

Abstract
The objective of this work was to examine the atazanavir–bilirubin relationship using a population-based approach and to assess the possible application of bilirubin as a readily available marker of atazanavir exposure. A model of atazanavir exposure and its concentration-dependent effect on bilirubin levels was developed based on 200 atazanavir and 361 bilirubin samples from 82 patients receiving atazanavir in the NORTHIV trial. The pharmacokinetics was adequately described by a one-compartment model with first-order absorption and lag-time. The maximum inhibition of bilirubin elimination rate constant (Imax) was estimated at 91% (95% CI, 87–94) and the atazanavir concentration resulting in half of Imax (IC50) was 0.30 μmol/L (95% CI, 0.24–0.37). At an atazanavir/ritonavir dose of 300/100 mg given once daily, the bilirubin half-life was on average increased from 1.6 to 8.1 h. A nomogram, which can be used to indicate suboptimal atazanavir exposure and non-adherence, was constructed based on model simulations.
Key words: atazanavir, bilirubin, HIV/AIDS, pharmacodynamics, pharmacokinetics
Introduction
The protease inhibitor (PI) atazanavir is currently recommended as one first line option for treatment of HIV-1-infected patients. Current treatment guidelines recommend minimal effective atazanavir trough concentrations (MEC) of 0.2 μmol/L (1,2). Elevated bilirubin levels are commonly observed in patients on an atazanavir-based treatment, although it is an uncommon cause of treatment discontinuation (1). Hyperbilirubinemia is attributed to a concentration-dependent atazanavir inhibition of the UGT1A1 enzyme responsible for bilirubin conjugation (3–5). The importance of UGT1A1 gene allele*28 for development of hyperbilirubinemia has been shown in several studies (6,7). Recent work has however revealed a complex interplay between multiple transporters affecting both atazanavir and bilirubin. Unconjugated bilirubin is to some extent transported into the hepatocytes by the organic anion-transporting polypeptide (OATP)–1B1. Atazanavir has been shown to be an inhibitor and a substrate of several OATPs, including 1B1 (8–11). Although the role of OATP1B1 may not be clear it seems to be of importance for bilirubin elevation and possibly atazanavir exposure (11). This has led to discussions on which mechanism is most important for the atazanavir-induced hyperbilirubinemia (12). Atazanavir is a substrate for CYP3A4 and CYP3A5 but also an inhibitor of CYP3A4 and p-glycoprotein (13,14). Polymorphism (3435C→T) in the multi-drug resistance gene 1 (MDR1) coding for p-glycoprotein (P-gp) has been associated with lower ATZ and bilirubin plasma concentrations (6,15) (Fig. 1a).
Fig. 1.

a Illustration of the bilirubin elimination process by the hepatocytes and the suggested associated proteins/transporters. Bilirubin can enter the hepatocytes passively (diffusion) and actively trough the organic anion-transporting polypeptide (OATP) 1B1. Bilirubin glucuronidation is mediated by glucuronosyltransferase (UGT) 1A1. The bilirubin glucuronide is excreted into bile canaliculi by multi-drug resistance protein (MRP) 2. Atazanavir is thought to enter the hepatocytes passively and to some extent actively by OATP1B1. Atazanavir inhibits UGT1A1 and possibly OATP1B1. Atazanavir is also substrate for p-glycoprotein (P-gp). b Schematic illustration of the final mathematical model describing the physiological process in (a). The bilirubin box represent the bilirubin concentration i plasma. Bilirubin is synthesized by a first-order process (k in) and eliminated by a second order process (k out). C p denotes the plasma concentration of atazanavir which inhibits the elimination (k out) of bilirubin. k a, lag-time, V c/F and CL/F are defined in Table I
A common observation in all clinical studies has been that increasing atazanavir plasma concentrations resulted in elevated bilirubin levels. Moreover, bilirubin levels were significantly higher in patients with successful virological suppression compared to those with virological failure (16–18). Similar direct relationships between atazanavir concentrations and virological outcome have not been demonstrated. This has lead to suggestions that bilirubin may be used as a marker of adherence to atazanavir therapy and possibly therapeutic outcome (16–19). High availability and low cost of bilirubin compared to atazanavir assays are some potential benefits of bilirubin as a marker of atazanavir exposure in therapeutic drug monitoring, offering clinicians a decision-making tool for change of therapy. Unfortunately, the lack of a quantitative assessment of the atazanavir–bilirubin relationship has prevented further explorations of bilirubin as a potential marker for adherence and ultimately treatment outcome.
The aim of this work was to develop a non-linear mixed effects model for the atazanavir exposure and bilirubin response relationship to be used for investigation of bilirubin as a marker of atazanavir exposure in HIV/AIDS patients.
Methods
The Data
The NORTHIV trial was a randomized open-label multicentre trial conducted in Sweden and Norway, in accordance with ICH-GCP and declaration of Helsinki as modified by the 48th world medical association, South Africa 1996. The NORTHIV study has been described in detail elsewhere (16,20). The present analysis included observations from 82 antiretroviral naïve HIV-serum-positive patients. The patients were administered 300 mg atazanavir and 100 mg ritonavir for once a day oral dosing. The nucleoside reverse transcriptase inhibitor backbone was chosen according to the Swedish guidelines (2,21,22) and was allowed to vary amongst the patients. In a few cases the dose was adjusted according to clinical practice. Plasma atazanavir concentrations were measured at weeks 4, 12, 48, 96 and 144. Bilirubin concentrations were collected at baseline and at time points matching atazanavir concentration measurements. In total 361 bilirubin observations and 200 atazanavir steady-state plasma samples were available. All patients receiving atazanavir in the study had normal bilirubin baseline concentrations (<20 μmol/L).
Data Analysis
Atazanavir concentrations were transformed to their natural logarithms and concentration time profiles were modelled by non-linear mixed effects modelling using NONMEM VI (ICON Development Solutions, Ellicot City, MD, USA) with first-order conditional estimation method with interaction (23). Model selection was based on mechanistic plausibility, precision of parameter estimates, the objective function value and diagnostic plots including visual predictive checks (1,000 replicates). The R-based programme Xpose 4, Census and PsN were used for handling of model files and graphics (24–26).
Pharmacokinetic and Pharmacodynamic Model Building
The search for a structural pharmacokinetic model for atazanavir started with a previously developed and validated model (27). The proposed model was a one-compartment structural model with lag time and first-order absorption and elimination rate. Lag time and the first-order absorption rate constant were fixed to previously reported estimates due to few samples collected in the absorption phase (27). Pharmacokinetic parameters were allometrically scaled by body weight (BW) and centred to the median BW (Eq. 1).
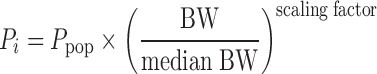 |
1 |
Where Pi is the individual parameter estimate and Ppop is the parameter estimate for a patient with median body weight (70 kg). The scaling factor was a priori set to 0.75 for clearance (CL/F) and to 1 for volume of distribution (V/F) (28). Inter-individual variability (IIV) was initially estimated for all the structural parameters of the model but only kept if they could be estimated with adequate precision. Correlations between variability components were also evaluated. Pharmacokinetic parameters were assumed to be log-normally distributed while the PD parameters were tested for normal or log-normal distribution. Proportional and slope intercept models were applied to explain the residual variability.
The pharmacokinetics of atazanavir and the bilirubin response were described in a sequential analysis where the effect of atazanavir exposure on bilirubin levels was described by an indirect response model with concentration-dependent inhibition of the fractural turnover rate (kout) (Eqs. 2 and 3; Fig. 1b) (29).
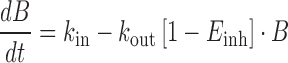 |
2 |
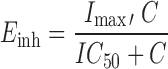 |
3 |
Where B is bilirubin, kin is the zero-order production rate of B and Einh is the fraction of kout that is inhibited at concentration C of atazanavir. Other pharmacodynamic models such as the direct effect model and the bio-phase model were also tested.
The bilirubin elimination constant (kout) was used to compute the half-life of bilirubin in presence and absence (Einh = 0) of atazanavir (Eq. 4).
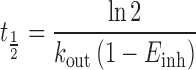 |
4 |
Simulations of the proposed model were performed in Berkley Madonna (Berkeley Madonna, Berkeley, CA, USA).
Results
The population pharmacokinetic model describing the time course of atazanavir plasma concentrations was based on 200 samples from 82 patients. Due to few samples obtained during the absorption phase, the lag-time (0.96 h) and absorption rate (3.4 h−1) were fixed to published values (27). The data was adequately described with a one-compartment model. Parameter estimates of the final pharmacokinetic model are found in Table I. Visual predictive checks of observed and predicted atazanavir concentrations as well as basic goodness of fit plots can be seen in Fig. 2a. In total 361 measurements of bilirubin were available. At baseline patients had a mean (±SD) bilirubin value of 7.8 (±3.3) μmol/L which increased to 34 (±18.6) μmol/L when the new steady state was reached. The effect of atazanavir on bilirubin elevation was described by an indirect response model, where atazanavir inhibits the bilirubin elimination. The maximum inhibition (Imax) of bilirubin elimination was estimated to 90.5% (95% CI, 87–94) while the concentration resulting in 50% of Imax (IC50) was estimated to 0.300 μmol/L (95% CI, 0.24–0.37). Parameter estimates for the final pharmacodynamic model are tabulated in Table I. Basic goodness of fit plots and a visual predictive check are shown in Fig. 2b. Other models, such as the bio-phase model and direct response model were discarded by goodness of fit criteria as specified in “Methods” section.
Table I.
Parameter Estimates of the Final Pharmacokinetic and Pharmacodynamic Models Describing Atazanavir and Its Influence on Bilirubin in HIV/AIDS Patients
| Estimate | IIV, %CV | ||
|---|---|---|---|
| Parameter | (95% CI) | (RSE%) | |
| PK model | Lag time (h) | 0.96a | |
| k a (h−1) | 3.4a | ||
| V/F (L) | 93.6 (62–125) | 53.1 (43.6) | |
| CL/F (L/h) | 6.47 (5.39–7.55) | 43.8 (19.5) | |
| Correlation | |||
| p(CL/F, V/F) | 0.290 | ||
| Residual error | |||
| σ prop (%) | 51.0 (42.7–59.3) | ||
| PD model | Baseline (μmol/L) | 7.69 (6.99–8.39) | 32.6 (20.2) |
| k out (h−1) | 0.420 (0.36–0.48) | ||
| Imax(%) | 91.0 (87–94) | ||
| IC50 (μmol/L) | 0.300 (0.24–0.37) | ||
| Residual error | |||
| σ prop (%) | 39.4 (35.5–43.3) | ||
| σ add (μmol/L) | 2.39 (1.96–2.82) | ||
k a absorption rate constant, V/F volume of distribution, CL/F clearance, p correlation coefficient, σ prop proportional residual variability, k out fractional turnover rate, I max maximum inhibition constant, IC 50 concentration resulting in 50% of I max, σ add additive residual error, IIV inter-individual variability
aFixed according to (27)
Fig. 2.

Goodness of fit plots of the pharmacokinetic atazanavir model a and the pharmacodynamic bilirubin model b. The four upper panels of a and b: population predictions versus observations (1). Ideally the nonparametric smoother (dashed line) should fall on the line of unity (solid line). (2) conditional weighted residuals versus time after dose (a) and time (b). Conditional weighted residuals should not show any trend over time, the nonparametric smoother (dashed line) should not give a pronounced slope. The two lower panels of a and b: visual predictive check using the final atazanavir population pharmacokinetic model (a, 3) and the pharmacodynamic bilirubin model (b, 3). The circles are observed concentrations, the black lines (simulated) and the grey (real) lines are the fifth, 5th, 50th and 95th percentiles of the data. The shaded areas are 95% confidence intervals around the simulated predictions
A 300 mg dose of ATZ(r), resulting in an average steady-state concentration of 2.75 μmol/L, reduced bilirubin kout by 82.0% and increased bilirubin’s half-life fivefold. Uninhibited, bilirubin displayed a half-life of 1.64 h which was in the same magnitude as the dominant beta phase half-life of radiolabeled bilirubin (1.16 h) (30). The inhibited half-life was estimated to be 8.2 h with a range of 5.4–10.8 h based on predicted average (2.75 μmol/L), maximum (4.5 μmol/L), and minimum (1.0 μmol/L) atazanavir concentrations at steady state for a typical individual.
Simulations of Non-adherence
Simulations of three different scenarios where one (scenario A), two (scenario B) or three (scenario C) consecutive steady-state doses of atazanavir are missed are shown in Fig. 3a–c. Missing one atazanavir dose (scenario A) resulted in decreased average bilirubin concentrations from 35 to 13 μmol/L. Similarly atazanavir concentrations decreased from an expected trough value of 1 to 0.2 μmol/L. In scenario B, two consecutive missed doses resulted in undetectable atazanavir levels while bilirubin levels almost returned to the baseline 8.2 umol/L. In scenario C, bilirubin returned completely to its baseline (7.8 μmol/L).
Fig. 3.

Simulations of the final model illustrating the influence of one (a), two (b) or thee (c) consecutively missed doses of atazanavir on the bilirubin plasma concentration in the typical individual. The black lines represent atazanavir plasma concentration (left axis) while the grey lines represent the bilirubin concentration (right axis)
Simulations of Bilirubin Steady-State Concentrations
Based on simulations of the proposed model, final bilirubin steady-state concentrations were predicted to be affected primarily by the baseline bilirubin levels and atazanavir exposure. Figure 4 shows the influence of atazanavir CL/F on bilirubin steady-state levels in three groups of individuals with low (5 μmol/L), typical (7.8 μmol/L) and high (17 μmol/L) bilirubin baseline. Given the same baseline, a higher CL/F and thus lower exposure will result in less pronounced bilirubin elevation. Steady state bilirubin concentrations were predicted at the minimal effective concentration of atazanavir (MEC = 0.2 μmol/L) for patients with various baseline bilirubin levels as shown in Fig. 5. The black area represents results from individuals with atazanavir exposure below MEC. The borders of the grey area represent how peak and trough steady-state bilirubin concentrations vary with bilirubin baseline concentrations for patients with atazanavir exposure at MEC. While the white area represents bilirubin concentration corresponding to atazanavir exposure over MEC. Based on Fig. 5, exposure over or at MEC can be expected in individuals that have steady-state bilirubin measurements outside of black area.
Fig. 4.

Simulations of the final model illustrating the influence of low, typical and high atazanavir clearance on bilirubin elevation in individuals with a low, typical and high bilirubin baseline. The black lines represent atazanavir plasma concentration (left axis) while the grey lines represent the bilirubin concentration (right axis)
Fig. 5.

Nomogram of the linear relationship between the baseline bilirubin concentrations and the final steady-state bilirubin concentrations after atazanavir therapy initiation. The black area represents bilirubin steady state levels associated with atazanavir exposure below the minimal effective concentration of 0.2 μmol/L. The borders of the grey area represent maximal (10 h after atazanavir dose) and the minimal (24 h after atazanavir dose) bilirubin steady-state concentrations associated with atazanavir exposure at MEC. The white area represents bilirubin levels assosiated with atazanavir concentrations over MEC
Discussion
Previous studies have shown a positive correlation between atazanavir exposure and hyperbilirubinemia. Moreover, in some cases hyperbilirubinemia have been correlated to virological outcomes (16–18). The primary focus of this work was to quantify the atazanavir–bilirubin relationship by a population-based modelling approach and to investigate potential uses of bilirubin as a marker of atazanavir exposure. The proposed model was based on the turnover concept, where a drug can inhibit or stimulate the production or elimination of a given variable, in this case bilirubin (29). The leading hypothesis behind atazanavir-induced hyperbilirubinemia is inhibition of bilirubin’s conjugation by UGT1A1 enzyme (4–7). Consequently, the use of an indirect response model, where the fractional turnover rate of bilirubin elimination is inhibited, is in many aspects mechanistic in nature. It is not clear whether the delayed bilirubin response can be attributed to the indirect effect of atazanavir on the inhibition of bilirubin removal or a combination of indirect effect and delayed atazanavir distribution to the active site (the hepatocytes). The number of transporters involved in the process suggests that distribution delay might have an impact. However, the data at hand did not support a combined indirect response and bio-phase model (31).
Model predictions of bilirubin kinetics are in agreement with what is known from radiolabeled bilirubin studies in man (30). Radiolabeled bilirubin is known to display tri-phasic disposition. The indirect response model assumes mono-exponential decline of bilirubin. Hence the estimated bilirubin half-life reflects the initial two phases of the radiolabeled bilirubin’s tri-phasic disposition. This is however not a major misspecification since calculations of harvested data from Berk et al. show that the first two phases constitute 77% of the total area under the curve, data not shown (30). Further, it was estimated that radiolabel bilirubin will reach 90% of its final steady-state concentrations within 8.3 h after a simulated atazanavir constant infusion. This is in good agreement with our estimation of the uninhibited half-life (1.6 h). Multi-phasic indirect response models have been proposed earlier (32). However, the application of a multi-phasic model was not feasible with the available data. The impact of an underestimation of bilirubin’s half-life would translate in an increased time to reach a new steady state after treatment initiation. Also smaller fluctuations between the maximum and the minimum bilirubin concentrations are to be expected. Nevertheless, this would only further increase bilirubins’ utilities as a marker since it would reflect atazanavir exposure over an even longer period of time. Also the need of accurate information of time after dosing would not be as important due to smaller fluctuations of bilirubin in plasma.
The data available could only support inter-individual variability on bilirubin baseline in the pharmacodynamic model. With more data available, the model can be extended to include IIV on additional pharmacodynamic parameters. Of special interest is variability in the IC50 value. A highly variable IC50 suggest that same plasma concentrations can result in different bilirubin response. This can be interpreted as variable intracellular concentrations. Variable intracellular concentrations can be explained by transport-dependent access to the hepatocytes and HIV-infected cells. Bilirubin would in that case be an indirect measure of the active atazanavir concentrations. But until variable IC50 values are confirmed, bilirubin can only be an indicator of atazanavir concentrations in plasma.
The final steady-state bilirubin levels were predicted to depend on the initial baseline level and atazanavir pharmacokinetics (Fig. 4). Atazanavir exposure governs time to new steady state as well as the final steady-state concentration. The baseline level on the other hand only affects the magnitude of the bilirubin steady-state concentration. These observations are consistent with well-known properties of the indirect response model and may be of importance if bilirubin would be used as a marker of atazanavir exposure (29). In atazanavir therapy it is desirable to achieve concentrations over the proposed minimum effective concentration of 0.2 μmol/L (1). The relative short half-life in combination with once daily administration of atazanavir results in similar steady-state exposure compared to single-dose exposure. This proposes a challenge in interpretation of therapeutic drug monitoring results where it is difficult to discriminate between patients taking their first dose of atazanavir (after a drug holiday) and patients with full adherence. Based on the simulations in Fig. 3, we cannot conclude if bilirubin is a practical marker of long-term atazanavir exposure. However, the utility of bilirubin as a cheaper and more readily available marker of short-term atazanavir exposure is still equally valid. The potential benefits of bilirubin as a measure of long-term adherence or exposure measure may motivate need for additional studies. Our proposed nomogram, illustrating the linear relationship between baseline and final steady-state concentrations, is designed to be of use to clinicians interested in using bilirubin as a measure of atazanavir exposure. In order to predict adequate atazanavir exposure in patients two separate measurements of bilirubin are needed (at baseline and at new steady state). The two measurements should be taken 2 weeks apart giving enough time for bilirubin to reach its new steady state. Bilirubin is reported to have 10.3% variance due to time of day. Bilirubin levels are however relatively stable between 9 am and 3 pm. Measurement after 3 pm are generally lower than those performed before. It would be advisable for all bilirubin samples to be collected between 9 am and 3 pm, avoiding a large part of the circadian fluctuations (33). If the two samples place a patient on the white area of the nomogram, it is an indication of atazanavir exposure over the minimum effective concentration (>0.2 μmol/L). While placing a patient on the black area means that the patient has been exposed to suboptimal atazanavir concentrations. The grey area indicates that the atazanavir concentration is at or above the MEC concentration. During the NORTHIV trial three patients were identified to have low atazanavir exposure <0.2 μmol/L at the 4 weeks follow-up visit. All three of these patients were in the black area of Fig. 5. Although all of the patients included in the atazanavir arm of the NORTHIV trial had normal bilirubin levels (<20 μmol/L) at baseline, we cannot exclude the possible inclusion of patients with undiagnosed Gilbert’s syndrome since data on UGT1A1 gene allele*28 was not available. Although the proposed model takes into account baseline bilirubin levels, it is difficult to predict the effect of Gilbert’s syndrome on the atazanavir inhibition of bilirubin conjugation. Variable intra- and inter-individual bilirubin concentrations may depend on several other factors such as fasting or smoking status or use of oral contraceptives in women (34,35). These and other known causes of bilirubin variation may need to be considered when interpreting results.
Even though the nomogram is not designed to provide dosing recommendations it offers the clinician a valuable tool to be used in combination with current clinical practice and aid in the decision-making process on whether to increase the dose or to evaluate adherence. In addition, bilirubin measurements may be of especial use in resource-limited settings due to the significantly lower cost for equipment and reduced need of highly trained personnel. The nomogram was generated based on data in the NORTHIV study which was not designed for the purpose of this exploratory work. External validation is therefore recommended before full-scale clinical implementation of the method. Preferably, the current model should be challenged in a prospective multiple atazanavir dose study based on optimal design theory.
Conclusions
A semi-mechanistic model describing atazanavir-induced hyperbilirubinemia has been developed. Based on the model, a nomogram has been constructed to predict suboptimal exposure in patients. Although, this nomogram needs external validation before full-scale clinical implementation, it could facilitate the use of bilirubin as a marker of sufficient atazanavir exposure.
Acknowledgements
The NORTHIV study was supported by grants from the Sahlgrenska Academy at the University of Gothenburg (ALFGBG-11067), the Research Foundation of Swedish Physicians Against AIDS, The Health & Medical Care Committee of the Region Västra Götaland (VGFOUREG 2591), the Swedish Research Council (2007-7092) and the Stockholm County Council ALF-grant (581513). We thank Dr. Filip Josephson for valuable discussions concerning bilirubin kinetics and Gilbert’s syndrome.
Disclosure
Daniel Röshammar is a current employee of AstraZeneca. However, this work is not sponsored by AstraZeneca or any other pharmaceutical company.
References
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. January 10, 2011; 1–166. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed January 2011.
- 2.Josephson F, Albert J, Flamholc L, Gisslén M, Karlström O, Moberg L, et al. Treatment of HIV infection: Swedish recommendations 2009. Scand J Infect Dis. 2009;41(11–12):788–807. doi: 10.3109/00365540903214322. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D, Chando T, Everett D, Patten C, Dehal S, Humphreys W. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metabolism and Disposition. 2005;33(11):1729. doi: 10.1124/dmd.105.005447. [DOI] [PubMed] [Google Scholar]
- 4.Smith D, Jeganathan S, Ray J. Atazanavir plasma concentrations vary significantly between patients and correlate with increased serum bilirubin concentrations. HIV Clinical Trials. 2006;7(1):34–38. doi: 10.1310/0KX0-H9VH-99EE-5D0L. [DOI] [PubMed] [Google Scholar]
- 5.Cleijsen R, Van de Ende M, Kroon F, Lunel F, Koopmans P, Gras L, et al. Therapeutic drug monitoring of the HIV protease inhibitor atazanavir in clinical practice. J Antimicrob Chemother. 2007;60(4):897. doi: 10.1093/jac/dkm298. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez-Nóvoa S, Martín-Carbonero L, Barreiro P, González-Pardo G, Jiménez-Nácher I, González-Lahoz J, et al. Genetic factors influencing atazanavir plasma concentrations and the risk of severe hyperbilirubinemia. AIDS. 2007;21(1):41. doi: 10.1097/QAD.0b013e328011d7c1. [DOI] [PubMed] [Google Scholar]
- 7.Monaghan G, Ryan M, Hume R, Burchell B, Seddon R. Genetic variation in bilirubin UDP-glucuronosyltransferase gene promoter and Gilbert’s syndrome. Lancet. 1996;347(9001):578–581. doi: 10.1016/S0140-6736(96)91273-8. [DOI] [PubMed] [Google Scholar]
- 8.Annaert P, Ye Z, Stieger B, Augustijns P. Interaction of HIV protease inhibitors with OATP1B1, 1B3, and 2B1. Xenobiotica. 2010;40(3):163–176. doi: 10.3109/00498250903509375. [DOI] [PubMed] [Google Scholar]
- 9.Cui Y, König J, Leier I, Buchholz U, Keppler D. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem. 2001;276(13):9626. doi: 10.1074/jbc.M004968200. [DOI] [PubMed] [Google Scholar]
- 10.Mediavilla M, Pascolo L, Rodriguez J, Guibert E, Ostrow J, Tiribelli C. Uptake of [3H] bilirubin in freshly isolated rat hepatocytes: role of free bilirubin concentration. FEBS Lett. 1999;463(1–2):143–145. doi: 10.1016/S0014-5793(99)01606-3. [DOI] [PubMed] [Google Scholar]
- 11.Hartkoorn R, Kwan W, Shallcross V, Chaikan A, Liptrott N, Egan D, et al. HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenetics and Genomics. 2010;20(2):112. doi: 10.1097/FPC.0b013e328335b02d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell S, de Morais S, Xu J. Inhibition of human organic anion transporting polypeptide OATP 1B1 as a mechanism of drug-induced hyperbilirubinemia. Chemico–Biological Interactions. 2004;150(2):179–187. doi: 10.1016/j.cbi.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Perloff ES, Duan SX, Skolnik PR, Greenblatt DJ, von Moltke LL. Atazanavir: effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metabolism and Disposition. 2005;33(6):764. doi: 10.1124/dmd.104.002931. [DOI] [PubMed] [Google Scholar]
- 14.Busti AJ, Hall RG, Margolis DM. Atazanavir for the treatment of human immunodeficiency virus infection. Pharmacotherapy. 2004;24(12):1732–1747. doi: 10.1592/phco.24.17.1732.52347. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez Novoa S, Barreiro P, Rendón A, Barrios A, Corral A, Jimenez-Nacher I. Plasma levels of atazanavir and the risk of hyperbilirubinemia are predicted by the 3435C T polymorphism at the multidrug resistance gene 1. Clinical Infectious Diseases. 2006;42(2):291–295. doi: 10.1086/499056. [DOI] [PubMed] [Google Scholar]
- 16.Josephson F, Andersson M, Flamholc L, Gisslén M, Hagberg L, Ormaasen V, et al. The relation between treatment outcome and efavirenz, atazanavir or lopinavir exposure in the NORTHIV trial of treatment-naïve HIV-1 infected patients. Eur J Clin Pharmacol. 2010;66(4):349–357. doi: 10.1007/s00228-009-0763-z. [DOI] [PubMed] [Google Scholar]
- 17.Petersen K, Riddle M, Jones L, Furtek K, Christensen A, Tasker S, et al. Use of bilirubin as a marker of adherence to atazanavir-based antiretroviral therapy. AIDS. 2005;19(15):1700. doi: 10.1097/01.aids.0000186826.60983.85. [DOI] [PubMed] [Google Scholar]
- 18.Karlström O, Josephson F, Sönnerborg A. Early virologic rebound in a pilot trial of ritonavir-boosted atazanavir as maintenance monotherapy. JAIDS Journal of Acquired Immune Deficiency Syndromes. 2007;44(4):417. doi: 10.1097/QAI.0b013e31802e2940. [DOI] [PubMed] [Google Scholar]
- 19.Morello JE, Cuenca L, Vispo E. Use of serum bilirubin levels as surrogate marker of early virological response to atazanavir-based antiretroviral therapy. AIDS research and human retroviruses. 2011;27(00):2–5. doi:10.1089/aid.2011.0019. [DOI] [PubMed]
- 20.Edén A, Andersson LM, Andersson Ö, Flamholc L, Josephson F, Nilsson S, et al. Differential effects of efavirenz, lopinavir/r, and atazanavir/r on the initial viral decay rate in treatment naïve HIV-1–infected patients. AIDS Research and Human Retroviruses. 2010;26(5):533–540. doi: 10.1089/aid.2009.0177. [DOI] [PubMed] [Google Scholar]
- 21.Josephson F, Albert J, Flamholc L, Gisslén M, Karlström O, Lindgren SR, et al. Antiretroviral treatment of HIV infection: Swedish recommendations 2007. Scand J Infect Dis. 2007;39(6–7):486–507. doi: 10.1080/00365540701383154. [DOI] [PubMed] [Google Scholar]
- 22.Gisslén M, Ahlqvist-Rastad J, Albert J, Blaxhult A, Hamberg AK, Lindbäck S, et al. Antiretroviral treatment of HIV infection: Swedish recommendations 2005. Scand J Infect Dis. 2006;38(2):86–103. doi: 10.1080/00365540500388834. [DOI] [PubMed] [Google Scholar]
- 23.Beal SL SL, Boeckmann AJ (eds) Icon Development Solutions,, MD U, editors. NONMEM users guides. Ellicot City,1986–2006.
- 24.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64. doi: 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 25.Wilkins JJ. NONMEMory: a run management tool for NONMEM. Comput Methods Programs Biomed. 2005;78(3):259–267. doi: 10.1016/j.cmpb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson L, Boffito M, Back D, Waters L, Else L, Davies G, et al. Population pharmacokinetics of ritonavir-boosted atazanavir in HIV-infected patients and healthy volunteers. Journal of Antimicrobial Chemotherapy. 2009;63(6):1233–1243. doi: 10.1093/jac/dkp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson B, Holford N. Mechanism-based concepts of size and maturity in pharmacokinetics. Pharmacol Toxicol. 2008;48(1):303. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 29.Jusko WJ, Ko HC. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther. 1994;56(4):406–419. doi: 10.1038/clpt.1994.155. [DOI] [PubMed] [Google Scholar]
- 30.Berk PD, Howe RB, Bloomer JR, Berlin NI. Studies of bilirubin kinetics in normal adults. J Clin Investig. 1969;48(11):2176. doi: 10.1172/JCI106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verotta D, Sheiner L. A general conceptual model for non-steady state pharmacokinetic/pharmacodynamic data. J Pharmacokinet Pharmacodyn. 1995;23(1):1–4. doi: 10.1007/BF02353780. [DOI] [PubMed] [Google Scholar]
- 32.Krzyzanski W, Jusko WJ. Indirect pharmacodynamic models for responses with multicompartmental distribution or polyexponential disposition. J Pharmacokinet Pharmacodyn. 2001;28(1):57–78. doi: 10.1023/A:1011517718990. [DOI] [PubMed] [Google Scholar]
- 33.Pocock S, Ashby D, Shaper A, Walker M, Broughton P. Diurnal variations in serum biochemical and haematological measurements. J Clin Pathol. 1989;42(2):172. doi: 10.1136/jcp.42.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manolio T, Burke G, Savage P, Jacobs D, Jr, Sidney S, Wagenknecht L, et al. Sex- and race-related differences in liver-associated serum chemistry tests in young adults in the CARDIA study. Clin Chem. 1992;38(9):1853. [PubMed] [Google Scholar]
- 35.White G, Jr, Nelson J, Pedersen D, Ash K. Fasting and gender (and altitude?) influence reference intervals for serum bilirubin in healthy adults. Clin Chem. 1981;27(6):1140. [PubMed] [Google Scholar]