Collagen IV, and Laminin (ECL matrix; Upstate Biotechnology, Temecula, CA). Differentiation was then induced by switching primary growth media to the differentiation media (see above). Cell morphology was monitored daily. For RT-PCR or qPCR analyses, MPCs were lysed in RNAzol RT reagent (Molecular Research Inc., Cincinnati, OH).
MicroRNA Array and Real-Time qPCR33
For the microRNA profile analysis, RNA enriched in small RNAs was extracted from the muscle using the mirVana miRNA isolation kit (Ambion, Austin, TX). After DNase treatment, the integrity of total RNA for each sample was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA): Only RNA samples meeting a minimum RNA integrity number value of 9 or larger (<10% degradation) were analyzed. MicroRNA profiling was performed by a Rodents miRNA Taqman Low Density Array (v1.0; containing 368 miRNAs; Applied Biosystems, Foster City, CA).
To measure pri-miR and pre-miR by RT-PCR, muscle RNA was extracted using RNAzol RT reagent (Molecular Research Inc.). For reverse transcription, 1 μg of RNA samples was used with oligo 9-mers primers and Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA). The expression of pri-miR and pre-miR was measured as described.17 PCR was performed with Platinum PCR SuperMix (Invitrogen) using the cycle parameters: 94°C for 2 minutes and 40 cycles at 94°C for 20 seconds, 55°C for 20 seconds, 72°C for 20 seconds with final extension at 72°C for 10 minutes. Products were separated in 2% (w/v) agarose gel electrophoresis and visualized by ethidium bromide fluorescence.
Individual microRNAs were measured by real-time qPCR. For synthesis cDNA, 1 μg of total RNA that was enriched in small RNAs was reverse transcripted using the NCode miRNA cDNA synthesis kit (Invitrogen). For qPCR, MiR-29a, MiR-29b, and MiR-29c primers were custom designed by Invitrogen. NCode express SYBR GreenER nuRBA qPCR kit (Invitrogen) was used with the following cycle parameters: 94°C for 2 minutes and 45 cycles at 95°C for 15 seconds and 60°C for 60 seconds. The Ct (threshold cycle) was defined as the number of cycles required for the fluorescence signal to exceed the detection threshold. Expression of individual microRNAs was standardized to the mouse U6 gene and calculated as the difference between the threshold values of the two genes (2Δct). Melting curve analyses were performed during real-time qPCR to analyze and verify the specificity of the reaction.
Luciferase Reporter Assay and Transfection
Signosis Inc. (Sunnyvale, CA) provided us with a custom-made vector (pLuc-3UTR-YY1) containing the firefly luciferase gene and the 3′-UTR(756-794) of YY1 (the miR-29 binding site of the 3′-UTR of YY1 is located at 774 to 780). The pLuc-3UTRmutant-YY1 vector was a gift from Dr. Huating Wang (The Ohio State University, Columbus, OH).12 The lentivector-based anti-microRNA expression vectors, pmiRZip29a and pmiRZip29b, were purchased from SBI system Biosciences (Mountain View, CA) and used to inhibit the activation of MiR-29a or/and MiR-29b in C2C12 cells.
For transfection, C2C12 cells in growth medium were seeded in 12-well plates and transfected using Effectene transfection reagent (Qiagen, Valencia, CA). In each well, 0.3 μg of firefly luciferase vector and 0.08 μg of the Renilla luciferase (control vector) were introduced. After 24 hours, the media was changed to differentiation medium and firefly and Renilla luciferase activities were measured consecutively by dual-luciferase assays (Promega) using TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA).34 We calculated the results as the ratio of firefly luciferase to renilla luciferase (×100). Results from control experiments (e.g., pLuc-3UTR-YY1 expressing cells that were treated with a control adenovirus) are expressed as 100%. Experimental results were calculated in the same fashion and expressed as a percent of control levels.
Western Blot and Antibodies
Hind-limb muscles were homogenized in RIPA buffer. Proteins in the soluble fraction of tissue homogenates were assessed by Western blotting.35 Primary antibodies included anti-YY1 (1:1000 dilution) from Santa Cruz (Santa Cruz, CA); anti-glyceraldehyde-3-phosphate dehydrogenase from Cell Signaling (Danvers, MA); and anti-emyHC from DSHB product (University of Iowa, IA).
Virus Reagents
The adenovirus construct, Ad-miR-29a/b/c, was produced by Dr. Fude Fang, and it was amplified as described.35,36 The adenovirus transduction unit (TU) was achieved by serial dilutions, and 5 μl of the concentrated viral preparation (107−9 TU) was added to 2-ml media and applied to 6-well plates; >90% of cells were transduced (based on green fluorescent protein expression). We used the Ad-miR-29 adenovirus to express miR-29a/b/c. Adenovirus transduction was accomplished when growth media was changed to differentiation media.
Statistical Analysis
All data are reported as the mean ± SEM. Comparison between groups was performed with the Kruskal-Wallis one-way ANOVA (nonparametric data); P < 0.05 was considered significant. MicroRNA array data were analyzed in GeneSpring GX 9.0, and preprocess was with GC Robust Multiarray Analysis (Mathworks, Natick, MA). Three steps were taken to process the probe-level data of the Rodents miRNA Taqman arrays: background correction, normalization, and probe-level summarization at the log2-scale.
DISCLOSURES
None.
Acknowledgments
We thank Dr. Huating Wang for kindly providing the pLuc-3UTRmutant-YY1 vector. This work was supported in part by the University Research Committee of Emory University and Norman S. Coplon extramural research grants from Satellite Health Research to both X.H.W. and L.Z. Additional support was NIH DK062081 to J.D.K. and NIH R37 DK37175 to W.E.M. and the Virology & Drug Discovery Core of the Emory Center for AIDS Research (P30 AI050409).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
REFERENCES
- 1. Carrero JJ, Chmielewski M, Axelsson J, Snaedal S, Heimburger O, Barany P, Suliman ME, Lindholm B, Stenvinkel P, Qureshi AR: Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin Nutr 27: 557–564, 2008 [DOI] [PubMed] [Google Scholar]
- 2. Hu J, Du J, Zhang L, Price SR, Klein JD, Wang XH: XIAP reduces muscle proteolysis induced by CKD. J Am Soc Nephrol 21: 1174–1183, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. May RC, Kelly RA, Mitch WE: Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J Clin Invest 79: 1099–1103, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang L, Wang XH, Wang H, Du J, Mitch WE: Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J Am Soc Nephrol 21: 419–427, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang XH, Du J, Klein JD, Bailey JL, Mitch WE: Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int 76: 751–759, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seale P, Rudnicki MA: A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev Biol 218: 115–124, 2000 [DOI] [PubMed] [Google Scholar]
- 7. Gros J, Manceau M, Thome V, Marcelle C: A common somitic origin for embryonic muscle progenitors and satellite cells. Nature 435: 954–958, 2005 [DOI] [PubMed] [Google Scholar]
- 8. Mitchell PO, Pavlath GK: A muscle precursor cell-dependent pathway contributes to muscle growth after atrophy. Am J Physiol Cell Physiol 281: C1706–C1715, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Vincent CK, Gualberto A, Patel CV, Walsh K: Different regulatory sequences control creatine kinase-M gene expression in directly injected skeletal and cardiac muscle. Mol Cell Biol 13: 1264–1272, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V: The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18: 2627–2638, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee TC, Shi Y, Schwartz RJ: Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal alpha-actin transcription in embryonic myoblasts. Proc Natl Acad Sci U S A 89: 9814–9818, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, Croce CM, Guttridge DC: NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14: 369–381, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP: The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 310: 1817–1821, 2005 [DOI] [PubMed] [Google Scholar]
- 14. Stefani G, Slack FJ: Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 9: 219–230, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Bartel DP: MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE: Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: Implications for muscle atrophy. J Am Soc Nephrol 17: 1388–1394, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, Fernandez-Zapico ME: Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem 110: 1155–1164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yaffe D, Saxel O: Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270: 725–727, 1977 [DOI] [PubMed] [Google Scholar]
- 19. Bondesen BA, Jones KA, Glasgow WC, Pavlath GK: Inhibition of myoblast migration by prostacyclin is associated with enhanced cell fusion. FASEB J 21: 3338–3345, 2007 [DOI] [PubMed] [Google Scholar]
- 20. Wang H, Hertlein E, Bakkar N, Sun H, Acharyya S, Wang J, Carathers M, Davuluri R, Guttridge DC: NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol 27: 4374–4387, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon S, Akopyan G, Garban H, Bonavida B: Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 25: 1125–1142, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Shi Y, Lee JS, Galvin KM: Everything you have ever wanted to know about Yin Yang 1. Biochim Biophys Acta 1332: F49–F66, 1997 [DOI] [PubMed] [Google Scholar]
- 23. Huerta-Yepez S, Vega M, Garban H, Bonavida B: Involvement of the TNF-alpha autocrine-paracrine loop, via NF-kappaB and YY1, in the regulation of tumor cell resistance to Fas-induced apoptosis. Clin Immunol 120: 297–309, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Patten M, Wang W, Aminololama-Shakeri S, Burson M, Long CS: IL-1 beta increases abundance and activity of the negative transcriptional regulator yin yang-1 (YY1) in neonatal rat cardiac myocytes. J Mol Cell Cardiol 32: 1341–1352, 2000 [DOI] [PubMed] [Google Scholar]
- 25. Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, Heimburger O, Cederholm T, Girndt M: IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia–the good, the bad, and the ugly. Kidney Int 67: 1216–1233, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE: IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang XH, Zhang L, Mitch WE, LeDoux JM, Hu J, Du J: Caspase-3 cleaves specific 19 S proteasome subunits in skeletal muscle stimulating proteasome activity. J Biol Chem 285: 21249–21257, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, Auricchio A, Musaro A, Bozzoni I: MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 12: 341–351, 2010 [DOI] [PubMed] [Google Scholar]
- 29. Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE: The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest 97: 1447–1453, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, Gregorevic P: TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem 286: 13805–13814, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, Wang DZ: MicroRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol 190: 867–879, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE: Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest 113: 115–123, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Hu Z, Hu J, Du J, Mitch WE: Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 147: 4160–4168, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Wang X, Chinsky JM, Costeas PA, Price SR: Acidification and glucocorticoids independently regulate branched-chain alpha-ketoacid dehydrogenase subunit genes. Am J Physiol Cell Physiol 280: C1176–C1183, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Zhou Q, Du J, Hu Z, Walsh K, Wang XH: Evidence for adipose-muscle cross talk: Opposing regulation of muscle proteolysis by adiponectin and fatty acids. Endocrinology 148: 5696–5705, 2007 [DOI] [PubMed] [Google Scholar]
- 36. He A, Zhu L, Gupta N, Chang Y, Fang F: Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3–L1 adipocytes. Mol Endocrinol 21: 2785–2794, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, Song Y, Min H, Wang X, Du J, Mitch WE: Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 25: 1653–1663, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]