

Abstract
Dysfunction and destruction of pancreatic islet β-cells is a hallmark of diabetes. Better understanding of cell signals regulating β-cell growth and antiapoptosis will allow development of therapeutic strategies for diabetes by preservation and expansion of β-cell mass. GH and IGF-I share a complicated physiological relationship and have both been implicated in β-cell function. GH and IGF-I exert their biological effects through binding to respective receptors (GHR and IGF-IR) and subsequently engaging downstream signaling pathways. However, their collaborative roles in modulation of β-cell mass and the underlying molecular mechanisms remain poorly understood. In this study, we demonstrate that cultured β-cells are appealing systems for investigating potential GH-IGF-I signaling cross talk. We uncover that GH specifically promotes formation of a protein complex containing GHR, Janus kinase 2 (a nonreceptor kinase coupled to GH/GHR signaling), and IGF-IR. More importantly, GH and IGF-I synergistically activate both signal transducer and activator of transcription 5 and Akt pathways. Concomitantly, β-cells proliferate more robustly and are better protected from serum deprivation-induced apoptosis when exposed to GH and IGF-I in combination vs. GH or IGF-I alone. The augmented proliferative effects by GH and IGF-I are confirmed in isolated islets. Taken together, our findings strongly suggest that there exists a novel signaling relationship between GH/GHR and IGF-I/IGF-IR systems in β-cells, i.e. IGF-IR may serve as a proximal component of GH/GHR signaling, contributing to enhancement of β-cell mass and function. In support of this, IGF-IR knockdown in β-cells resulted in the desensitization of acute GH-induced signal transducer and activator of transcription 5 activation.
Impaired insulin secretion resulting from dysfunction and destruction of insulin-producing β-cells in the pancreatic islets of Langerhans plays a central role in the development of hyperglycemia in diabetes mellitus (1). Type 2 diabetes (the most common type) is characterized by progressive β-cell dysfunction and a reduction in β-cell mass (2, 3). It is well known that β-cell mass is governed by a constant balance between β-cell growth (replication from preexisting β-cells and neogenesis from precursor cells) and β-cell death (apoptosis and necrosis) (4, 5). The β-cell mass changes in response to metabolic status and insulin demand. Increasing evidence suggests that growth factors and hormones, such as IGF-I and GH, are important mediators of β-cell growth, survival, differentiation, and insulin secretion (6–9). However, the underlying signal transduction pathways, especially their potential cross talk, remain poorly understood. Thus, better understanding of the signaling aspects contributing to β-cell proliferation and antiapoptosis will be essential to allow the development of therapeutic strategies for diabetes by preservation and expansion of β-cell mass.
IGF-I (also known as somatomedin-C) is synthesized largely in the liver but also in other target tissues. Classically, IGF-I is considered a major physiological effector of GH. IGF-I exerts its biological actions by interacting with type 1 IGF-I receptor (IGF-IR), which is a cell surface heterotetramer with intrinsic kinase activity embedded in its β-subunit cytoplasmic domains (10) and is a member of receptor tyrosine kinase family (11). IGF-I binding causes IGF-IR intrinsic kinase activation and subsequent activation of downstream signaling cascades, mainly including the Shc/Ras/MAPK kinase/ERK pathway and the insulin receptor substrate (IRS)/phosphoinositide 3-kinase (PI3K)/Akt pathway (11). In general, these pathways are thought to be critical for cell proliferation and antiapoptosis (12, 13). Previous studies in mouse models and cultured insulinoma cells suggested that the IGF-I-mediated IRS-2/PI3K/Akt pathway plays an important role in maintaining β-cell function (14, 15). In particular, the activation of the PI3K/Akt pathway is not only essential for IGF-I and glucose-induced β-cell proliferation (although the Ras/ERK pathway is likely involved) (16) but also important for promoting β-cell survival and maintaining β-cell mass (17).
GH is produced in the anterior pituitary gland and is an important regulator of growth and metabolism (18). Nearly all known GH actions require its specific binding to the GH receptor (GHR), a transmembrane glycoprotein member of the cytokine receptor superfamily (19). GHR lacks intrinsic kinase activity but is physically and functionally coupled to Janus kinase (JAK) 2, a nonreceptor cytoplasmic tyrosine kinase of the JAK family. GH engagement enhances the association between GHR and JAK2, causes JAK2 activation and phosphorylation of GHR, and elicits several intracellular signaling cascades, including the signal transducer and activator of transcription 5 (STAT5), Ras/ERK, and PI3K/Akt pathways (19, 20). Compared with IGF-I signaling, much less is known about how GH signaling regulates β-cell function. Although IGF-I expression in the pancreas in vivo mainly occurs in endothelial cells and proliferating duct cells (21), several studies have shown that IGF-I appears to be specifically localized to α-cells in islets (22–24). Thus, GH is more likely to exert a direct effect independent of IGF-I on β-cell proliferation (25). A previous study in rat insulinoma INS-1 cells showed that GH promoted cell proliferation in a glucose-dependent manner, which was enhanced further by IGF-I (26). Given that neither glucose nor the combination of glucose and IGF-I activated JAK2 and STAT5 in these cells, it was concluded that although dependent on glucose, GH promoted INS-1 cell growth directly via the JAK2/STAT5 pathway with no cross talk to IGF-I signaling (26).
Although IGF-I can be an effector of GH action, signaling synergy by these two growth factors has been reported in several cell systems (27–29), suggesting their collaboration or cross talk. In particular, we previously showed that GH induces formation of a protein complex containing GHR, JAK2, and IGF-IR in mouse preadipocytes (29), implicating that IGF-IR may serve as a proximal component in GH/GHR signaling. Furthermore, two recent studies in mouse primary osteoblasts in which the IGF-IR gene was disrupted via Cre/loxP have demonstrated some direct actions of GH independent of IGF-IR and participation of IGF-IR in aspects of GH signaling (30, 31). Given the importance of physiological interrelationships between GH and IGF-I and their respective roles in pancreatic islet β-cell function, here we used multiple rodent β-cell lines that retain many key functional features of normal islets (32, 33) as model systems to further explore the potential GH-IGF-I signaling cross talk and its impact on β-cell mass. Our new data suggest that there exists physical and functional interaction between GH/GHR and IGF-I/IGF-IR signaling systems in β-cells, and this could underlie their synergistic effects on β-cell proliferation and survival.
Results
GH specifically induces formation of GHR-JAK2-IGF-IR protein complex in β-cells
Here we used rat INS-1 and mouse MIN6 and BTC6 cells, which have been widely accepted as model systems for studying β-cell signaling and function (32, 33) to examine the GH and IGF-I signaling. Because human GH interacts with both GHR and prolactin receptor in rodents (8), we used bovine GH (bGH) that only binds to GHR (34) throughout the study. As shown in Fig. 1A, all three β-cell lines expressed GHR, JAK2, and IGF-IR, which were readily detected by immunoblotting with anti-GHR, anti-JAK2, and anti-IGF-IR antibodies, respectively. However, their expression levels, in particular GHR and IGF-IR, differed among the cell lines and seemed to be the highest in INS-1 cells. The appearance of multiple (diffuse) bands of GHR and IGF-IR is due to the glycosylation (29) that apparently varies between species (mouse vs. rat). To test their hormonal responsiveness, the cells were exposed to vehicle (−), GH or IGF-I for 15 min, and cell extracts were immunoblotted with anti-phosphorylated (p) STAT5 and anti-pAkt antibodies. Our results demonstrated that GH and IGF-I acutely activated the STAT5 and Akt pathways, respectively (Fig. 1, B and C). Taken together, these data indicated that all three cell lines tested here were responsive to both GH and IGF-I, making them appealing systems to study potential cross talk between the GH and IGF-I signaling systems.
Fig. 1.
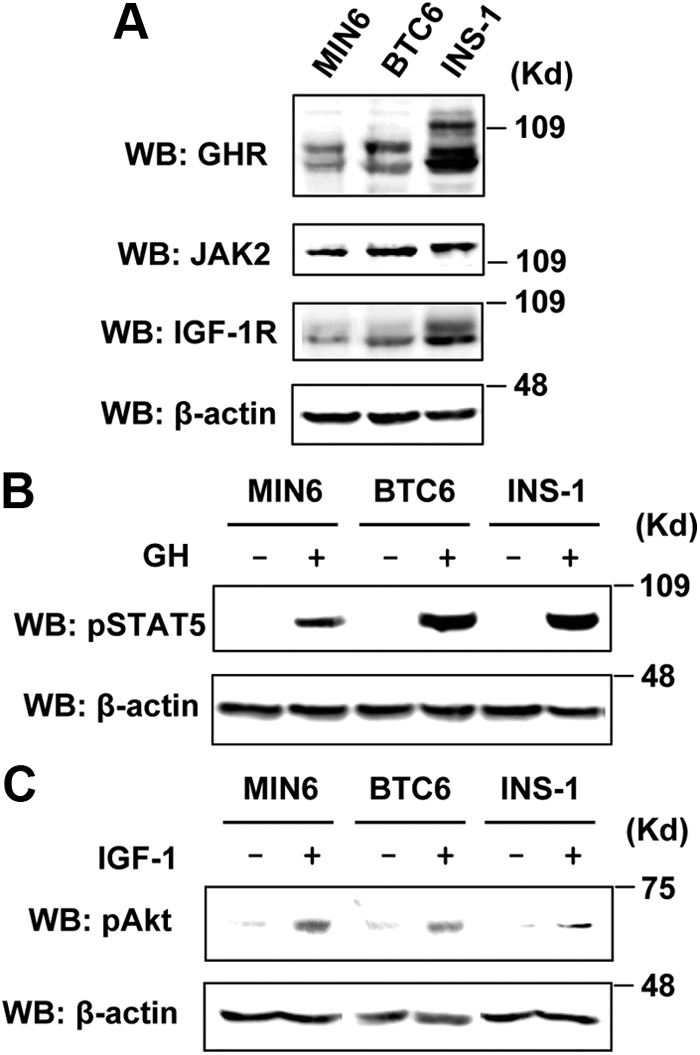
Rodent β-cell lines endogenously express GHR, JAK2, and IGF-IR and are responsive to GH and IGF-I. A, Comparison of expression levels of GHR, JAK2, and IGF-IR among MIN6, BTC6, and INS-1 cells by immunoblotting with anti-GHR, anti-JAK2, and anti-IGF-IR antibodies, respectively. WB, Western blot. B and C, Serum-starved cells were stimulated with vehicle (−), bGH (500 ng/ml), or IGF-I (20 ng/ml) for 15 min. Cell extracts were analyzed by immunoblotting with anti-pSTAT5, anti-pAkt, or anti-β-actin (loading), as indicated.
To examine the effects of GH and IGF-I on tyrosine phosphorylation of GHR and IGF-IR, we performed immunoprecipitation (IP) with anti-GHR or anti-IGF-IR antibody and then immunoblotting with an antiphosphotyrosine (pTyr) antibody using detergent extracts from MIN6 cells stimulated with vehicle, GH, or IGF-I (Fig. 2A, upper panel). As expected, anti-GHR precipitation revealed GH-induced tyrosine phosphorylation of a collection of proteins within the 100- to 120-kDa range (Fig. 2A, upper panel, lane 2), which included a lower diffuse band (bracket) partially overlapping with a sharper upper band (arrow). These patterns resulted from GH stimulation are consistent with GH-induced association of tyrosine-phosphorylated GHR and JAK2, as seen in other GH-responsive cell types (29, 35). In contrast, IGF-I stimulation did not result in the formation of this GHR-JAK2 complex (Fig. 2A, upper panel, lane 3). Likewise, IGF-I treatment caused enhanced tyrosine phosphorylation of IGF-IR β-chain migrating at approximately 95 kDa (Fig 2A, lane 6 vs. lane 4, arrowhead). Notably, the IGF-IR was basally activated, possibly due to the chronic insulin secretion by the insulinoma cells (Fig. 2A, upper panel, lane 4). Interestingly, we observed a diffuse phosphoprotein complex of 100–120 kDa (partially overlapping with the IGF-IR β-chain) present in the anti-IGF-IR precipitates from the GH-treated cells (Fig. 2A, upper panel, lane 5). The size, appearance, and pattern of migration on SDS-PAGE of this GH-induced protein complex detected in the anti-IGF-IR precipitates (Fig. 2A, upper panel, lane 5 vs. lane 2) suggested that it might contain GHR and JAK2. This phenomenon was also observed in BTC6 and INS-1 cells (data not shown). Reprobing the same blots with anti-GHR or anti-IGF-IR verified equal protein precipitates in each lane (Fig. 2A, lower panels).
Fig. 2.

GH specifically induces formation of GHR-JAK2-IGF-IR protein complex in β-cells. A, GH-induced phosphoprotein complex in the anti-IGF-IR precipitates. Serum-starved MIN6 cells were stimulated with vehicle (−), bGH (500 ng/ml), or IGF-I (20 ng/ml) for 15 min. Cell extracts were immunoprecipitated with anti-GHR (lanes 1–3) or anti-IGF-IR (lanes 4–6). The immunoprecipitates were analyzed by immunoblotting with anti-pTyr (lanes 1–6), anti-GHR (lanes 1–3), or anti-IGF-IR (lanes 4–6). The GHR, JAK2, and IGF-IR bands are indicated by the bracket, arrow, and arrowhead, respectively. WB, Western blot. B, JAK2 is a component of the GH-induced protein complex from the anti-IGF-IR precipitates. MIN6 and BTC6 cells treated as in A were subjected to IP with anti-IGF-IR antibody followed by immunoblotting with anti-JAK2 and anti-IGF-IR, respectively. C, GHR is also a component of the GH-induced protein complex from the anti-IGF-IR precipitates. Serum-starved INS-1 cells were stimulated with vehicle (−) or bGH (500 ng/ml) for 15 min. Cell extracts were subjected to IP with anti-IGF-IR followed by immunoblotting (lanes 1 and 2) or direct immunoblotting (lanes 3 and 4) with anti-GHR, anti-JAK2, and anti-IGF-IR, respectively. D, Reverse coimmunoprecipitation experiment. INS-1 cells as treated in C were subjected to IP with anti-GHR followed by immunoblotting (lanes 1 and 2) or direct immunoblotting (lanes 3 and 4) with anti-IGF-IR, anti-JAK2, and anti-GHR, respectively.
Further co-IP experiments (IP with anti-IGF-IR followed by immunoblotting with an anti-JAK2 antibody) in both BTC6 and MIN6 cells confirmed that the protein complex seen in the anti-IGF-IR precipitates in response to GH stimulation indeed contained JAK2 (Fig. 2B, upperpanel, lanes 2 and 5). In contrast, IGF-I treatment did not cause the physical association of IGF-IR with JAK2 (Fig. 2B, upper panel, lanes 3 and 6). Under these experimental conditions in BTC6 and MIN6 cells, we failed to detect the GHR in the anti-IGF-IR precipitates from the GH-treated cells using our anti-GHR antibody (data not shown). This could be due to the low abundance of endogenous GHR in the two mouse lines (Fig. 1A). Because rat INS-1 cells expressed a higher level of GHR (Fig. 1A), we pursued similar co-IP experiments (IP with anti-IGF-IR followed by immunoblotting with anti-GHR) in this cell line. Our results indicated that GHR was indeed a component of this protein complex (Fig. 2C, upper panel, lane 2). Reprobing with anti-JAK2 confirmed the presence of JAK2 in the complex (Fig. 2C, middle panel, lane 2), which is in consistent with the results from both BTC6 and MIN6 cells (Fig. 2B). We also performed reverse co-IP experiments (IP with anti-GHR followed by immunoblotting with anti-IGF-IR) in INS-1, given its higher GHR and IGF-IR abundance compared with the two mouse lines (Fig. 1A). The results revealed that IGF-IR was coprecipitated with anti-GHR upon GH stimulation (Fig. 2D, upper panel, lane 2 vs. lane 1). JAK2 was also detected in the anti-GHR precipitates from GH-treated cells (Fig. 2D, middle panel, lane 2 vs. lane 1).
We further asked whether rodent GH had similar effects as the bGH did in cultured rodent β-cells. In a representative experiment, INS-1 cells were treated with vehicle (−) or rat GH (+) for 15 min, and cell extracts were used for co-IP assays. The results showed that rat GH promoted the physical association of GHR/JAK2 with IGF-IR (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Thus, based on all these data (Fig. 2, A–D and Supplemental Fig. 1), we concluded that GH specifically induced the formation of the GHR-JAK2-IGF-IR protein complex in β-cells.
We also sought to estimate how much GHR and IGF-IR were associated with each other upon GH stimulation. Immunoblots from multiple independent experiments as shown in Fig. 2, C and D, were subjected to densitometric analysis. The actual inputs of total cell extracts used in co-IP (0.75–2 mg) vs. loaded directly onto SDS-PAGE (50–100 μg) in each experiment were calculated. By analyzing these parameters in combination, our estimations were that 7.3 ± 0.7% of GHR proteins present in the cell extracts were detected in the anti-IGF-IR precipitates (n = 5, as exemplified in Fig. 2C) and that 2.4 ± 0.8% of IGF-IR proteins were detected in the anti-GHR precipitates (n = 3, as exemplified in Fig. 2D). The relative low percentages of GHR and IGF-IR coassociation from their respective free pools could reflect the efficacy of each antibody for immunoprecipitation and/or the nature of in vitro co-IP assays. Regardless, specific association between GHR and IGF-IR can be detected by reciprocal co-IP.
It is well known that the active signaling conformation of the GHR upon GH engagement is as a dimer bound to a single GH molecule (GHR/GH = 2:1) (36). Accumulating evidence also suggests that GHR is likely a predimer in the absence of ligand and active assembly arises by the GH-induced GHR conformational change, which could allow more productive interaction between GHR and JAK2 and resultant initiation of the GH/GHR signaling cascades (37–39). A class of recombinant human GH antagonists harboring mutations at residues known to be critical for the active GHR dimer conformation has been developed (40, 41). G120K is one of them and can antagonize the ability of GH to induce GHR disulfide linkage (a reflection of the GHR conformation change) and tyrosine phosphorylation of both GHR and JAK2 in cell culture (42) and in the liver of mice (43). Here we tested whether G120K could antagonize the ability of bGH to promote the GHR-JAK2-IGF-IR complex formation in β-cells (Fig. 3). As expected, G120K did not itself induce GHR and JAK2 tyrosine phosphorylation (Fig. 3, A and B, upper panels, lane 4); however, it antagonized the ability of bGH to induce these effects (Fig. 3, A and B, upper panels, lane 3 vs. 2), demonstrating that G120K can act as a bGH antagonist in β-cells. More importantly, in the same experimental setting, G120K prevented the bGH-induced formation of GHR-JAK2-IGF-IR complex (Fig. 3C, upper and middle panels, lane 3 vs. lane 2). These results further suggested that this protein complex formation was strictly dependent on GH engagement and a proper GHR conformation.
Fig. 3.
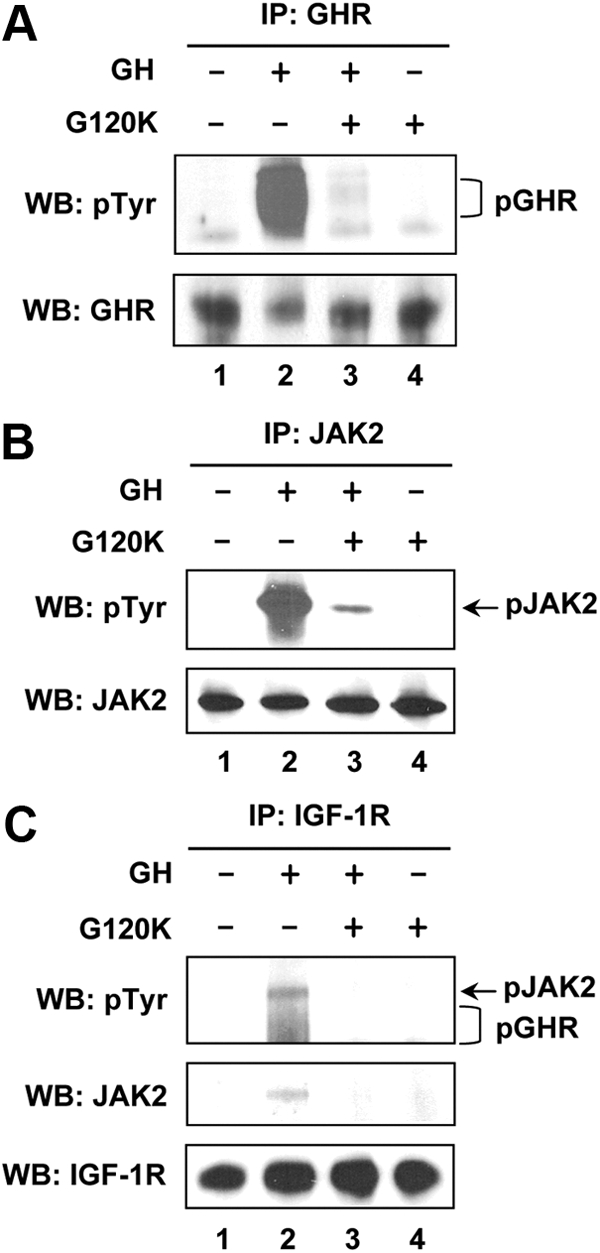
GH antagonist, G120K, prevents the GH-induced GHR-JAK2-IGF-IR complex formation. Serum-starved BTC6 cells were stimulated with vehicle (−) or bGH (500 ng/ml) in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of G120K (2500 ng/ml) for 15 min. Cell extracts were subjected to IP with anti-GHR (A), anti-JAK2 (B), and anti-IGF-IR (C), respectively, followed by immunoblotting with anti-pTyr, anti-GHR, anti-JAK2, or anti-IGF-IR, as indicated. The phosphorylated forms of GHR and JAK2 are indicated by a bracket and arrow, respectively. WB, Western blot.
GH and IGF-I synergize in acute β-cell signaling
Previous studies have suggested that GH and IGF-I may act synergistically in promoting gene activation, cell signaling, and proliferation (27–29). Here we observed that GH promoted the formation of a GHR-JAK2-IGF-IR complex in β-cells (Figs. 2 and 3), making them appealing systems for assessing the signaling outcomes of the GH and IGF-I interplay by comparing the acute signaling in response to GH or IGF-I alone with that in response to their combination (GH/IGF-I cotreatment). Using MIN6 cells, we first examined pSTAT5 levels by immunoblotting. As expected, GH stimulation resulted in robust STAT5 activation (Fig. 4A, upper panel, lane 2). Interestingly, although IGF-I itself did not cause any pSTAT5 signal (Fig. 4A, upper panel, lane 3), it enhanced GH-induced STAT5 activation when used in combination with GH (Fig. 4A, upper panel, lane 4 vs. lane 2). The pSTAT5 levels were measured densitometrically from eight such experiments and plotted in Fig. 4B. IGF-I cotreatment significantly augmented GH-induced STAT5 activation by approximately 50% (P < 0.05). Similar results were obtained in INS-1 cells (data not shown).
Fig. 4.
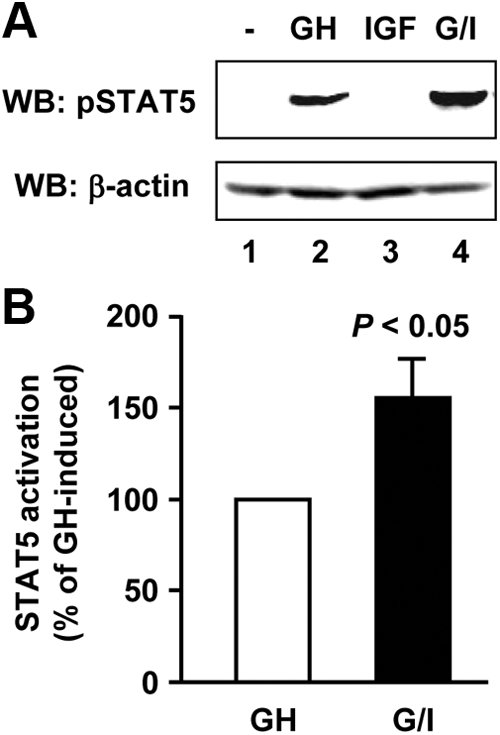
GH and IGF-I synergize in STAT5 activation. A, Serum-starved MIN6 cells were treated with vehicle (−), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) for 15 min. Cell extracts were analyzed by immunoblotting with anti-pSTAT5 and anti-β-actin, respectively. WB, Western blot. B, Densitometric analysis of pooled data, as in A, from eight independent experiments. Comparison of the level of STAT5 activation induced by bGH and IGF-I cotreatment (referred to as G/I) with that induced by bGH alone (set as 100%) is shown. Data are mean ± sem (n = 8). P < 0.05 (G/I vs. GH).
We next assessed the Akt activation. In β-cells, GH and IGF-I each acutely induced Akt activation (Fig. 5A, upper panel, lanes 1 and 3 vs. lane 4). Notably, GH/IGF-I cotreatment caused greater pAkt signal than each single treatment did (Fig. 5A, upper panel, lane 2 vs. lanes 1 and 3). This was confirmed by densitometric analysis of data from five such experiments (Fig. 5B). To determine whether such augmentation achieved by the cotreatment reflected simply the summation of GH-induced plus IGF-I-induced pAkt or rather reflected a synergistic effect (greater than the summation), we used the same analytic methodology as we used in previous studies (29, 44–46). The reanalyzed results are displayed in Fig. 5C, in which the white bar indicates the sum of pAkt signals elicited by GH alone plus IGF-I alone (referred to as GH + IGF, set to 100%) and the black bar (referred to as G/I) represents the actual pAkt level achieved by the costimulation. The Akt activation induced by GH/IGF-I cotreatment was 155 ± 19% (P < 0.05), indicating signaling synergy.
Fig. 5.
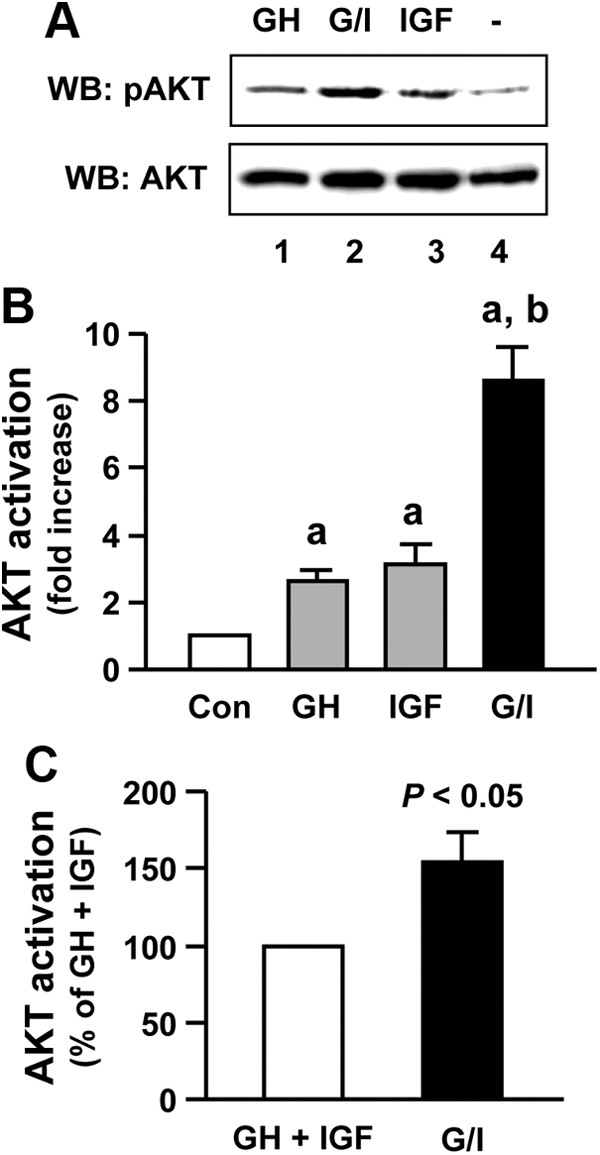
GH and IGF-I synergize in Akt activation. A, Serum-starved MIN6 cells were treated with vehicle (−), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) for 15 min. Cell extracts were analyzed by immunoblotting with anti-pAkt and anti-total Akt, respectively. WB, Western blot. B, Densitometric analysis of pooled data, as in A, from five independent experiments. Fold increase in Akt activation is shown for each condition with the basal (vehicle control) set as 1. Data are mean ± sem (n = 5). a, P < 0.01 (compared with control); b, P < 0.01 (compared with either GH or IGF). C, Reanalyzed results of pooled data of Akt activation. In this display, the sum of pAkt levels induced by bGH alone plus IGF-I alone (referred to as GH + IGF) is set as 100% and compared with the pAkt level induced by bGH and IGF-I cotreatment (referred to as G/I). Data are mean ± sem (n = 5). P < 0.05 (G/I vs. GH + IGF).
GH and IGF-I cotreatment augments β-cell growth and antiapoptosis
Cell growth and survival are two important aspects that significantly impact β-cell mass and function. Our findings of GH-IGF-I signaling synergy further prompted us to evaluate these two cellular processes. We first performed 5-bromo-2′-deoxyuridine (BrdU) incorporation assay to measure DNA synthesis, a marker of cell proliferation (47). In these experiments, cells with incorporated BrdU in their nuclei (proliferating cells) were detected by immunostaining with an anti-BrdU antibody (red), and total cell numbers were measured by nuclear counterstaining with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; blue). In the representative experiment in INS-1 cells shown in Fig. 6A, the basal BrdU-positive (red) cell number was, as expected, very low in serum-free medium (control). The numbers were markedly increased after 24 h treatment with either GH or IGF-I. Notably, more BrdU-positive cells were seen in the cotreatment of GH and IGF-I (G/I) for 24 h. A total of approximately 1500 cells of INS-1 and MIN6, respectively, were counted under each condition. The ratio of the number of BrdU-positive cells (red) to the number of total cell (blue) in each field (as shown in Fig. 6A) was considered as cell proliferation rate and plotted in Fig. 6B for experiments in both INS-1 and MIN6 cells. In both, GH and IGF-I cotreatment (G/I) significantly augmented the β-cell proliferation when compared with individual hormone treatments.
Fig. 6.

GH and IGF-I cotreatment augments β-cell growth. A, Representative images from BrdU incorporation assays. INS-1 cells were treated with vehicle (control), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) in serum-free medium for 24 h, in which BrdU was added in the last 4 h of treatment (chase phase). Cells with incorporated BrdU (proliferating cells) were detected by immunostaining with an anti-BrdU monoclonal antibody (red). Total cell numbers were measured by DAPI staining (blue). Scale bar, 50 μm. B, Cell proliferation rates determined by BrdU incorporation assays. The BrdU chasing periods in the INS-1 and MIN6 cells were 4 and 1.5 h, respectively. A total of approximately 1500 cells from 10 random imaging fields under each condition, as in A, were counted and the ratios of BrdU-positive cells (red) to the total cells (blue) (representing cell proliferation rates) were plotted with the control set as 1. Data are mean ± sem (n = 10). *, P < 0.05; **, P < 0.01. C, Control; G, GH; I, IGF-I.
We next examined the effects by regimens of GH, IGF-I, or their combination on cell death by Hoechst staining, a well-established method for measurement of cell apoptosis (48, 49). Hoechst is a fluorescent dye, which penetrates into intact cells and is taken by the nuclei. The apoptotic nuclei exhibit as bright blue due to their nuclear condensation, whereas viable cells are dark blue, as we previously demonstrated (50). In Fig. 7A, a substantial fraction of apoptotic cells (bright blue fluorescence) was observed after MIN6 cells were serum starved for 24 h (control). The addition of either GH or IGF-I to the serum-free medium prevented the cells from undergoing apoptosis because much less bright blue fluorescence was seen in these regimens. Interestingly, addition of GH and IGF-I (G/I) simultaneously further reduced the proportion of apoptotic cells. A total of approximately 1500 cells, as in Fig. 7A, were counted under each condition, and the percentage of apoptotic cells (bright blue) from the total cells (the sum of bright blue and dark blue cells), which represents the apoptosis rate, is displayed in Fig. 7B. G/I cotreatment significantly enhanced the antiapoptotic effect when compared with either GH or IGF-I treatments alone. We also measured the cell death rate by propidium iodide (PI) staining, as we described previously (50). PI is a fluorescent dye that is taken only by late apoptotic and necrotic cells. The PI-positive (red) cells under the four conditions are displayed in Fig. 7C, and statistical results are shown in Fig. 7D. In MIN6 cells, G/I cotreatment diminished serum withdrawal-induced necrosis (including late apoptosis) to a greater extent than each single hormonal regiment did. Similar results of Hoechst and PI staining were also obtained in INS-1 cells (data not shown). To corroborate this finding, we performed terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL) staining, which measures nuclear DNA fragmentation (detailed in Materials and Methods), an important biochemical hallmark of apoptosis in many cell types. As shown in Fig. 7, E and F, G/I cotreatment significantly protected INS-1 cells from serum deprivation-induced apoptosis better than either GH or IGF-I treatment alone did (P < 0.01).
Fig. 7.

GH and IGF-I cotreatment augments β-cell antiapoptosis. A and B, Hoechst staining of apoptotic cells. MIN6 cells were treated with vehicle (control), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) in serum-free medium for 24 h, in which Hoeschst 33258 was added in the last 15 min of treatment. The apoptotic nuclei exhibit as bright blue due to their nuclear condensation, whereas viable cells are dark blue, shown as representative images in A. Scale bar, 50 μm. A total of approximately 1500 cells from five random imaging fields under each condition, as in A, were counted, and the apoptosis rates were plotted with the control set as 100% (B). Data are mean ± sem (n = 5). *, P < 0.05; **, P < 0.01. C, Control. C and D, PI staining of necrotic and late apoptotic cells. MIN6 cells were treated with vehicle (control), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) in serum-free medium for 48 h, in which PI was added in the last 15 min of treatment. PI-stained necrotic and late apoptotic cells (red) are shown as representative images in C. Scale bar, 50 μm. A total of approximately 500 cells from eight random imaging fields under each condition, as in C, were counted, and cell death (necrosis and late apoptosis) rates were plotted with the control set as 100% (D). Data are mean ± sem (n = 8). *, P < 0.05; **, P < 0.01. E and F, TUNEL staining of apoptotic cells. INS-1 cells were treated with vehicle (control), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) in serum-free medium for 24 h. Apoptotic cells (green) were detected using the DeadEnd fluorometric TUNEL system (Promega) (detailed in Materials and Methods), shown as representative images in E. Scale bar, 50 μm. A total of approximately 2500 cells from five random imaging fields under each condition, as in E, were counted, and cell apoptosis rates were plotted with the control set as 100% (F). Data are mean ± sem (n =5). **, P < 0.01.
Knockdown of IGF-IR in β-cells desensitizes GH-induced STAT5 activation
Our findings of GH-induced GHR-JAK-IGF-IR complex formation and GH-IGF-I signaling synergy in β-cells (Figs. 2–7) have implicated a close structural-functional relationship, i.e. IGF-IR may serve as a proximal component in GH/GHR signaling. To address this, we knocked down the endogenous IGF-IR in MIN6 cells by RNA interference (RNAi). To improve the transfection efficiency in primary-like β-cells, we used a lentiviral based short hairpin RNA (shRNA) delivery system. Transient transfection with a set of plasmids, pGIPZ-IGF-IR shRNAmir-1, 2, and 3 (targeting mouse IGF-IR), or pGIPZ-NS (nonsilencing control) (detailed in Materials and Methods), indicated that shRNAmir-1 markedly reduced the IGF-IR expression in MIN6 cells, whereas the NS control had no apparent effect (Fig. 8A). Thus, the shRNAmir-1 and NS control were used to produce lentivirus to further transduce MIN6 cells, as indicated by simultaneous TurboGFP expression (Fig. 8B). The stable knockdown of IGF-IR after puromycin selection was confirmed by immunoblotting with anti-IGF-IR (Fig. 8C). We next assessed the GH-dependent activation of STAT5 in these cells. As shown in Fig. 8D, GH but not IGF-I stimulation resulted in robust STAT5 activation in both parental and NS control cells (Fig. 8D, upper panel, lanes 2 and 8). Notably, however, the GH-induced STAT5 activation was substantially diminished in the IGF-IR knockdown cells (Fig. 8D, upper panel, lane 5). Densitometric analysis of pooled data from three independent experiments indicated that knockdown of IGF-IR significantly reduced the GH-dependent STAT5 activation by approximately 50% (P < 0.05) (Fig. 8E).
Fig. 8.

Knockdown of endogenous IGF-IR desensitizes GH-induced STAT5 activation in β-cells. A, The efficacy of knockdown. MIN6 cells were transiently transfected with pGIPZ-IGF-IR shRNAmir-1, 2, or 3, or pGIPZ-NS (nonsilencing control) plasmid DNA for 72 h. Cell extracts were analyzed by immunoblotting with anti-IGF-IR and anti-β-actin, respectively. Mock, Transfection control without shRNAmir. WB, Western blot. B, Infection of MIN6 cells. The lentiviruses containing IGF-IR shRNAmir-1 or NS shRNAmir were produced in HEK293T cells and used to transduce MIN6 cells. Images were taken 6 d after infection. Upper panels, TurboGFP; lower panels, phase contrast. C, Stable knockdown of IGF-IR (IGF-IR KD). The lentivirally infected cells were selected with puromycin for 4 wk. Cell extracts were immunoblotted with anti-IGF-IR and anti-β-actin, respectively. D and E, IGF-IR knockdown desensitizes GH-induced STAT5 activation. Serum-starved MIN6, IGF-IR KD, and NS control cells were stimulated with vehicle (−), bGH (500 ng/ml), or IGF-I (20 ng/ml) for 15 min. Cell extracts were immunoblotted with anti-pSTAT5 and anti-β-actin, respectively (D). Pooled data of anti-pSTAT5, as in D, from three independent experiments were densitometrically analyzed and statistical results are plotted (E). Data are mean ± sem (n = 3). *, P < 0.05 (IGF-IR KD vs. either MIN6 or NS control).
Augmented cell proliferation by GH and IGF-I in isolated islets
Although cultured rodent pancreatic insulinoma cell lines retain many key functional features of normal islets and have been proven to be a valuable tool for studying islet β-cell signaling (32, 33), whether some cellular processes in these immortalized cell lines, especially cell proliferation, represent the actual scenario in pancreatic islets remains a concern. To address this, we isolated and purified islets from mice (detailed in Materials andMethods) (Fig. 9A). To examine whether GH and IGF-I cotreatment augmented islet cell proliferation as we have already observed in β-cell lines (Fig. 6), single islet cells were cultured for 4 d in serum-free medium containing vehicle (control), GH, IGF-I, or G/I (cotreatment), and the dividing cells were visualized by labeling with BrdU during the last 24-h culture (Fig. 9B). A total of approximately 1000 islet cells were counted under each condition. The relative proliferation rates were plotted in Fig. 9C. Our results indicated that G/I cotreatment significantly augmented the islet cell proliferation when compared with either GH or IGF-I treatment alone (P < 0.01) (Fig. 9C).
Fig. 9.

GH and IGF-I cotreatment augments islet cell proliferation. A, Purified mouse islets. Scale bar, 50 μm. B, Representative images from BrdU incorporation assays. Single-islet cells were treated with vehicle (control), bGH (500 ng/ml), or IGF-I (20 ng/ml) or cotreated with bGH plus IGF-I (G/I) in serum-free medium for 96 h, in which BrdU was added in the last 24 h of treatment (chase phase). Cells with incorporated BrdU (proliferating cells) were detected by immunostaining with an anti-BrdU monoclonal antibody (red). Total cell numbers were measured by DAPI staining (blue). Scale bar, 10 μm. C, Cell proliferation rates determined by BrdU incorporation assays. A total of approximately 1000 cells from six random imaging fields under each condition, as in B, were counted, and the ratios of BrdU-positive cells (red) to the total cells (blue) (representing cell proliferation rates) were plotted with the control set as 1. Data are mean ± sem (n = 6). **, P < 0.01. C, Control.
Discussion
Pancreatic islet β-cell mass is tightly controlled by a dynamic balance between cell growth and death. Disruption of such a balance by autoimmune-mediated β-cell destruction or by failure of the β-cell plasticity to compensate for metabolic demand leads to the development of type 1 or type 2 diabetes. Thus, better understanding how growth factors and hormones (e.g. GH and IGF-I) collaborate to signal to regulate β-cell mass and function will provide the basis for development of more effective diabetic therapies. In this study, we demonstrate that: 1) cultured islet β-cells are appealing systems for investigating GH and IGF-I signaling and their cross talk; 2) GH specifically promotes the formation of GHR-JAK2-IGF-IR protein complex in a number of β-cell lines; 3) when exposed to both GH and IGF-I simultaneously, β-cells signal better (i.e. augmented activation of STAT5 and Akt), proliferate more vitally, and are better protected from stress (e.g. serum withdrawal)-induced apoptosis, compared with their exposure to GH or IGF-I alone; 4) knockdown of endogenous IGF-IR by RNAi desensitizes GH signaling in β-cells; and 5) consistent with their actions in cultured β-cell lines, GH and IGF-I collaborate to augment cell proliferation in isolated islets.
GH and IGF-I are key somatogenic and metabolic hormones in humans and animals. Although their complicated physiological relationship has been studied for several decades, the molecular mechanisms underlying their collaborative roles, especially in pancreatic islet β-cells, remain poorly understood. Classically, IGF-I is a GH effector. However, not all of GH's effects are exerted through stimulation of IGF-I from liver and peripheral tissues. For example, GH can promote skeletal muscle cell fusion, an essential process of muscle growth, independent of IGF-I (51). In pancreatic islets, IGF-I expression appears to be specifically in α-cells (22–24). Thus, it is now believed that GH most likely mediates a direct effect on β-cell proliferation via the JAK2/STAT5 signaling cascade (26, 52). GH and IGF-I bind to their respective receptors, GHR and IGF-IR, to engage diverse signaling pathways. We have previously uncovered that, in mouse preadipocytes, acute GH stimulation specifically induces formation of a protein complex including GHR, JAK2, and IGF-IR. The complex assembly does not seem to require phosphorylation of the components but is rather governed by a proper GHR conformation upon GH engagement. These intriguing findings raise a possibility that IGF-IR may be a proximal cofactor in GH/GHR signaling. Indeed, genetic deletion of IGF-IR in mouse primary osteoblasts impairs some of the GH-directed signaling (30, 31). In our current study, we demonstrate that GH promotes the GHR-JAK2-IGF-IR complex formation in cultured islet β-cells, which can be detected by reciprocal coimmunoprecipitation. In addition, the G120K antagonizing effect on GH-mediated complex assembly observed in β-cells suggests that GHR conformational change upon GH engagement may be a prerequisite for such physical interaction.
Although we can not completely rule out a requirement for tyrosine phosphorylation of any partners of such a GHR-JAK2-IGF-IR protein complex, our previous and current data do not favor such a requirement based on several facts. First, the complex formation is strictly dependent on GH engagement to GHR but not IGF-I binding to IGF-IR (subsequent activation/phosphorylation of IGF-IR) in both preadipocytes (29) and islet β-cells (this study). Second, the kinase inhibitor staurosporine, which is known to uncouple GH/GHR engagement from GH-induced GHR/JAK2 tyrosine phosphorylation by inhibiting phosphorylation of both GHR and JAK2 (29, 42), does not prevent GH-induced physical association between GHR and IGF-IR (29). Furthermore, deletion of IGF-IR in primary osteoblasts reduces GH-mediated STAT5 signaling, which can be partially rescued by reexpression of a truncated IGF-IR lacking the intracellular domain of its β-chain that contains the kinase domain (30). In contrast, IGF-IR tyrosine kinase inhibitor has no effect on GH-induced STAT5 signaling (30). Finally, IGF-IR knockdown in β-cells diminishes GH-dependent STAT5 activation (this study; see below). Collectively these results suggest that an IGF-IR component(s) other than the kinase domain impacts GH action and GH-induced GHR-JAK2-IGF-IR complex formation. Thus, it is reasonable to speculate that the extracellular domain of IGF-IR may affect GHR signaling conformation and could be essential for GHR/JAK2 and IGF-IR physical interaction.
To probe the potential signaling impact of GHR-JAK2-IGF-IR complex formation in response to GH in β-cells, we assessed the acute signaling aspects of GH and IGF-I cotreatment vs. single-hormone treatments. We found that the GH-induced STAT5 activation was significantly augmented by simultaneous costimulation with IGF-I, although IGF-I itself did not activate STAT5. Furthermore, knockdown of IGF-IR in β-cells by RNAi desensitized GH-induced STAT5 activation. Thus, the results from the current and previous studies (29–31) strongly support the notion that IGF-IR could serve as a proximal cofactor in GH/GHR signaling, as exemplified by the GHR/JAK2/STAT5 pathway. A growing body of evidence has suggested a crucial role of the IRS/PI3K/Akt pathway in regulating β-cell mass and function (14, 15, 17, 53). In examining the Akt signaling, we observed synergistic Akt activation elicited by GH and IGF-I cotreatment compared with the summation of the individual hormone effects, further suggesting that there exists a cross talk between GH and IGF-I in β-cells.
We note that our findings of Akt signaling differ from those reported by Cousin et al. (26). In that study, using the physical association of the 85-kDa subunit of PI3K (p85 PI3K) with IRS-1 or IRS-2 (revealed by co-IP) as a readout, the investigators concluded that glucose and IGF-I, but not rat GH, activated the IRS/PI3K pathway in INS-1 cells, although the effect of GH and IGF-I cotreatment was not examined (26). In contrast, we found that IGF-I and GH each activated Akt (the downstream effector of PI3K), and more importantly they synergized in Akt signaling (revealed by direct immunoblotting), which is largely consistent with our antiapoptosis data (further discussed below). The discrepancy in these results could be due to different experimental conditions used. In the study of Cousin et al. (26), cells were quiescent by serum and glucose deprivation and then exposed to GH or IGF-I with the addition of glucose (0–15 mm) for 10 min. However, in our case, cells were only serum starved in normal culture media containing glucose (11.2 mm for INS-1 and 25 mm for both MIN6 and BTC6) and then stimulated with GH and IGF-I for 15 min. Whether, or to what extent, glucose exposure dictates GH and IGF-I responsiveness and impacts the GH-induced GHR-JAK2-IGF-IR complex formation is currently under further investigation in our laboratory.
It is known that β-cell mass is determined and maintained by a dynamic balance of cell proliferation and cell death. Insufficient understanding of the signals regulating the growth and survival of adult β-cells remains one of the main challenges in diabetes research. Previous studies have shown that GH and IGF-I each can independently promote β-cell mitogenesis and prevent apoptosis in vitro (16, 26, 54–57). However, very little is known about the collaborative roles (signaling cross talk) of GH and IGF-I in regulating β-cell mass. Existing data suggest that GH-mediated JAK2/STAT5 and IGF-I (and perhaps GH, as demonstrated in this study)-mediated IRS/PI3K/Akt signaling can directly contribute to β-cell proliferation and apoptosis (16, 26). We are intrigued by our exciting findings that GH and IGF-I cotreatment augments β-cell growth and antiapoptosis, which could be ultimate manifestations of synergistic activation of STAT5 and Akt observed in these cells. Although it is difficult to assign a specific pathway to β-cell growth and/or survival at this point, our findings in β-cells are consistent with the general view of STATs and Akt as potent mediators of mitogenic (58, 59) and antiapoptotic (13, 60) signals, respectively. Future studies will further elucidate the precise roles of GH-IGF-I cross talk in the regulation of β-cell mass and function.
The classical somatomedin hypothesis of GH action implies that GHR acts in series with IGF-IR (61). Based on our data in several cell types (this study and Refs. 29–31), we propose that there also exists a parallel signaling relationship between these receptors (Fig. 10). Under resting conditions, GHR (most likely predimerized) and IGF-IR are present on the cell plasma membrane (Fig. 10A). GH engagement causes GHR conformational changes, allowing achievement of an active signaling status (with enhanced GHR-JAK2 interaction). This, in turn, allows the association of GHR/JAK2 with IGF-IR (Fig. 10B). Further IGF-I binding to IGF-IR within such a complex facilitates, stabilizes, enhances the GHR signaling (augmenting STAT5 activation), and simultaneously activates IGF-IR signaling (yielding synergistic Akt activation), which in part contributes to increased β-cell proliferation and survival under the condition of GH and IGF-I cotreatment (Fig. 10C). We realize that our findings in β-cells regarding physical and functional interaction between the GH/GHR and IGF-I/IGF-IR signaling systems are largely limited to several rodent cultured insulinoma lines at this point. However, these β-cell models are particularly appealing in that they retain many key functional features of normal islets containing authentic β-cells and have been extremely valuable tools for studying the complex signaling network and molecular mechanisms underlying β-cell dysfunction and destruction (32, 33). As a proof of concept, in the current study, we demonstrate that GH and IGF-I collaborate to augment cell proliferation in isolated rodent islets as they act in cultured β-cell lines.
Fig. 10.

Model for physical and functional interaction of GH and IGF-I signaling elements in β-cells. A parallel signaling arrangement of GHR and IGF-IR is proposed, based on our experimental data in several cell systems (this study and Refs. 29–31). Under resting conditions, GHR (most likely predimerized) and IGF-IR are present on the cell plasma membrane (A). GH engagement causes GHR conformational changes, allowing achievement of an active signaling status (with enhanced GHR-JAK2 interaction). This, in turn, allows the association of GHR/JAK2 with IGF-IR (B). Further IGF-I binding to the IGF-IR within such a complex facilitates, stabilizes, and enhances the GHR signaling (augmenting STAT5 activation) and simultaneously activates IGF-IR (yielding synergistic Akt activation), which in part contributes to enhanced β-cell proliferation and enhanced survival under the condition of GH and IGF-I cotreatment (C). P, Phosphorylation.
GH and IGF-I signaling play critical roles in many physiological and pathophysiological conditions including diabetes. In concert with recent observations that GH increases β-cell proliferation in transplanted human and fetal rat islets (62), our novel findings reported herein may have important biological and therapeutic implications, such as preservation or augmentation of β-cell mass and function. Our current work also lays a solid foundation for future studies of GH-IGF-I signaling cross talk in primary islets and animal models, which will further advance our knowledge in the field.
Materials and Methods
Materials
Recombinant bGH, recombinant rat GH, and recombinant human IGF-I were obtained through the National Hormone and Peptide Program (Dr. A. F. Parlow, Harbor-UCLA Medical Center, Torrance, CA). Recombinant human GH-G120K was kindly provided by Sensus Corp. (Austin, TX). The DeadEnd fluorometric TUNEL system was purchased from Promega (Madison, WI). BrdU, DAPI, Hoechst 33258, PI, and other routine reagents were purchased from Sigma (St. Louis, MO), unless otherwise noted.
Antibodies
Anti-IGF-IRβ (Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal anti-pTyr clone 4G10 (Millipore/Upstate Biotechnology, Lake Placid, NY), anti-pSTAT5 (Invitrogen, Carlsbad, CA), anti-pAkt (Cell Signaling, Beverly, MA), anti-Akt (Cell Signaling), anti-β-actin (Sigma), and the monoclonal anti-BrdU clone BU-33 (Sigma) antibodies were commercially available. The rabbit polyclonal antisera, anti-JAK2, and anti-GHR were described previously (29). Tetramethyl rhodamine isothiocyanate-conjugated goat antimouse IgG was from Jackson ImmunoResearch Laboratories (West Grove, PA). The secondary antibodies used in immunoblotting were purchased from Pierce (Rockford, IL).
Cell culture, cell starvation, growth factor and antagonist treatment, and protein extraction
Mouse MIN6 and BTC6 cells were cultured in DMEM with 4.5 g/liter glucose, supplemented with 10% fetal bovine serum (FBS) (Mediatech, Manassas, VA) and 50 μm β-mercaptoethanol. Rat INS-1 cells were grown in RPMI 1640 supplemented with 10% FBS, 10 mm HEPES, and 50 μm β-mercaptoethanol. Cell starvation, growth factor and antagonist treatment, and protein extraction were performed as previously described (29, 44, 63). Briefly, cells were starved in serum-free medium containing 0.5% BSA for 16 h and then stimulated with vehicle (control), bGH (500 ng/ml), IGF-I (20 ng/ml), or bGH plus IGF-I (cotreatment) for 15 min at 37 C. When applicable, the GH antagonist G120K (2500 ng/ml) was added simultaneously with bGH (500 ng/ml). The cells were harvested and lysed in the lysis buffer [1% Triton X-100, 10% glycerol, 150 mm NaCl, 50 mm Tris-HCl (pH 8.0), 2 mm EDTA, 100 mm NaF, 1 mm sodium orthovanadate, 1 mm phenylmethylsufonyl fluoride, 10 mm benzamidine, 5 μg/ml aprotinin, and 5 μg/ml leupeptin] (29). Total protein concentrations of detergent extracts were determined using bicinchoninic acid reagents (Pierce). The cell lysates were subjected to immunoprecipitation or were directly electrophoresed and immunoblotted, as indicated below.
Mouse islet isolation
Pancreatic islets were isolated from 4-wk-old C57BL/6 mice (Charles River Laboratories, Wilmington, MA) by collagenase V (Sigma) digestion and Ficoll (Sigma) density gradient purification, as described elsewhere (64) (protocol is available via http://www.jove.com/index/details.stp?ID=255). The whole islets were further purified by hand picking under a stereomicroscope and cultured in RPMI 1640 medium supplemented with 10% FBS, 10 mm HEPES, 1 mm sodium pyruvate, 1× nonessential amino acids, and 50 μm β-mercaptoethanol for 1 d before assays. All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee.
Immunoprecipitation, immunoblotting, and densitometry
For IP, cell lysates (0.75–2 mg) were mixed with anti-IGF-IRβ (5–10 μl), anti-GHR (3 μl), or anti-JAK2 (3 μl) antibody at 4 C overnight with continuous agitation. Protein A-Sepharose beads (Amershan Pharmacia Biotechnology, Arlington Heights, IL) were added and incubated at 4 C for an additional 2 h. The beads were washed three times with the lysis buffer adjusted to 0.5% Triton X-100. Laemmli sample buffer eluates were analyzed by immunoblotting using standard protocols (29, 44). Briefly, IP eluates or cell lysates (50 μg) were resolved by SDS-PAGE, and the proteins were transferred onto Hybond enhanced chemiluminescence nitrocellulose membranes (Amershan Pharmacia Biotechnology). The membranes were blocked with a buffer of 20 mm Tris-HCl (pH 7.6), 150 mm NaCl, and 0.1% Tween 20 containing 1% BSA and incubated with antibodies as specified in each experiment. After three washes with the buffer of 20 mm Tris-HCl (pH 7.6), 150 mm NaCl, and 0.1% Tween 20, the membranes were incubated with appropriate secondary antibodies. The bound antibodies were detected with SuperSignal chemiluminescent substrate (Pierce). Digital images of immunoblots were acquired with a Kodak Multimodal Imager 4000MM (Kodak, Rochester, NY) and quantified densitometrically using Kodak molecular imaging software (version 4.0) (63, 65).
BrdU incorporation assays
BrdU incorporation assay to measure DNA synthesis was performed as described elsewhere (47). For β-cell lines, MIN6 or INS-1 cells were preplated on coverslips precoated with gelatin (2%) and allowed to grow in serum-free medium containing vehicle (control), bGH (500 ng/ml), IGF-I (20 ng/ml), or bGH plus IGF-I (cotreatment) for 24 h. The last 1.5 h (for MIN6) or 4 h (for INS-1) treatments were performed in the presence of BrdU (20 μm). The cells with incorporated BrdU (proliferating cells) were detected by immunostaining with anti-BrdU antibody and total cell numbers were measured by nuclear counterstaining with DAPI (0.5 μg/ml).
For isolated islets, the hand-picked islets were washed once with PBS and then dispersed into single cells by incubation with 0.025% trypsin for 7–10 min at 37 C. The trypsin digestion was terminated by the addition of RPMI 1640 medium containing 10% FBS. Single-islet cells were obtained by pipetting up and down several times and collected by centrifugation. The cells were resuspended in RPMI 1640 medium supplemented with 1% BSA, 10 mm HEPES, 1 mm sodium pyruvate, 1× nonessential amino acids and 50 μm β-mercaptoethanol and seeded in the Lab-Tek 8-Chamber Slide (Nagle Nunc International, Rochester, NY) precoated with fibronectin (20 μg/ml) at a density of 9 × 104 cells/well. The cells were allowed to grow in the serum-free medium containing vehicle (control), bGH (500 ng/ml), IGF-I (20 ng/ml), or bGH plus IGF-I (cotreatment) for 4 d, and the dividing cells were visualized by labeling with BrdU (10 μm) during the last 24-h culture.
After immunostaining with anti-BrdU antibody and DAPI counterstaining, the coverslips or chamber slides were mounted and fluorescent images were captured with a Zeiss Imager Z1 upright microscope (Carl Zeiss, Thornwood, NY) (63). Cells were counted from six to 10 random fields under each condition, and the cell proliferation rate was calculated as the ratio of BrdU-positive cells to the total cells.
Hoechst and PI staining
Apoptosis and necrosis (including late apoptosis) were visualized and quantified using Hoechst 33258 and PI staining, respectively, as we demonstrated previously (50). Hoechst is a fluorescent dye, which penetrates into intact cells and is taken by the nuclei. The apoptotic nuclei exhibit as bright blue due to their nuclear condensation, whereas viable cells are dark blue. PI is a fluorescent dye that is taken only by late apoptotic and necrotic cells. Cells were grown in serum-free medium containing vehicle (control), bGH (500 ng/ml), IGF-I (20 ng/ml), or bGH plus IGF-I (cotreatment) for 24 h (for measuring apoptosis) or 48 h (for measuring necrosis). During the last 15 min of treatment, Hoechst 33258 (10 μg/ml) and PI (15 μg/ml) were added. Fluorescent images were acquired with a Zeiss Axiovert 200M inverted microscope. Quantification of bright blue (apoptotic), PI-positive/red (late apoptotic and necrotic), and total cells from five to eight random fields were counted. The cell death rate was expressed as the percentage of apoptotic or necrotic cells from the total cells.
TUNEL staining
TUNEL is a common method for detecting nuclear DNA fragmentation that results from apoptosis. The assay was performed using the DeadEnd fluorometric TUNEL system according to the manufacturer's recommendations (Promega). In brief, cells were seeded onto glass coverslips precoated with gelatin (2%) in growth medium containing 10% FBS until they were fully attached. The cells were allowed to grow in serum-free medium containing vehicle (control), bGH (500 ng/ml), IGF-I (20 ng/ml), or bGH plus IGF-I (cotreatment) for 24 h. At the end of the incubation, the cells were washed with ice-cold PBS, fixed with 4% paraformaldehyde at 4 C for 30 min, and permeabilized in 0.2% Triton X-100 in PBS for 5 min at room temperature. The permeabilization solution was removed and the TUNEL reaction mixture (50 μl) containing nucleotide mix and recombinant terminal deoxynucleotidyl transferase was then added to the cells. After incubation at 37 C for 1 h, the cells were washed with PBS and counterstained with DAPI (0.5 μg/ml) for 10 min. The incidence of apoptosis was assessed by fluorescence microscopy. The cells with TUNEL-positive nuclei were considered apoptotic. DAPI staining was used to determine the total number of cells in a field. A minimum of five fields per condition was used to calculate the percentage of apoptosis.
Knockdown of IGF-IR by lentiviral based delivery of shRNAmir
A set of shRNAmir (microRNA adapted shRNA) targeting the mouse IGF-IR gene and nonspecific control shRNAmir (all precloned in the lentiviral vector pGIPZ containing a Turbo GFP marker) were purchased from Open Biosystems (Huntsville, AL). These plasmids were transiently transfected into MIN6 cells, respectively, using Arrest-In reagents (Open Biosystems) according to the manufacturer's suggestions. Knockdown efficacy was evaluated by immunoblotting with anti-IGF-IR antibody at 72 h after transfection. The shRNAmir-1 exhibited the highest knockdown efficiency and was thus used to produce lentivirus in HEK293T cells using the Trans-Lentivral pGIPZ packaging system (Open Biosystems). Viral supernatants were collected at 48 h after transfection and used to infect MIN6 cells. The cells expressing shRNAmir can be visualized by TurboGFP. Stable knockdown of IGF-IR was achieved by selection of the cells with puromycin (2 μg/ml) for at least 14 d and further confirmed by immunoblotting.
Statistical analysis
All statistical data were from multiple experiments or measurements and presented as mean ± sem. The significance of differences was estimated using unpaired t test and P < 0.05 was considered significant (63, 65).
Supplementary Material
Acknowledgments
We thank Dr. Gregory Szot (University of California, San Francisco, Diabetes Research Center) for the expert advice on islet isolation. We also thank Dr. Anath Shalev (Comprehensive Diabetes Center University of Alabama at Birmingham) for helpful discussion and suggestions.
This work was supported by American Heart Association Beginning Grant-in-Aid Award 10BGIA4050019, Science Foundation Arizona Competitive Advantage Award CAA0259-08, St. Joseph's Foundation Startup Fund (to Y.H.), and National Institutes of Health Grant R01 DK46395 (to S.J.F.).
Parts of the work were presented orally at the 88th Annual Meeting of The Endocrine Soceity, Boston, Massachusetts, and the 90th Annual Meeting of The Endocrine Society, San Francisco, California.
Disclosure Summary: F.M., Z.W., C.S., Y.G., J.L., S.J.F., J.B., and Y.H. have nothing to disclose.
Footnotes
- bGH
- Bovine GH
- BrdU
- 5-bromo-2′-deoxyuridine
- DAPI
- 4′,6-diamidino-2-phenylindole dihydrochloride
- FBS
- fetal bovine serum
- GHR
- GH receptor
- G/I
- cotreatment of GH and IGF-I
- IGF-IR
- IGF-I receptor
- IP
- immunoprecipitation
- IRS
- insulin receptor substrate
- JAK
- Janus kinase
- p
- phosphorylated
- PI
- propidium iodide
- PI3K
- phosphoinositide 3-kinase
- pTyr
- phosphotyrosine
- RNAi
- RNA interference
- shRNA
- short hairpin RNA
- STAT5
- signal transducer and activator of transcription 5
- TUNEL
- terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling.
References
- 1. de Koning EJ, Bonner-Weir S, Rabelink TJ. 2008. Preservation of β-cell function by targeting β-cell mass. Trends Pharmacol Sci 29:218–227 [DOI] [PubMed] [Google Scholar]
- 2. DeFronzo RA. 2004. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am 88:787–835 [DOI] [PubMed] [Google Scholar]
- 3. Prentki M, Nolan CJ. 2006. Islet β cell failure in type 2 diabetes. J Clin Invest 116:1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rhodes CJ. 2005. Type 2 diabetes—a matter of cell life and death? Science 307:380–384 [DOI] [PubMed] [Google Scholar]
- 5. Karaca M, Magnan C, Kargar C. 2009. Functional pancreatic β-cell mass: involvement in type 2 diabetes and therapeutic intervention. Diabetes Metab 35:77–84 [DOI] [PubMed] [Google Scholar]
- 6. Rhodes CJ. 2000. IGF-I and GH post-receptor signaling mechanisms for pancreatic β-cell replication. J Mol Endocrinol 24:303–311 [DOI] [PubMed] [Google Scholar]
- 7. Nielsen JH, Serup P. 1998. Molecular basis for islet development, growth, and regeneration. Curr Opin Endocrinol Diabetes 5:97–107 [Google Scholar]
- 8. Nielsen JH, Galsgaard ED, Moldrup A, Friedrichsen BN, Billestrup N, Hansen JA, Lee YC, Carlsson C. 2001. Regulation of cell mass by hormones and growth factors. Diabetes 50:S25–S29 [DOI] [PubMed] [Google Scholar]
- 9. Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, Garcia-Ocaña A. 2006. Growth factors and β cell replication. Int J Biochem Cell Biol 38:931–950 [DOI] [PubMed] [Google Scholar]
- 10. Favelyukis S, Till JH, Hubbard SR, Miller WT. 2001. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 8:1058–1063 [DOI] [PubMed] [Google Scholar]
- 11. Laviola L, Natalicchio A, Giorgino F. 2007. The IGF-I signaling pathway. Curr Pharm Des 13:663–669 [DOI] [PubMed] [Google Scholar]
- 12. Krishna M, Narang H. 2008. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 65:3525–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burks DJ, White MF. 2001. β-Cell mass and function in type 2 diabetes: IRS proteins and β-cell function. Diabetes 50:S140–S145 [DOI] [PubMed] [Google Scholar]
- 15. Elghazi L, Bernal-Mizrachi E. 2009. Akt and PTEN: β-cell mass and pancreas plasticity. Trends Endocrinol Metab 20:243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hügl SR, White MF, Rhodes CJ. 1998. Insulin-like growth factor I (IGF-I)-stimulated pancreatic β-cell growth is glucose-dependent. J Biol Chem 273:17771–17779 [DOI] [PubMed] [Google Scholar]
- 17. Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, Birnbaum MJ. 2001. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat Med 7:1133–1137 [DOI] [PubMed] [Google Scholar]
- 18. Isaksson OG, Edén S, Jansson JO. 1985. Mode of action of pituitary growth hormone on target cells. Annu Rev Physiol 47:483–499 [DOI] [PubMed] [Google Scholar]
- 19. Frank SJ, Messina JL. 2002. Growth hormone receptor. In: Oppenheim JJ, Feldman M. eds. Cytokine reference on-line. London, UK: Academic Press, Harcourt; 1–21 [Google Scholar]
- 20. Carter-Su C, Schwartz J, Smit LS. 1996. Molecular mechanism of growth hormone action. Annu Rev Physiol 58:187–207 [DOI] [PubMed] [Google Scholar]
- 21. Smith F, Rosen K, Villa-Komarof L, Weir GC, Bonner-Weir S. 1990. Enhanced IGF-I expression in regenerating rat pancreas is localized to capillaries and proliferating duct cells. Diabetes 39(Suppl 1):66A [Google Scholar]
- 22. Maake C, Reinecke M. 1993. Immunohistochemical localization of insulin-like growth factor 1 and 2 in the endocrine pancreas of rat, dog, and man, and their coexistence with classical islet hormones. Cell Tissue Res 273:249–259 [DOI] [PubMed] [Google Scholar]
- 23. Jevdjovic T, Maake C, Eppler E, Zoidis E, Reinecke M, Zapf J. 2004. Effects of insulin–like growth factor-I treatment on the endocrine pancreas of hypophysectomized rats: comparison with growth hormone replacement. Eur J Endocrinol 151:223–231 [DOI] [PubMed] [Google Scholar]
- 24. Jevdjovic T, Maake C, Zwimpfer C, Krey G, Eppler E, Zapf J, Reinecke M. 2005. The effect of hypophysectomy on pancreatic islet hormone and insulin-like growth factor I content and mRNA expression in rat. Histochem Cell Biol 123:179–188 [DOI] [PubMed] [Google Scholar]
- 25. Billestrup N, Nielsen JH. 1991. The stimulatory effect of growth hormone, prolactin, and placental lactogen on β-cell proliferation is not mediated by insulin-like growth factor-I. Endocrinology 129:883–888 [DOI] [PubMed] [Google Scholar]
- 26. Cousin SP, Hügl SR, Myers MG, Jr, White MF, Reifel-Miller A, Rhodes CJ. 1999. Stimulation of pancreatic β-cell proliferation by growth hormone is glucose-dependent: signal transduction via Janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) with no crosstalk to insulin receptor substrate-mediate mitogenic signalling. Biochem J 344:649–658 [PMC free article] [PubMed] [Google Scholar]
- 27. Ashcom G, Gurland G, Schwartz J. 1992. Growth hormone synergizes with serum growth factors in inducing c-fos transcription in 3T3–F442A cells. Endocrinology 131:1915–1921 [DOI] [PubMed] [Google Scholar]
- 28. Edmondson SR, Russo VC, McFarlane AC, Wraight CJ, Werther GA. 1999. Interactions between growth hormone, insulin-like growth factor I, and basic fibroblast growth factor in melanocyte growth. J Clin Endocrinol Metab 84:1638–1644 [DOI] [PubMed] [Google Scholar]
- 29. Huang Y, Kim SO, Yang N, Jiang J, Frank SJ. 2004. Physical and functional interaction of growth hormone and insulin-like growth factor-1 signaling elements. Mol Endocrinol 18:1471–1485 [DOI] [PubMed] [Google Scholar]
- 30. Gan Y, Zhang Y, Digirolamo DJ, Jiang J, Wang X, Cao X, Zinn KR, Carbone DP, Clemens TL, Frank SJ. 2010. Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling. Mol Endocrinol 24:644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, Frank SJ, Clemens TL. 2007. Mode of growth hormone action in osteoblasts. J Biol Chem 282:31666–31674 [DOI] [PubMed] [Google Scholar]
- 32. McClenaghan NH. 2007. Physiological regulation of the pancreatic β-cell: functional insights for understanding and therapy of diabetes. Exp Physiol 92:481–496 [DOI] [PubMed] [Google Scholar]
- 33. Hohmeier HE, Newgard CB. 2004. Cell lines derived from pancreatic islets. Mol Cell Endocrinol 228:121–128 [DOI] [PubMed] [Google Scholar]
- 34. Goffin V, Shiverick KT, Kelly PA, Martial JA. 1996. Sequence-function relationships within the expanding family of prolactin, growth hormone, placental lactogen, and related proteins in mammals. Endocr Rev 17:385–410 [DOI] [PubMed] [Google Scholar]
- 35. He K, Wang X, Jiang J, Guan R, Bernstein KE, Sayeski PP, Frank SJ. 2003. Janus kinase 2 determinants for growth hormone receptor association, surface assembly, and signaling. Mol Endocrinol 17:2211–2227 [DOI] [PubMed] [Google Scholar]
- 36. de Vos AM, Ultsch M, Kossiakoff AA. 1992. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science 255:306–312 [DOI] [PubMed] [Google Scholar]
- 37. Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, Waters MJ. 2005. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol 12:814–821 [DOI] [PubMed] [Google Scholar]
- 38. van den Eijnden MJ, Lahaye LL, Strous GJ. 2006. Disulfide bonds determine growth hormone receptor folding, dimerisation and ligand binding. J Cell Sci 119:3078–3086 [DOI] [PubMed] [Google Scholar]
- 39. Frank SJ, Fuchs SY. 2008. Modulation of growth hormone receptor abundance and function: roles for the ubiquitin-proteasome system. Biochim Biophys Acta 1782:785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA. 1992. Rational design of potent antagonists to the human growth hormone receptor. Science 256:1677–1680 [DOI] [PubMed] [Google Scholar]
- 41. Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ. 2002. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev 23:623–646 [DOI] [PubMed] [Google Scholar]
- 42. Zhang Y, Jiang J, Kopchick JJ, Frank SJ. 1999. Disulfide linkage of growth hormone (GH) receptors (GHR) reflects GH-induced GHR dimerization. Association of JAK2 with the GHR is enhanced by receptor dimerization. J Biol Chem 274:33072–33084 [DOI] [PubMed] [Google Scholar]
- 43. Thirone AC, Carvalho CR, Saad MJ. 2002. G120K-PEG, a human GH antagonist, decreases GH signal transduction in the liver of mice. Mol Cell Endocrinol 192:65–74 [DOI] [PubMed] [Google Scholar]
- 44. Huang Y, Kim SO, Jiang J, Frank SJ. 2003. Growth hormone-induced phosphorylation of epidermal growth factor (EGF) receptor in 3T3–F442A cells. Modulation of EGF-induced trafficking and signaling. J Biol Chem 278:18902–18913 [DOI] [PubMed] [Google Scholar]
- 45. Huang Y, Li X, Jiang J, Frank SJ. 2006. Prolactin modulates phosphorylation, signaling and trafficking of epidermal growth factor receptor in human T47D breast cancer cells. Oncogene 25:7565–7576 [DOI] [PubMed] [Google Scholar]
- 46. Li X, Huang Y, Jiang J, Frank SJ. 2011. Synergy in ERK activation by cytokine receptors and tyrosine kinase growth factor receptors. Cell Signal 23:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luther MJ, Davies E, Muller D, Harrison M, Bone AJ, Persaud SJ, Jones PM. 2005. Cell-to-cell contact influences proliferative marker expression and apoptosis in MIN6 cells grown in islet-like structures. Am J Physiol Endocrinol Metab 288:E502–E509 [DOI] [PubMed] [Google Scholar]
- 48. Woo D. 1995. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med 333:18–25 [DOI] [PubMed] [Google Scholar]
- 49. He J, Bazan HE. 2006. Synergistic effect of platelet-activating factor and tumor necrosis factor-α on corneal myofibroblast apoptosis. Invest Ophthalmol Vis Sci 47:883–891 [DOI] [PubMed] [Google Scholar]
- 50. Maurice JM, Gan Y, Ma FX, Chang YC, Hibner M, Huang Y. 2010. Bupivacaine causes cytotoxicity in mouse C2C12 myoblast cells: involvement of ERK and Akt signaling pathways. Acta Pharmacol Sin 31:493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sotiropoulos A, Ohanna M, Kedzia C, Menon RK, Kopchick JJ, Kelly PA, Pende M. 2006. Growth hormone promotes skeletal muscle cell fusion independent of insulin-like growth factor 1 up-regulation. Proc Natl Acad Sci USA 103:7315–7320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hansen LH, Wang X, Kopchick JJ, Bouchelouche P, Nielsen JH, Galsgaard ED, Billestrup N. 1996. Identification of tyrosine residues in the intracellular domain of the growth hormone receptor required for transcriptional signaling and Stat5 activation. J Biol Chem 271:12669–12673 [DOI] [PubMed] [Google Scholar]
- 53. Dickson LM, Rhodes CJ. 2004. Pancreatic β-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab 287:E192–E198 [DOI] [PubMed] [Google Scholar]
- 54. Sekine N, Ullrich S, Regazzi R, Pralong WF, Wollheim CB. 1996. Postreceptor signaling of growth hormone and prolactin and their effects in the differentiated insulin-secreting cell line, INS-1. Endocrinology 137:1841–1850 [DOI] [PubMed] [Google Scholar]
- 55. Nielsen JH, Linde S, Welinder BS, Billestrup N, Madsen OD. 1989. Growth hormone is a growth factor for the differentiated pancreatic β-cell. Mol Endocrinol 3:165–173 [DOI] [PubMed] [Google Scholar]
- 56. Harrison M, Dunger AM, Berg S, Mabley J, John N, Green MH, Green IC. 1998. Growth factor protection against cytokine-induced apoptosis in neonatal rat islets of Langerhans: role of Fas. FEBS Lett 435:207–210 [DOI] [PubMed] [Google Scholar]
- 57. Ling Z, Hannaert JC, Pipeleers D. 1994. Effect of nutrients, hormones and serum on survival of rat islet β cells in culture. Diabetologia 37:15–21 [DOI] [PubMed] [Google Scholar]
- 58. Ihle JN. 2001. The Stat family in cytokine signaling. Curr Opin Cell Biol 13:211–217 [DOI] [PubMed] [Google Scholar]
- 59. Levy DE, Darnell JE., Jr 2002. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662 [DOI] [PubMed] [Google Scholar]
- 60. Sale EM, Sale GJ. 2008. Protein kinase B: signalling roles and therapeutic targeting. Cell Mol Life Sci 65:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. 2001. The somatomedin hypothesis: 2001. Endocr Rev 22:53–74 [DOI] [PubMed] [Google Scholar]
- 62. Höglund E, Mattsson G, Tyrberg B, Andersson A, Carlsson C. 2009. Growth hormone increases β-cell proliferation in transplanted human and fetal rat islets. JOP 10:242–248 [PubMed] [Google Scholar]
- 63. Gan Y, Shi C, Inge L, Hibner M, Balducci J, Huang Y. 2010. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene 29:4947–4958 [DOI] [PubMed] [Google Scholar]
- 64. Szot GL, Koudria P, Bluestone JA. 2007. Murine pancreatic islet isolation. J Vis Exp 7:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma F, Zhang D, Yang H, Sun H, Wu W, Gan Y, Balducci J, Wei YQ, Zhao X, Huang Y. 2009. Endothelial cell-specific molecule 2 (ECSM2) modulates actin remodeling and epidermal growth factor receptor signaling. Genes Cells 14:281–293 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.