

Abstract
Sodium–glucose co-transporter-2 (SGLT2) inhibitors are an emerging class of agents for use in the treatment of type 2 diabetes mellitus (T2DM). Inhibition of SGLT2 leads to improved glycemic control through increased urinary glucose excretion (UGE). In this study, a biologically based pharmacokinetic/pharmacodynamic (PK/PD) model of SGLT2 inhibitor-mediated UGE was developed. The derived model was used to characterize the acute PK/PD relationship of the SGLT2 inhibitor, dapagliflozin, in rats. The quantitative translational pharmacology of dapagliflozin was examined through both prospective simulation and direct modeling of mean literature data obtained for dapagliflozin in healthy subjects. Prospective simulations provided time courses of UGE that were of consistent shape to clinical observations, but were modestly biased toward under prediction. Direct modeling provided an improved characterization of the data and precise parameter estimates which were reasonably consistent with those predicted from preclinical data. Overall, these results indicate that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be reasonably well predicted from preclinical data through rational accounting of species differences in pharmacokinetics, physiology, and SGLT2 pharmacology. Because these data can be generated at the earliest stages of drug discovery, the proposed model is useful in the design and development of novel SGLT2 inhibitors. In addition, this model is expected to serve as a useful foundation for future efforts to understand and predict the effects of SGLT2 inhibition under chronic administration and in other patient populations.
Key words: diabetes, glucosuria, pharmacodynamics, pharmacokinetics, SGLT2, translational pharmacology, UGE
INTRODUCTION
It has been estimated that more than 25 million Americans (8.3% of the population) have type 2 diabetes (T2DM) (1). In 2007 alone, the costs associated with diabetes amounted to greater than 280,000 lives and 170 billion dollars (2). As such, T2DM remains one of the largest therapeutic areas for research and development in the pharmaceutical industry. This therapeutic area is also one of the most competitive, with the majority (i.e. 57%) of medicinal chemistry patent applications between 2008 and 2010 centered upon just eight molecular targets (3). Under these circumstances, clinical attrition of a lead molecule due to suboptimal pharmacokinetics, efficacy, or safety can be disastrous, as there are often multiple competitor programs in play against the same target. As such, early decisions regarding chemical optimization and candidate selection are critically important to success. To this end, considerable efforts have been made to develop and apply biologically based mathematical models in support of quantitative human pharmacokinetic predictions from in vitro and in vivo data generated preclinically (4–17). It is now commonplace for such predictive pharmacokinetic models to be used in the design and selection of drug candidates across the pharmaceutical industry. The decline in pharmacokinetic attrition observed in the pharmaceutical industry can be attributed, in part, to these efforts (18). In contrast, to date, far less attention has been paid to the development and application of biologically based mathematical models to support quantitative pharmacology predictions from in vitro and in vivo data generated preclinically. Such models are expected to be useful in reducing the relatively high rate of late-stage safety and efficacy-based attrition (18). This is expected to be particularly enabling in highly competitive areas, like diabetes, where the margin for error in the design and advancement of the best clinical candidate is relatively low.
Within the T2DM therapeutic area, the sodium–glucose co-transporter 2 (SGLT2) target represents one of the most competitive areas of R&D (3–19). Glucose entering the glomerulus through passive filtration is reabsorbed by the combined actions of SGLT2 and SGLT1 in the S1 and S3 segments of the proximal tubule, respectively. Genetic functional data suggest that the urinary glucose excretion (UGE) associated with impaired SGLT2 function represents a safe and effective approach for managing the hyperglycemia associated with T2DM (20–22). Other attractive features of this mechanism include its non-insulin-dependent nature and the potential for weight loss due to the caloric loss associated with UGE. As such, it is one of the top eight targets being pursued in the pharmaceutical industry and there are several novel SGLT2 inhibitors currently in clinical development (3–19). While several compounds have attrited in phase I and II, clinical results with the most advanced SGLT2 inhibitor, dapagliflozin, indicate that clinically significant HbA1c reductions are possible with this target (23,24). To date, dapagliflozin is also the only SGLT2 inhibitor for which extensive preclinical and clinical data have been disclosed. These data provide the opportunity to examine the quantitative translational pharmacology of SGLT2 inhibition. As such, the objective of this work is to quantitatively examine the translational pharmacology of SGLT2 inhibition between the preclinical and clinical setting using a simple, physiologically based pharmacokinetic/pharmacodynamic (PK/PD) model. The results of this work indicate that the acute clinical pharmacology of SGLT2-mediated UGE can be reasonably well predicted from preclinical data using the proposed PKPD model. This work highlights several important principles of general relevance to the quantitative scaling of preclinical information in support of the design and advancement of clinical candidates with the highest likelihood of clinical success.
MATERIALS AND METHODS
Materials
(2S,3R,4R,5S,6R)-2-[4-Chloro-3-(4-ehoxybenzyl)phenyl]-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol, referred to as dapagliflozin hereafter, was synthesized as described previously (25).
Preclinical Studies
All animal care and in vivo procedures were approved and conducted in accordance with guidelines of the Pfizer Animal Care and Use Committee. Male Sprague–Dawley rats (~300 g) were singly housed in metabolic cages for urine collection over 24 h (n = 4–5 per group). A separate set of male Sprague–Dawley rats with indwelling jugular vein catheters were used for determination of pharmacokinetics (n = 3 per group). Dapagliflozin was administered via oral gavage (1 ml/300 g body weight) at single doses of 0.1, 1, 3, and 10 mg/kg (n = 4–5 per group). A subset of rats (n = 5) was given control vehicle (0.5% methylcellulose and 0.1% Tween 80). Blood (0.3 mL) was collected from jugular vein catheters at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h, centrifuged and the resultant plasma kept frozen at −20°C until analysis for dapagliflozin concentration via LC-MS/MS. Urine was collected following treatment administration over a duration of 24 h. Urinary glucose concentration was measured by UV absorbance spectrophotometry at 340 nm using a Roche Hitachi 917 spectrophotometer (Diamond Diagnostics, Holliston, MA, USA). The total amount of glucose excreted in the urine (UGE) was calculated as the product of urine concentration and urine volume.
Bioanalysis
Aliquots (50 μL) of plasma samples and standards were subjected to protein precipitation with acetonitrile containing an internal standard. Samples were vortexed and centrifuged to obtain supernatant which was analyzed by LC-MS/MS. Analyst (version 1.4.1) was used to measure peak areas, and peak area ratios of analyte to internal standard were calculated. A calibration curve was constructed from the peak area ratios of the standards to the internal standard by applying a weighted linear (1/x2) regression. The linear range of the standard curve was 5.00 to 5,000 ng/mL. LC-MS/MS conditions were as follows: Mass Spectrometer + Source Type was Sciex API 4,000 – Turbo Spray; HPLC pump was Shimadzu; Autosampler was CTC PAL Autosampler; injection volume was 10 μL; a gradient was used with mobile phase A, 10 mM ammonium acetate and 1% isopropyl alcohol in water; B, acetonitrile; flow rate 0.500 mL/min (column 2.0 × 30 mm, 5 μm LUNA C18 column (phenomenex)). An initial condition of 90% A, 10% B was held for 0.25 min, ramped to 10% A, 90% B over 1 min, returned to the initial condition in a single step and held for 0.25 min prior to the next injection. Detection mode was negative.
Literature Reports on Human Studies
Mean human plasma dapagliflozin concentrations following single doses of 2.5, 10, 50, 100, and 250 mg and associated urinary glucose excretion over 120 h in healthy subjects were obtained from a literature report (26) via digitization using Grab It! XP 10 (DataTrend Software Inc.). Digitized data were electronically overlaid on figures from the original report to ensure concordance.
Rat PK/PD Model
The concentration-time profiles of dapagliflozin in rats were described using a one-compartment linear disposition model with first-order absorption:
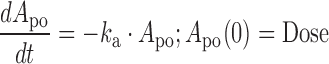 |
1 |
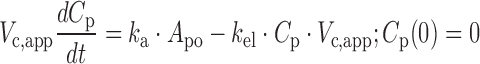 |
2 |
where ka is the first-order absorption rate constant after oral dosing and kel is the first-order elimination rate constant. Equation 1 describes the amount of drug (Apo) at the site of absorption after oral dosing. The administered dose represents the initial condition at the site of absorption. Plasma drug concentration and apparent volume of distribution in the central compartment are denoted by Cp (initial condition = 0) and Vc,app, respectively. The dynamic cumulative UGE response to SGLT2 inhibition was modeled as a function of the inhibitor concentration, C, as previously described (27):
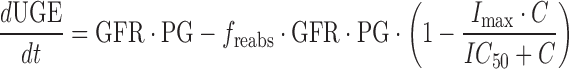 |
3 |
where GFR represents glomerular filtration rate and PG represents the plasma glucose concentration. The product of these parameters represents the rate of glucose filtration by the kidney. The freabs parameter represents the fraction of filtered glucose which is reabsorbed in the untreated condition. The Imax and IC50 parameters represent the maximum fractional inhibition of glucose reabsorption by the inhibitor and the concentration of inhibitor which achieves half-maximal inhibition of glucose reabsorption, respectively. For purposes of modeling, the physiologically relevant parameters of glucose filtration and reabsorption were fixed to independent estimates obtained from the literature. GFR was set to 0.55 ml/h/g consistent with that previously reported in rats (28). PG was fixed to a mean experimental estimate obtained in an independent, in-house study of 30 male Sprague–Dawley rats weighing 290 ± 50 g (1.41 + 0.54 mg/ml determined spectrophotometrically). This value is consistent with plasma glucose values reported in normal rats (29–31). The freabs parameter was fixed to 0.996 as previously estimated and is commensurate with near complete glucose reabsorption in normal rats (27). As such, only Imax and IC50 were estimated in the modeling process.
Human PK/PD Model
The reported mean concentration-time profiles of dapagliflozin in healthy subjects were described using a two-compartment linear disposition model with first-order absorption:
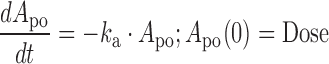 |
4 |
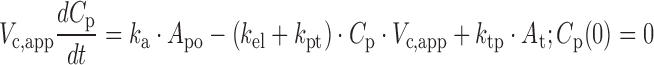 |
5 |
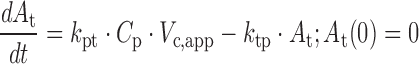 |
6 |
where ka is the first-order absorption rate constant after oral dosing, kpt and ktp are the first-order rate constants for non-specific distribution of the drug to and from a peripheral compartment, and kel is the first-order elimination rate constant. Equation 4 describes the amount of drug (Apo) at the site of absorption after oral dosing and Eq. 6 describes the amount of drug in the peripheral compartment (At). Plasma drug concentration and apparent volume of distribution in the central compartment are denoted by Cp and Vc,app, respectively. Subsequent prediction and modeling of human UGE employed the pharmacokinetic parameters estimated in this step.
A slightly modified form of Eq. 3 was used to both predict and model the pharmacodynamics of SGLT2 inhibitor-mediated UGE in healthy subjects:
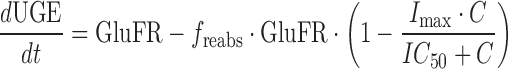 |
7 |
where GluFR denotes glucose filtration rate as determined by the product of GFR and PG. GluFR was fixed to a population mean value of 7.5 g/h commensurate with typical mean plasma glucose concentrations (i.e. 100 mg/dl, 5 mM) and GFR (i.e. 125 ml/min) in healthy subjects (32–34). This simplification was performed in order to allow for a fixed 20% inter-cohort coefficient of variation around the assumed population mean GluFR in modeling the human data. As neither this parameter nor its composite parts (PG and GFR) were reported in the literature, this allowance provides for a reasonable degree of variability in this physiological parameter around the assumed population mean value across dose groups. In the modeling process, Imax, IC50, and post hoc estimates of GluFR in each dose group were estimated. In addition to the modeling described above, prospective simulations of human UGE were performed from pharmacodynamic parameters estimated in rats. For this analysis, GluFR was assumed to be 7.5 g/h. The Imax and freabs parameters were assumed to translate 1:1 between rats and humans. Lastly, the IC50 parameter estimated in rats was translated to an expected human value by accounting for species differences in unbound fraction and in vitro potency as follows:
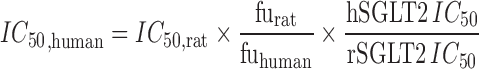 |
where hSGLT2 IC50 and rSGLT2 IC50 denote the potency against human and rat SGLT2 detemined in vitro.
Data Analysis
The PK/PD model was implemented in NONMEM version 5, release 1.1 using the first-order method of approximation (35). Analysis was conducted in a sequential manner whereby pharmacokinetic parameters estimated in step 1 were fixed to population mean estimates during estimation of pharmacodynamic parameters in the second step. Residual error was characterized according to a proportional error model:
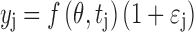 |
8 |
where f(θ,tj) is the predicted value of the data from the model given the population mean PK and PD parameters (θ) and time (tj). yj denotes the jth observation and εj accounts for the residual between the predicted and observed values. Goodness-of-fit was analyzed using the objective function, visual inspection of curve fits, predicted and observed plots, residual plots, and an assessment of parameter estimates and associated correlations of parameter estimates.
RESULTS
Rat Pharmacokinetics and Pharmacodynamics
Dapagliflozin exposure was dose proportional between 0.1 and 10 mg/kg. The proposed one-compartment model with first-order absorption well characterized each dose level (Fig. 1a), and uncertainty (SEM%) associated with the parameter estimates was low (Table I). In addition, the proposed PK/PD model reasonably well fitted the observed 24-h UGE (Fig. 1b). Both the data and model indicate less than dose proportional increase in UGE at doses above 1 mg/kg. However, slightly greater increases in UGE were observed at the 3 and 10 mg/kg dose levels than were fitted by the model. Incorporation of a hill coefficient in the model did not improve the fit to the data further (data not shown). The Imax parameter was estimated to be 0.32 and the associated uncertainty was low (Table I). A larger degree of uncertainty (51% SEM) was associated with the IC50 parameter estimate of 29 ng/ml (2.9 nM unbound).
Fig. 1.

Single-dose plasma pharmacokinetics a and UGE pharmacodynamics b of dapagliflozin at four dose levels in rats. Lines of a and shaded bars of b represent model fits. Open symbols of a represent individual observations at doses of 0.1 (triangle), 1 (square), 3 (triangle), and 10 mg/kg (circle). Open bars of b (mean ± S.D.) represent mean experimental observations (±S.D.). UGE over 24 h in the vehicle group was 4.1 ± 0.6 g
Table I.
Estimated PK/PD Model Parameters for Dapaglifozin in Rats
| Parameter (units) | Definition | Final estimate | % SEM |
|---|---|---|---|
| Pharmacokinetic parameters | |||
| k el (1/hr) | First-order elimination rate constant | 0.13 | 7.6 |
| k a (1/hr) | First-order absorption rate constant | 0.67 | 11.9 |
| V c,app (L/kg) | Apparent volume of distribution | 1.5 | 6.7 |
| Pharmacodynamic parameters | |||
| IC 50, total (ng/ml) | Total plasma concentration associated with half-maximal inhibition of SGLT2-mediated glucose reabsorption | 29 | 51 |
| IC 50,unbound (nM) | Unbound plasma concentration associated with half-maximal inhibition of SGLT2-mediated glucose reabsorption | 2.9a | – |
| I max | Maximum fractional inhibition of glucose reabsorption | 0.32 | 6.3 |
aCalculated based on a molecular weight of 409 Da and a rat unbound plasma fraction of 0.040 estimated by standard equilibrium dialysis techniques
Human Pharmacokinetics and Pharmacodynamics
Previously reported dapagliflozin pharmacokinetics were well characterized by the proposed linear two-compartment model (Fig. 2), and the uncertainty associated with parameter estimates was low (Table II). As described in the MATERIALS AND METHODS section, prospective predictions of human UGE assumed a 1:1 translation of Imax and freabs from rats (0.32 and 0.996, respectively). An expected human IC50 of 6.6 ng/ml was obtained by accounting for species differences in unbound fraction and in vitro potency as follows:
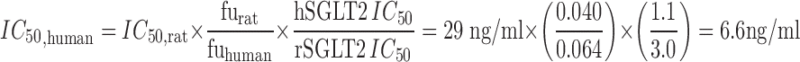 |
Fig. 2.

Mean dapagliflozin pharmacokinetics in healthy subjects following single a and multiple daily doses b as obtained from the literature (26). Symbols represent doses of 2.5 (plus sign), 10 (triangle), 50 (diamond), 100 (circle), and 250 mg (multiplication sign). Lines represent model fits
Table II.
Estimated PK/PD Model Parameters for Dapagliflozin in Healthy Subjects
| Parameter (units) | Definition | Final estimate | % SEM |
|---|---|---|---|
| Pharmacokinetic parameters | |||
| k el (1/hr) | First-order elimination rate constant | 0.26 | 7.7 |
| k a (1/hr) | First-order absorption rate constant | 2.7 | 14.8 |
| k pt (1/hr) | First-order rate constant for distribution of drug from plasma to tissues | 0.14 | 14.3 |
| k tp (1/hr) | First-order rate constant for distribution of drug from tissues to plasma | 0.09 | 22.2 |
| V c,app (L) | Apparent central volume of distribution | 70 | 8.5 |
| Pharmacodynamic parameters | |||
| IC 50, total (ng/ml) | Total plasma concentration associated with half-maximal inhibition of SGLT2- mediated glucose reabsorption | 3.5 | 17.1 |
| IC 50,unbound (nM) | Unbound plasma concentration associated with half-maximal inhibition of SGLT2- mediated glucose reabsorption | 0.55a | – |
| I max | Maximum fractional inhibition of glucose reabsorption | 0.35 | 5.7 |
aCalculated based on a molecular weight of 409 Da and a human unbound plasma fraction of 0.064 estimated by standard equilibrium dialysis techniques. GluFR was assumed to be 7.5 g/h with a 20% inter-cohort coefficient of variation. GluFR was estimated to be 6.6, 7.7, 6.8, 8.6, and 8.6 g/h in the 2.5, 10, 50, 100, and 250 mg dose cohorts
Assuming these parameter values, prospective predictions of human UGE were of consistent shape, but biased toward under-prediction (Fig. 3a). Overall, this bias was modest in nature, with the error in UGE prediction at 120 h being 23%, 28%, 7%, 28%, and 23% at dapagliflozin doses of 2.5, 10, 50, 100, and 250 mg, respectively. Not surprisingly, modeling of the data provided a better characterization, with no obvious bias across doses (Fig. 3b). The Imax and IC50 parameters were estimated to be 0.35 and 3.5 ng/ml (0.55 nM unbound), and uncertainty associated with the parameter estimates was low (Table II). Post hoc estimates of glucose filtration rate in the 2.5, 10, 50, 100, and 250 mg dose groups were 6.6, 7.7, 6.8, 8.6, and 8.6 g/h, respectively. These estimates correspond to daily glucose filtrations of 158, 185, 163, 206 and 206 g, respectively.
Fig. 3.

Model-based prediction a and description b of mean UGE pharmacodynamics of dapagliflozin in healthy subjects following single doses as obtained from the literature (26). Symbols represent doses of 0 (asterisk), 2.5 (plus sign), 10 (triangle), 50 (diamond), 100 (circle), and 250 mg (multiplication sign). Lines represent model predictions a and fits b
DISCUSSION
Preclinical Pharmacokinetics and Pharmacodynamics
The proposed biologically based PK/PD model provided a reasonable characterization of observed dapagliflozin-mediated UGE in rats. Modest UGE under predictions at high doses and a 51% SEM on the IC50 estimate may be due, in part, to the relatively sparse, single time point, UGE data available for PK/PD modeling in the rat. Given the observed exposures of dapagliflozin in this study, it is also feasible that some modest degree of SGLT1 inhibition may confound the analysis of rat data. As reported previously, the IC50 of dapagliflozin for rat SGLT1 is 620 nM (36). Model-predicted maximum, average and minimum dapagliflozin concentrations at the highest dose tested (10 mg/kg) were 4,458 ng/ml (436 nM unbound), 2,055 ng/ml (201 nM unbound), and 409 ng/ml (40 nM unbound), respectively. As such, the fractional inhibition of SGLT1 at 10 mg/kg could vary between 6% and 40% over the 24-h UGE determination. Although relatively less important to glucose reabsorption in normal conditions (~10% vs. 90% for SGLT2) (37), SGLT1 may play a more important fractional role as SGLT2 is increasingly inhibited. Factoring in the relative contribution of these two transporters under various degrees of pharmacological inhibition will require a more mechanistic model and is outside the scope of the current work. Nevertheless, additional confidence that the PK/PD model predominantly represents SGLT2 inhibition can be derived from the concordance between the estimated unbound IC50 and that previously reported for rSGLT2 in vitro (2.9 vs. 3 nM, respectively) (36). In addition, we have recently reported a similar in vitro–in vivo concordance of IC50 in applying this PK/PD model to new SGLT2 inhibitor, PF-04971729 (27). It is interesting to note that the Imax of dapagliflozin was estimated to be 0.32, indicating that maximum SGLT2 inhibition is associated with only a 32% reduction in glucose reabsorption. Again, this observation is consistent with the potential that other transporters (e.g., SGLT1) may play an increasing important role in glucose reabsorption as SGLT2 is inhibited.
Human Pharmacokinetics and Pharmacodynamics
Though generally consistent, prospective prediction of human UGE from the pharmacodynamic parameters obtained in rats were modestly biased toward under prediction (Fig. 3a). In contrast, the proposed biologically based PK/PD model for SGLT2 inhibitor-mediated UGE provided an excellent characterization of mean UGE in healthy subjects taken from the literature (Fig. 3b). Unlike the rat studies, an extensive amount of pharmacokinetic and UGE data were available to support precise parameter estimation. In addition, the improved selectivity of dapagliflozin for human SGLT2 over human SGLT1 (1.1 vs. 1,391 nM) (36), removes the potentially cofounding influence of SGLT1 inhibition at the doses examined herein. The estimated Imax of 0.35 was similar to that estimated in rats (0.32) and that reported previously in humans (26). In addition, as was observed in rats, the unbound IC50 estimate was very similar to that previously reported for dapagliflozin against hSGLT2 in vitro (0.55 vs. 1.1 nM, respectively) (36). Post hoc estimates of GluFR were slightly different among dose groups with no obvious trend. These results suggest that the observed bias in prospective predictions is likely a function of slight errors in the assumed values of IC50, Imax, and GluFR. Overall, these results indicate an excellent quantitative translation of dapagloflozin pharmacology across species.
Implications for Applied Translational Research
This work suggests that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be predicted from preclinical data using the proposed biologically based PK/PD model of UGE with sufficient accuracy to support decision making (compound rank ordering, candidate selection, early clinical trial design). More specifically, this work suggests that SGLT2 inhibitor-mediated UGE can be predicted in humans from the following data which can be obtained preclinically: (1) a prediction of human pharmacokinetics, (2) parameters describing the normal physiological process of glucose filtration and reabsorption (GFR, plasma glucose concentration, freabs), and (3) parameters describing the intrinsic efficacy (Imax) and potency (IC50) of the SGLT2 inhibitor for UGE. Key principles and limitations underlying how each of these key areas of translation was handled in the current work are discussed below.
Translational Pharmacokinetics
The prediction of human pharmacokinetics will usually serve as the foundation upon which pharmacodynamics are predicted. Many reports have demonstrated that reasonably accurate pharmacokinetic predictions from preclinical data are feasible and common in the pharmaceutical industry (4–17). In a previous report, we have utilized these approaches to support a pharmacodynamic prediction for a new SGLT2 inhibitor, PF-04971729 (27). Since the focus of the current work is upon an examination of translational pharmacology, human pharmacokinetics of dapagliflozin were simply obtained from the literature (26).
Translational Physiology
Physiologically, both the circulating concentration of glucose and the rate at which the circulation is filtered will govern the rate at which glucose is filtered by the kidney. This basic concept is codified in the model as the product of GFR and plasma glucose concentration. GFR is an independent physiological parameter that can be experimentally estimated at the subject level or approximated at the level of a population (e.g., mean GFR in healthy subjects). Plasma glucose concentration can also be determined either experimentally at the subject level or approximated at the level of a population (e.g., mean plasma glucose concentration in healthy subjects). As described in the MATERIALS AND METHODS section, GFR and plasma glucose values were chosen to be consistent with population mean values as obtained from the literature for the current exercise. While this simplification is appropriate to support the goals of the current study-level analysis, inter-patient variability in these parameters would be important to understand in many cases as this would certainly contribute to variability in response among individuals. This may be particularly important in the diabetic population where there is a large degree of inter-patient variability in glycemic control and renal function. Another simplification was made in the current analysis such that these physiological parameters were fixed to stationary (time-invariant) values. Although GFR does follow a circadian pattern, the variation around the mean value over 24 h appears to be small in healthy rats and humans (28–38). In addition, although plasma glucose profiles would be expected to vary with time and meals, there also appears to be little variation around the mean value in healthy rats and humans (29–31,39). As such, fixing GFR and plasma glucose to mean and stationary parameters for characterization of healthy rats and humans is likely a reasonable approximation. Because these simplifying assumptions are less accurate in diabetic patients, particular attention should be paid to the time dependency of these parameters when extending this model to this patient population. Another potentially confounding variable with regard to the stationarity of plasma glucose concentrations is the effect of SGLT2 inhibitors to lower plasma glucose through UGE. However, as the amount of glucose excreted following a single dose of an SGLT2 inhibitor is small in healthy subjects, acute changes in plasma glucose are neither expected nor observed (26). In contrast, treatment of diabetic patients clearly lowers blood glucose concentration in studies of longer duration (23,24–40). As such, treatment-related changes in plasma glucose with time should be considered when extending this model to the chronic treatment of diabetics. This would require an accounting for the impact of UGE on the whole body physiology of glucose homeostasis. Lastly, the freabs parameter is an empirical descriptor of the fraction of filtered glucose that is reabsorbed. This descriptor is likely determined by the dynamic interplay of physiological rates (e.g., filtrate flow), SGLT1 and SGLT2 transport kinetics (e.g., Vmax/Km), plasma glucose concentrations and target biology (e.g. dynamics of SGLT2 expression). Accordingly, this parameter would be expected to vary greatly from patient to patient according to the level of disease and treatment. As such, codifying this aspect of the physiology may be critically important in extending this model to diabetics under chronic treatment. However, this is unlikely to significantly confound the current analysis as glucose is almost completely reabsorbed in healthy rats and humans.
Translational Pharmacology
As pharmacokinetics and physiology are major determinants of SGLT2 inhibitor-mediated UGE, the translation of the primary pharmacology data can only be examined once these aspects have been accounted for as described above. In the current exercise, translational pharmacology can be examined with respect to efficacy (the maximal inhibitor-induced UGE) and potency (the concentration of inhibitor which produces half-maximal UGE). The current work suggests that the in vivo potency of SGLT2 inhibitors can be reasonably well predicted from in vitro assays and mechanism-based PK/PD modeling of UGE data obtained in rats. One important principle in examining the in vivo translation of potency from in vitro systems is proper accounting for the unbound fraction of drug between systems. This is important as typically only the unbound fraction is available to interact with the target. This is particularly true for SGLT2 as: (1) only unbound drug is available for filtration to the site of the transporter and (2) the in vitro assays are conducted under protein-free conditions. The importance of this correction can be seen with the current results. For example, the estimated rat in vivo IC50 of dapagliflozin is 29 ng/ml. Converting this to molar units and correcting for an unbound plasma fraction of 0.040, one obtains an unbound IC50 estimate of 2.9 nM (vs. 3.0 nM in vitro). Likewise, converting the estimated human in vivo IC50 (3.5 ng/ml) using unbound fraction in human plasma (0.064), one obtains an unbound IC50 estimate of 0.55 nM (vs. 1.1 nM in vitro). These results suggest that the potency of SGLT2 inhibitors in humans can be reasonably well predicted from in vitro assays by accounting for unbound fraction. However, in the current exercise, human potency was predicted via scaling of the IC50 estimated obtained from PKPD modeling of data obtained in rats. In this approach, differences in unbound fraction and in vitro potency are considered in relative terms in order to afford scaling of an estimate of IC50 obtained in rats. This approach may be useful when unknown factors are suspected of confounding the absolute translation of in vitro potency or unbound fraction. Again, this approach yields a predicted IC50 that is comparable to that estimated from the human data (6.6 vs. 3.5 ng/ml, respectively).
With regard to efficacy, these results similarly indicate the utility of preclinical data for predicting maximum UGE. The estimated Imax of dapagliflozin from the data in healthy subjects was 0.35. This is consistent with previous reports which indicate dapagliflozin is capable of acutely reducing glucose reabsorption up to approximately 30% in healthy subjects (26). This is also similar to the 34.5–43.2% maximum inhibition of glucose reabsorption produced by sergliflozin, another SGLT2 inhibitor, in non-diabetic subjects (41). Similarly, we estimate the Imax of dapagliflozin be 0.32 in rats. This estimate is also generally consistent with the value of 0.49 we have previously reported for another SGLT2 inhibitor, PF-04971729, in rats (27). These small differences across compounds and species suggest that maximum UGE is a property of the system, which is reasonably well conserved across rats and humans. As such, we speculate that the Imax parameter employed in the current model predominately represents an empirical descriptor of the biological system rather than the maximal level of SGLT2 inhibition. On a mechanistic level, this inhibitory limitation of about one third may reflect the activity of SGLT1 transporters located further downstream in the distal tubule of the nephron. Although outside the scope of the current work, a model that explicitly accounts for these aspects may be useful in providing a better understanding of these observations.
CONCLUSION
A simple biologically based PK/PD model has been developed, which was useful in characterizing the PK/PD data of dapagliflozin in rats and humans. This model provides a structure by which species differences in pharmacokinetics, physiology, and pharmacology can be accounted in order to support clinical pharmacology predictions from preclinical data. Application of this model to preclinical and clinical dapagliflozin data indicates the general predictive utility of in vitro pharmacology assays and rat pharmacology. These findings suggest that this model would be useful in supporting the design of SGLT2 inhibitors (early in drug discovery) that are capable of producing a robust proof-of-mechanism (i.e., UGE in the clinic). Future extensions of this model may also provide: (1) a more granular representation of glucose filtration and reuptake, (2) the ability to take additional endpoints of relevance to SGLT2 inhibition (e.g., HbA1c) into consideration, (3) the ability to characterize different patient populations, and (4) the ability to capture dynamic changes with chronic therapy.
Acknowledgments
The authors would like to thank Neeta Amin and Gianluca Nucci for their expert internal review of this manuscript.
References
- 1.CDC. National diabetes fact sheet, 2011. 2011.
- 2.Anonymous Economic costs of diabetes in the U.S. In 2007. Diabetes Care. 2008;31(3):596–615. doi: 10.2337/dc08-9017. [DOI] [PubMed] [Google Scholar]
- 3.Carpino PA, Goodwin B. Diabetes area participation analysis: a review of companies and targets described in the 2008–2010 patent literature. Expert Opin Ther Pat. 2010;20(12):1627–1651. doi: 10.1517/13543776.2010.533171. [DOI] [PubMed] [Google Scholar]
- 4.Hosea NA, Collard WT, Cole S, Maurer TS, Fang RX, Jones H, et al. Prediction of human pharmacokinetics from preclinical information: comparative accuracy of quantitative prediction approaches. J Clin Pharmacol. 2009;49(5):513–533. doi: 10.1177/0091270009333209. [DOI] [PubMed] [Google Scholar]
- 5.Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24(1):53–75. doi: 10.2133/dmpk.24.53. [DOI] [PubMed] [Google Scholar]
- 6.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RAHJ. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 7.Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45(5):511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 8.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27(11):1350–1359. [PubMed] [Google Scholar]
- 9.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, Macintyre F, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283(1):46–58. [PubMed] [Google Scholar]
- 10.Emoto C, Murayama N, Rostami-Hodjegan A, Yamazaki H. Methodologies for investigating drug metabolism at the early drug discovery stage: prediction of hepatic drug clearance and P450 contribution. Curr Drug Metab. 2010;11(8):678–685. doi: 10.2174/138920010794233503. [DOI] [PubMed] [Google Scholar]
- 11.Blanchard N, Hewitt NJ, Silber P, Jones H, Coassolo P, Lave T. Prediction of hepatic clearance using cryopreserved human hepatocytes: a comparison of serum and serum-free incubations. J Pharm Pharmacol. 2006;58(5):633–641. doi: 10.1211/jpp.58.5.0008. [DOI] [PubMed] [Google Scholar]
- 12.McGinnity DF, Collington J, Austin RP, Riley RJ. Evaluation of human pharmacokinetics, therapeutic dose and exposure predictions using marketed oral drugs. Curr Drug Metab. 2007;8(5):463–479. doi: 10.2174/138920007780866799. [DOI] [PubMed] [Google Scholar]
- 13.Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. II. Volume of distribution and mean residence time. Drug Metab Dispos. 2004;32(6):612–619. doi: 10.1124/dmd.32.6.612. [DOI] [PubMed] [Google Scholar]
- 14.Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. I. Clearance. Drug Metab Dispos. 2004;32(6):603–611. doi: 10.1124/dmd.32.6.603. [DOI] [PubMed] [Google Scholar]
- 15.Tang H, Mayersohn M. A global examination of allometric scaling for predicting human drug clearance and the prediction of large vertical allometry. J Pharm Sci. 2006;95(8):1783–1799. doi: 10.1002/jps.20481. [DOI] [PubMed] [Google Scholar]
- 16.Mahmood I. Interspecies pharmacokinetic scaling: principles, applications, and limitations. Pharmacokinet Drug Dev. 2004;1:423–444. [Google Scholar]
- 17.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47(9):1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 18.Kola I, Landis J. Opinion: Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711–716. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 19.Washburn WN. Evolution of sodium glucose co-transporter 2 inhibitors as anti-diabetic agents. Expert Opin Ther Pat. 2009;19(11):1485–1499. doi: 10.1517/13543770903337828. [DOI] [PubMed] [Google Scholar]
- 20.van den Heuvel LP, Assink K, Willemsen M, Monnens L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2) Hum Genet. 2002;111(6):544–547. doi: 10.1007/s00439-002-0820-5. [DOI] [PubMed] [Google Scholar]
- 21.Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14(11):2873–2882. doi: 10.1097/01.ASN.0000092790.89332.D2. [DOI] [PubMed] [Google Scholar]
- 22.Calado J, Soto K, Clemente C, Correia P, Rueff J. Novel compound heterozygous mutations in SLC5A2 are responsible for autosomal recessive renal glucosuria. Hum Genet. 2004;114(3):314–316. doi: 10.1007/s00439-003-1054-x. [DOI] [PubMed] [Google Scholar]
- 23.List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32(4):650–657. doi: 10.2337/dc08-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9733):2223–2233. doi: 10.1016/S0140-6736(10)60407-2. [DOI] [PubMed] [Google Scholar]
- 25.Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–1149. doi: 10.1021/jm701272q. [DOI] [PubMed] [Google Scholar]
- 26.Komoroski B, Vachharajani N, Boulton D, Kornhauser D, Geraldes M, Li L, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85(5):520–526. doi: 10.1038/clpt.2008.251. [DOI] [PubMed] [Google Scholar]
- 27.Mascitti V, Maurer TS, Robinson RP, Bian J, Boustany-Kari CM, Brandt T, et al. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors. J Med Chem. 2011; in press, published online at http://pubs.acs.org/doi/pdfplus/10.1021/jm200049r. [DOI] [PubMed]
- 28.Chamberlain RM, Shirley DG. Time course of the renal functional response to partial nephrectomy: measurements in conscious rats. Exp Physiol. 2007;92(1):251–262. doi: 10.1113/expphysiol.2006.034751. [DOI] [PubMed] [Google Scholar]
- 29.Shih K-C, Liu L-Y, Kwok C-F, Hwu C-M, Juan C-C, Hsu Y-P, et al. Effect of reversing dark-light cycles on normal diurnal variation and related metabolic disturbance in rats. Chin J Physiol. 2007;50(2):69–76. [PubMed] [Google Scholar]
- 30.Jolin T, Montes A. Daily rhythm of plasma glucose and insulin levels in rats. Horm Res. 1973;4(3):153–156. doi: 10.1159/000178303. [DOI] [PubMed] [Google Scholar]
- 31.Matsuo T, Izumori K. Effects of dietary D-psicose on diurnal variation in plasma glucose and insulin concentrations of rats. Biosci Biotechnol Biochem. 2006;70(9):2081–2085. doi: 10.1271/bbb.60036. [DOI] [PubMed] [Google Scholar]
- 32.Rohlfing CL, Wiedmeyer H-M, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA1c: Analysis of glucose profiles and HbA1c in the Diabetes Control and Complications Trial. Diabetes Care. 2002;25(2):275–278. doi: 10.2337/diacare.25.2.275. [DOI] [PubMed] [Google Scholar]
- 33.Nathan DM, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine RJ. Translating the A1C assay into estimated average glucose values. Diabetes Care. 2008;31(8):1473–1478. doi: 10.2337/dc08-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeWoskin RS, Thompson CM. Renal clearance parameters for PBPK model analysis of early lifestage differences in the disposition of environmental toxicants. Regul Toxicol Pharmacol. 2008;51(1):66–86. doi: 10.1016/j.yrtph.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Beal SL, Boeckmann AJ, Sheiner LB. NONMEM Users Guides, Version 5 edition. San Francisco: University of California at San Francisco; 1999. [Google Scholar]
- 36.Han S, Hagan DL, Taylor JR, Xin L, Meng W, Biller SA, et al. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes. 2008;57(6):1723–1729. doi: 10.2337/db07-1472. [DOI] [PubMed] [Google Scholar]
- 37.Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280(1):F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- 38.Voogel AJ, Koopman MG, Hart AA, van Montfrans GA, Arisz L. Circadian rhythms in systemic hemodynamics and renal function in healthy subjects and patients with nephrotic syndrome. Kidney Int. 2001;59(5):1873–1880. doi: 10.1046/j.1523-1755.2001.0590051873.x. [DOI] [PubMed] [Google Scholar]
- 39.Mazze RS, Strock E, Wesley D, Borgman S, Morgan B, Bergenstal R, et al. Characterizing glucose exposure for individuals with normal glucose tolerance using continuous glucose monitoring and ambulatory glucose profile analysis. Diabetes Technol Ther. 2008;10(3):149–159. doi: 10.1089/dia.2007.0293. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Feng Y, List J, Kasichayanula S, Pfister M. Dapagliflozin treatment in patients with different stages of type 2 diabetes mellitus: effects on glycaemic control and body weight. Diabetes, Obes Metab. 2010;12(6):510–516. doi: 10.1111/j.1463-1326.2010.01216.x. [DOI] [PubMed] [Google Scholar]
- 41.Hussey EK, Dobbins RL, Stoltz RR, Stockman NL, O’Connor-Semmes RL, Kapur A, et al. Multiple-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy overweight and obese subjects: a randomized double-blind study. J Clin Pharmacol. 2010;50(6):636–646. doi: 10.1177/0091270009352185. [DOI] [PubMed] [Google Scholar]