

Abstract
Porphyromonas gingivalis is strongly correlated with chronic periodontitis. Its chronic persistence in the periodontium depends on its ability to evade host immunity without inhibiting the overall inflammatory response, which is actually beneficial for this and other periodontal bacteria. Indeed, the inflammatory exudate (gingival crevicular fluid) is a source of essential nutrients, such as peptides and hemin-derived iron. In this review, I discuss how P. gingivalis can promote its adaptive fitness through instigation of subversive crosstalk signaling. These interactions involve Toll-like receptor-2, complement receptor 3, C5a anaphylatoxin receptor, and CXC-chemokine receptor 4. Their exploitation by P. gingivalis allows the pathogen to escape elimination, obtain nutrients, and collaterally inflict periodontal tissue injury.
Keywords: Porphyromonas gingivalis, complement, Toll-like receptors, immune evasion, inflammation
Introduction
Human periodontitis is likely the most common infection-driven chronic inflammatory disease and is characterized by destruction of the supporting tissues of the teeth, including resorption of alveolar bone 1). Periodontal tissue degradation results primarily from unwarranted inflammatory host responses to a group of subgingival gram-negative anaerobic bacteria 2). Several oral microbes have been implicated in periodontitis including the so-called ‘red-complex’ group comprising Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola 3). The presence of P. gingivalis is strongly correlated with disease and is thought of as a keystone species, i.e., an organism that ‘serves an essential function for the entire community, similar to a differentiated cell serving a function for an entire tissue’ 4,5). This review summarizes evidence supporting that P. gingivalis can manipulate the host response in ways that can benefit companion species of the same microbial community and, moreover, amplify destructive inflammation. Before presenting the latest advances in this field, it would be necessary to provide a brief background on P. gingivalis virulence traits.
P. gingivalis is a gram-negative anaerobic and asaccharolytic rod which, besides being a predominant contributor to human periodontitis, is also implicated as an accessory factor in certain systemic conditions, such as atherosclerosis, aspiration pneumonia, and perhaps rheumatoid arthritis 1,6,7). This pathogen is perhaps the most intensively studied oral organism at the molecular level and its pathogenicity is attributed to an array of potential virulence factors, such as cysteine proteinases (gingipains), hemagglutinins, lipopolysaccharide (LPS), and fimbriae, i.e., adhesive hair-like appendages emanating from the bacterial cell surface 8). These molecules are thought to facilitate P. gingivalis initial colonization, retention, and growth within the gingival crevice 9,10).
However, the capacity of any pathogen to secure an appropriate niche and persist requires more than simply possessing virulence factors for tissue adherence and nutrient procurement. Successful human pathogens have also evolved strategies to escape protective immunity, often by manipulating key components of innate immunity, such as the Toll-like receptor and complement systems 11,12). Such subversive strategies enable these pathogens to disable the overall host response, since complement and TLRs play instructive roles in the development of adaptive immunity 13,14).
To establish a chronic infection in the hostile host environment of the gingival crevice, it is imperative that P. gingivalis find ways to evade or subvert host defense mechanisms aiming to eliminate it. For example, P. gingivalis expresses heterogeneous and atypical LPS molecules that either act as potent TLR4 antagonists or are immunologically inert 15). This allows the pathogen to evade or proactively inhibit a variety of potential TLR4-mediated antimicrobial functions, such as inhibition of expression of antimicrobial peptides (β defensins) in human epithelial cells 4). Since P. gingivalis releases LPS-bearing membrane vesicles that can readily diffuse in the crevice or even penetrate gingival tissue 16), the TLR4 antagonistic LPS of P. gingivalis can inhibit TLR4-mediated antimicrobial responses against other bacteria in the same mixed-species biofilm 4). This is just one example of how P. gingivalis can undermine innate immune responses for the microbial community at large.
In contrast to TLR4, P. gingivalis cannot antagonize TLR2 at the receptor level. However, this oral pathogen has evolved the ability to instigate subversive crosstalk interactions between TLR2 and other innate receptors for blunting the TLR2 antimicrobial response. These novel mechanisms are the focus of this review. General mechanisms by which P. gingivalis may undermine innate immunity are summarized in Table 1. Below we describe how this pathogen specifically exploits crosstalk pathways of innate immunity to promote its survival.
Table 1.
Subversion of innate immunity by P. gingivalis
| Mechanism | Effector molecules | Refs. | |
|---|---|---|---|
| 1 | Inhibition of complement activation through digestion of the central complement component (C3) | Gingipains, especially HRgpA and RgpB | 36,37) |
| 2 | Inherent resistance to complement-mediated lysis | LPS with anionic polysaccharide repeat units (A-LPS) | 38,39) |
| 3 | Hijacking complement regulatory proteins (C4b-binding protein) | HrgpA | 40) |
| 4 | Shedding and proteolysis of complement regulatory protein CD46 from oral epithelial cells | Kgp | 41) |
| 5 | TLR4 evasion by expressing dephosphorylated and tetra-acylated lipid A | Lipid A 1-deacylase and 4′-phosphatases and deacylase | 15) |
| 6 | TLR4 antagonism by expressing monophosphorylated tetra-acylated lipid A | Lipid A 4′-phosphatase and deacylase (lipid A 1-phosphatase suppressed by hemin) | 15,42) |
| 7 | Upregulation of negative regulators of TLR signaling (IRAK-M) in monocytes | LPS | 43) |
| 8 | Degradation of TLR coreceptors (CD14), cytokines (IL-12, IL-1β, IL-6, IFN-γ), or antimicrobial peptides (e.g., LL-37) | Gingipains | 28) |
| 9 | Inhibition of phagocyte killing via instigation of C5aR-TLR2 crosstalk | HRgpA, RgpB | 27,29) |
| 10 | Inhibition of phagocyte killing via instigation of CXCR4-TLR2 crosstalk | Fimbriae | 26) |
| 11 | Suppression of TLR2-induced IL-12 via CR3 binding | Fimbriae | 24) |
| 12 | Promotion of intracellular survival via CR3-mediated entry | Fimbriae | 23) |
| 13 | Counteraction of oxidative damage; resistance to environmental oxidative stress and oxidative killing by phagocytes | Rubrerythrin (nonheme iron protein), alkyl hydroperoxide reductase, FeoB2 (ferrous iron transport protein) | 44,45) |
Specifically, I will discuss the ability of P. gingivalis to promote its adaptive fitness through instigation of crosstalk interactions between TLR2 and three important innate receptors, the complement anaphylatoxin C5a receptor (C5aR), the complement receptor 3 (CR3), and the CXC-chemokine receptor 4 (CXCR4).
Induction and exploitation of the TLR2-CR3 crosstalk pathway
The TLR system senses P. gingivalis primarily through TLR2 both in vitro and in vivo, whereas TLR4 is not playing an important role 17,18). As alluded to above, this is because the pathogen expresses atypical lipopolysaccharide molecules. Specifically, the bacterium utilizes lipid A 1-and 4-phosphatases and a deacylase which in concert generate a tetra-acylated and dephosphorylated lipid A structure that is biologically inert 15). At the same time, this modification confers protection against polymyxin B and perhaps other cationic anti-microbial peptides 15). Moreover, high concentrations of hemin (as can be found in inflamed periodontal sites) suppress the lipid A 1-phosphatase activity and lead to generation of a mono-phosphorylated lipid A, which acts as a TLR4 antagonist 15). Although P. gingivalis does not directly antagonize TLR2, it has evolved strategies to exploit TLR2 signaling to its own advantage.
Following activation by P. gingivalis, TLR2 induces two distinct downstream signaling cascades 19). One of the pathways is MyD88-dependent and leads to induction of mostly nuclear factor-κB-dependent proinflammatory and antimicrobial responses. The other is a proadhesive pathway that leads to the induction of the high-affinity conformation of CR3 20). Specifically, P. gingivalis induces TLR2 inside-out signaling, which proceeds through Rac1, phospatidylinositol-3-kinase (PI3K), and cytohesin-1, which acts as the ultimate effector that transactivates CR3 19,21) (Fig. 1).
Figure 1. P. gingivalis subverts crosstalk pathways between TLRs and other innate immune receptors.
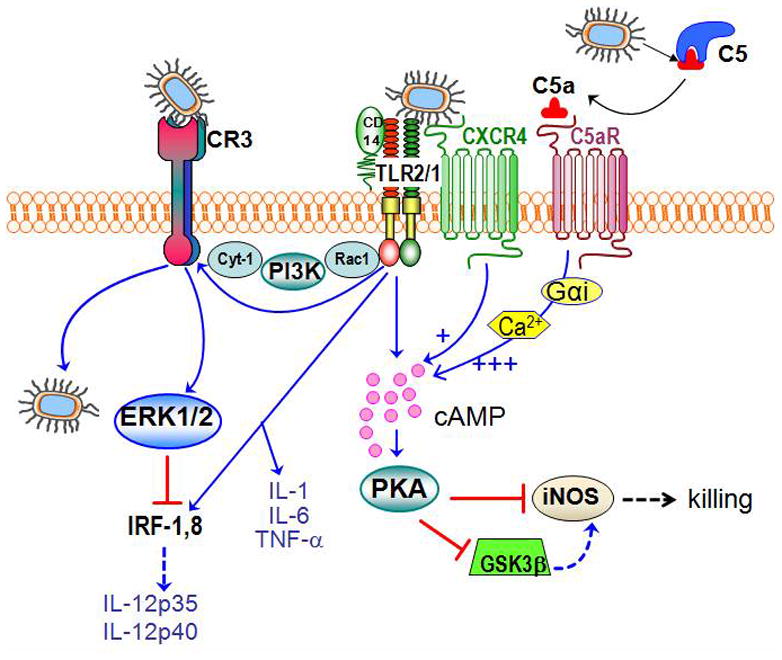
In macrophages, P. gingivalis is recognized by the CD14/TLR2/TLR1 receptor complex 17). This interaction activates inside-out signaling, propagated by Rac1, PI3K, and cytohesin-1 (Cyt-1), which induces the high-affinity conformation of CR3 20,35). CR3 then binds and internalizes P. gingivalis; this is a relatively safe portal of entry since CR3 is not linked to vigorous microbicidal mechanisms. The CR3-P. gingivalis interaction also leads to induction of ERK1/2 signaling. This in turn downregulates IL-12 p35 and p40 mRNA expression 24), possibly through suppression of a critical transcription factor (the interferon regulatory factors 1; IRF-1), required for IL-12 expression 14). This suppressive effect is specific for IL-12 and does not affect induction of other proinflammatory cytokines (e.g., IL-1β, IL-6, and TNF-α). Inhibition of bioactive IL-12 by this mechanism results in impaired immune clearance of P. gingivalis in vivo 24). Moreover, P. gingivalis uses its gingipains to attack C5 and release biologically active C5a 27,29). Upon C5aR binding, C5a stimulates Gαi-dependent intracellular Ca2+ signaling which synergistically enhances the otherwise weak cAMP responses induced by TLR2/TLR1 activation alone. Maximal cAMP induction is achieved by the participation of another G protein-coupled receptor, the CXCR4, which interacts directly by P. gingivalis and coassociates with both TLR2 and C5aR in lipid rafts 26,27). The ensuing activation of the cAMP-dependent protein kinase A (PKA) pathway inactivates glycogen synthase kinase-3β (GSK3β) and impairs the inducible nitrogen synthase (iNOS)-dependent killing of the pathogen in macrophages in vitro and in vivo 27).
P. gingivalis can then bind transactivated CR3 by means of its fimbriae and induces its phagocytic uptake by macrophages 22). However, CR3 is not linked to vigorous microbicidal mechanisms, possibly because this receptor is heavily committed with phagocytosis of iC3b-coated apoptotic cells, which are not normally recognized as danger 14). Consistent with this, CR3-mediated phagocytosis does not promote the killing of P. gingivalis 23). In fact, P. gingivalis enhances its in vivo survival by exploiting CR3; conversely, pharmacological inhibition or genetic ablation of CR3 greatly facilitate its killing 24).
Additional CR3-dependent mechanisms, however, may contribute to this in vivo evasion of immune clearance. In this regard, CR3 ligation by P. gingivalis induces outside-in signaling and extracellular-signal-regulated kinase 1/2 activation, which in turn selectively suppresses IL-12 production through inhibition of mRNA expression of the IL-12 p35 and p40 subunits 24). In vivo, CR3-deficient mice elicit higher levels of IL-12 (and secondarily increased interferon-γ production) and clear infection with P. gingivalis more efficiently than wild-type controls 24). In this evasion strategy, P. gingivalis appears to have co-opted a natural immunosuppressive mechanism, since induction of IL-12 is similarly inhibited during phagocytosis of apoptotic cells by macrophages 14). From a translational viewpoint, pharmacological blockade of CR3 suppresses P. gingivalis-induced periodontal bone loss in a mouse model 24). In the context of the periodontitis-atherosclerosis connection and the observation of viable P. gingivalis in atherosclerotic plaques 1,7), it is intriguing to hypothesize that the intracellular persistence of P. gingivalis in macrophages 23) might allow this organism to exploit these cells as ‘Trojan horses’ to relocate to systemic tissues and subsequently infect permissive cells (e.g., endothelial cells). Although this remains to be tested, the capacity of P. gingivalis for cell exit and infection of new host cells has been demonstrated 25).
Hijacking of complement-TLR2 crosstalk signaling
Although P. gingivalis cannot antagonize TLR2 at the receptor level as it does with TLR4 (see above), it has evolved the ability to intercept and undermine a subset of TLR2 signaling events for corrupting innate immunity 23,24,26,27). This section will discuss how this oral bacterium exploits complement and its capacity to crosstalk with the TLR system for inhibiting specific aspects of TLR2 immunity.
It has been firmly established that P. gingivalis degrades C3 and inhibits the complement cascade regardless of the initiation pathway involved 28). Strikingly, however, the gingipain enzymes of this bacterium (specifically HRgpA and RgpB) act in a C5 convertase-like manner to generate biologically active C5a. In fact, P. gingivalis can rapidly generate high levels of C5a (> 30 nM) in heat-inactivated human serum 27). This seems counterproductive for the survival of this pathogen, since C5a is probably the most potent effector of the complement cascade and generally promotes host defense. For example, C5a induces chemotactic recruitment and activation of leukocytes 14). Stunningly, however, P. gingivalis exploits C5a to undermine TLR2 immunity: Mechanistically, upon C5aR binding, C5a stimulates Gαi-dependent intracellular Ca2+ signaling which synergistically enhances an otherwise weak cAMP response induced by P. gingivalis-induced TLR2 activation alone. In this crosstalk pathway, sustained elevated production of cAMP leads to the activation of the cAMP-dependent protein kinase A (PKA) which inactivates the glycogen synthase kinase-3β (GSK3β) and impairs nitric oxide-dependent killing of P. gingivalis in macrophages 27) (Fig. 1).
The above discussed in vitro evasion mechanism is supported by in vivo observations. Mice with genetic deficiency of C5aR, or with pharmacological inhibition of the same receptor, elicit higher levels of nitric oxide and clear P. gingivalis more effectively than untreated normal controls 27). Moreover, the P. gingivalis-induced C5aR-TLR2 crosstalk also regulates cytokine production in favor of the pathogen 29). Specifically, this oral bacterium proactively and selectively inhibits TLR2-induced IL-12p70, whereas the same C5aR-TLR2 crosstalk upregulates other inflammatory and bone-resorptive cytokines (IL-1β, IL-6, and TNF-α). In vivo, the ability of P. gingivalis to manipulate TLR2 activation via the C5a-C5aR axis allows it to escape IL-12p70-dependent immune clearance and to cause inflammatory bone loss in a murine model of experimental periodontitis 29). Therefore, P. gingivalis targets C5aR not only to promote its adaptive fitness but also to cause periodontal disease. This has profound implications for the treatment of periodontal disease given the current availability of safe and effective C5aR antagonists 14). Specifically, C5aR antagonists have the potential to both promote the killing of P. gingivalis and to inhibit the host inflammatory response.
Because the C5aR-TLR2 crosstalk inhibits only a subset of TLR2 signaling events, C5aR was characterized as a ‘TLR modulatory receptor’ to differentiate it from ‘TLR inhibitory receptors’. Activation of such receptors, for example the IL-10 receptor and the TGF-β receptor, can block most, if not all, inflammatory responses 30). In fact, it would not be in P. gingivalis ‘best interest’ to induce a generalized immunosuppression. This is because inflammation brings in nutrients (present in the gingival crevicular fluid) and thus contributes to its growth and survival 10). In this regard, P. gingivalis-generated C5a may cause vasodilation and stimulation of inflammatory exudate for acquisition of essential nutrients like hemin 31). Interestingly, unlike C5a, the C5b remnant is readily degraded by P. gingivalis gingipains to ostensibly prevent activation of the terminal complement pathway and formation of the membrane attack complex.
On the basis of these novel findings, there is sufficient rationale for the use of C5aR antagonists to control P. gingivalis infections and to also inhibit periodontal inflammation.
Instigation of CXCR4-TLR2 subversive crosstalk
In the absence of C5aR signaling, P. gingivalis uses an alternative mechanism to interfere with TLR2-dependent antimicrobial responses in macrophages. In this case, the pathogen induces a crosstalk between TLR2 and CXCR4 26). Specifically, the concomitant activation of CXCR4 and TLR2 by the fimbriae of P. gingivalis induces cAMP-dependent PKA signaling, which in turn suppresses TLR2-dependent nitric oxide in response to the pathogen 26) (Fig. 1).
The biological relevance of the CXCR4-TLR2 crosstalk was confirmed in vivo. Specifically, mice treated with a selective CXCR4 antagonist display increased production of nitric oxide and enhanced ability to clear infection with P. gingivalis compared to untreated control mice 26). Intriguingly, CXCR4-TLR crosstalk interactions have been recently described also in zebrafish. Here, CXCR4 interfered with toxic effects of LPS activation of TLR4 32).
There is adequate evidence that P. gingivalis can integrate the CXCR4-TLR2 and C5aR-TLR2 crosstalk pathways for enhanced immune subversion. In this regard, confocal microscopy and fluorescence resonance energy transfer analysis indicates that all three receptors, CXCR4, TLR2, and C5aR, co-associate in the lipid rafts of P. gingivalis-challenged macrophages 27). Although the C5aR-TLR2 crosstalk can proceed independently of CXCR4 and potently upregulate cAMP, maximal cAMP induction requires cooperation of all three receptors 27). The following integrative model is proposed to describe combined CXCR4- and C5aR-mediated manipulation of TLR2 by P. gingivalis: The bacterium induces a weak cAMP response by acting on TLR2 alone, whereas activation of CXCR4 or C5aR signaling alone fails to induce cAMP. On the other hand, P. gingivalis-induced TLR2 signaling with concomitant activation of C5aR and CXCR4 synergistically enhances cAMP-PKA signaling that inactivates GSK3β and impairs iNOS-dependent killing (Fig. 1).
Summary and conclusions
In the course of evolution successful pathogens have ‘learned’ to breach innate defense systems such as complement and TLRs 11,33) but also, as exemplified here with P. gingivalis, to exploit their communication hubs 34). These subversive strategies of P. gingivalis (Table 1 & Fig. 1) may explain, at least in part, its ability to persist and establish chronic infections in the periodontium. A keystone pathogen is expected to modify the immune selective pressure in ways that stabilize the microbial community in which it resides. In this regard, P. gingivalis’ tactics to undermine innate immunity may promote the survival of other members of the periodontal biofilm community 5,10). This capacity is reinforced by the fact that P. gingivalis releases easily diffusible membrane vesicles that contain key virulence factors like gingipains, LPS, and fimbriae, which can thus become available to other bacteria within the same biofilm 4,16). Stimulation of inflammatory responses that do not kill (e.g., C5a-induced inflammation) can moreover result in acquisition of essential nutrients (e.g., gingival crevicular fluid-derived peptides and hemin, a source of iron) 31). Therefore, it is reasonable to suggest that periodontal pathogens have evolved in ways that allow them to not only endure inflammation but also exploit it for promoting their survival and, collaterally, inflicting periodontal tissue damage. In conclusion, pathogen manipulation of periodontal innate immunity may perturb otherwise homeostatic host-bacterial interactions, thereby leading to non-protective and non-resolving chronic inflammation in the periodontium. On the other hand, elucidation of the mechanisms by which periodontal bacteria interfere with immune clearance mechanisms and induce nonproductive inflammation, would facilitate the rational design of therapeutic interventions in periodontitis.
Acknowledgments
The author is supported by grants from the U.S. National Institutes of Health DE015254, DE018292, DE017138, and DE021580.
References
- 1.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 2.Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: rethinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res. 2008;87:817–828. doi: 10.1177/154405910808700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 4.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 5.Darveau RP. The oral microbial consortium’s interaction with the periodontal innate defense system. DNA Cell Biol. 2009;28:389–395. doi: 10.1089/dna.2009.0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010;6:727–730. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
- 7.Kebschull M, Demmer RT, Papapanou PN. “Gum bug leave my heart alone”--Epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010;89:879–902. doi: 10.1177/0022034510375281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshimura F, Murakami Y, Nishikawa K, Hasegawa Y, Kawaminami S. Surface components of Porphyromonas gingivalis. J Periodontal Res. 2008;44:1–12. doi: 10.1111/j.1600-0765.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 9.Lamont RJ, Jenkinson HF. Subgingival colonization by Porphyromonas gingivalis. Oral Microbiol Immunol. 2000;15:341–349. doi: 10.1034/j.1399-302x.2000.150601.x. [DOI] [PubMed] [Google Scholar]
- 10.Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637–645. doi: 10.1016/j.micinf.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 13.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 14.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4′-phosphatase activities. Cell Microbiol. 2009;11:1587–1599. doi: 10.1111/j.1462-5822.2009.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amano A, Takeuchi H, Furuta N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010;12:791–798. doi: 10.1016/j.micinf.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S-I, Ratti P, Schifferle RE, Lyle EA, Triantafilou M, Triantafilou K, Yoshimura F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- 18.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 19.Hajishengallis G, Wang M, Liang S. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J Immunol. 2009;182:6690–6696. doi: 10.4049/jimmunol.0900524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- 21.Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J Immunol. 2006;176:7645–7656. doi: 10.4049/jimmunol.176.12.7645. [DOI] [PubMed] [Google Scholar]
- 22.Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect Immun. 2006;74:5658–5666. doi: 10.1128/IAI.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Shakhatreh MAK, James D, Liang S, Nishiyama S-i, Yoshimura F, Demuth DR, Hajishengallis G. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- 24.Hajishengallis G, Shakhatreh MAK, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- 25.Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897–2903. doi: 10.1099/mic.0.2008/021220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A. 2008;105:13532–13537. doi: 10.1073/pnas.0803852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, Triantafilou M, Triantafilou K, Lambris JD, Hajishengallis G. Microbial hijacking of complement-Toll-like receptor crosstalk. Sci Signal. 2010;3:ra11. doi: 10.1126/scisignal.2000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potempa J, Pike RN. Corruption of innate immunity by bacterial proteases. J Innate Immun. 2009;1:70–87. doi: 10.1159/000181144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, Li F, Tzekou A, Lambris JD, Hajishengallis G. The C5a receptor impairs IL-12–dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2011;186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kagan JC. “Complementing” toll signaling. Sci Signal. 2010;3:pe15. doi: 10.1126/scisignal.3120pe15. [DOI] [PubMed] [Google Scholar]
- 31.Krauss JL, Potempa J, Lambris JD, Hajishengallis G. Complementary Tolls in the periodontium: how periodontal bacteria modify complement and Toll-like receptor responses to prevail in the host. Periodontol 2000. 2010;52:141–162. doi: 10.1111/j.1600-0757.2009.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novoa B, Bowman TV, Zon L, Figueras A. LPS response and tolerance in the zebrafish (Danio rerio) Fish Shellfish Immunol. 2009;26:326–331. doi: 10.1016/j.fsi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy CR, Mocarski ES. Pathogen subversion of cell-intrinsic innate immunity. Nat Immunol. 2007;8:1179–1187. doi: 10.1038/ni1528. [DOI] [PubMed] [Google Scholar]
- 34.Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nature Rev Immunol. 2011 doi: 10.1038/nri2918. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hajishengallis G, Harokopakis E. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion? Front Biosci. 2007;12:4547–4557. doi: 10.2741/2409. [DOI] [PubMed] [Google Scholar]
- 36.Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. 2007;178:7242–7250. doi: 10.4049/jimmunol.178.11.7242. [DOI] [PubMed] [Google Scholar]
- 37.Potempa M, Potempa J, Kantyka T, Nguyen KA, Wawrzonek K, Manandhar SP, Popadiak K, Riesbeck K, Eick S, Blom AM. Interpain A, a cysteine proteinase from Prevotella intermedia, inhibits complement by degrading complement factor C3. PLoS Pathog. 2009;5:e1000316. doi: 10.1371/journal.ppat.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slaney JM, Gallagher A, Aduse-Opoku J, Pell K, Curtis MA. Mechanisms of resistance of Porphyromonas gingivalis to killing by serum complement. Infect Immun. 2006;74:5352–5361. doi: 10.1128/IAI.00304-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rangarajan M, Aduse-Opoku J, Paramonov N, Hashim A, Bostanci N, Fraser OP, Tarelli E, Curtis MA. Identification of a second lipopolysaccharide in Porphyromonas gingivalis W50. J Bacteriol. 2008;190:2920–2932. doi: 10.1128/JB.01868-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, Riesbeck K, Blom AM. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol. 2008;181:5537–5544. doi: 10.4049/jimmunol.181.8.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahtout H, Chandad F, Rojo JM, Grenier D. Porphyromonas gingivalis mediates the shedding and proteolysis of complement regulatory protein CD46 expressed by oral epithelial cells. Oral Microbiol Immunol. 2009;24:396–400. doi: 10.1111/j.1399-302X.2009.00532.x. [DOI] [PubMed] [Google Scholar]
- 42.Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175:4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- 43.Domon H, Honda T, Oda T, Yoshie H, Yamazaki K. Early and preferential induction of IL-1 receptor-associated kinase-M in THP-1 cells by LPS derived from Porphyromonas gingivalis. J Leukoc Biol. 2008;83:672–679. doi: 10.1189/jlb.0607432. [DOI] [PubMed] [Google Scholar]
- 44.Mydel P, Takahashi Y, Yumoto H, Sztukowska M, Kubica M, Gibson FC, 3rd, Kurtz DM, Jr, Travis J, Collins LV, Nguyen KA, Genco CA, Potempa J. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006;2:e76. doi: 10.1371/journal.ppat.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheets SM, Robles-Price AG, McKenzie RM, Casiano CA, Fletcher HM. Gingipain-dependent interactions with the host are important for survival of Porphyromonas gingivalis. Front Biosci. 2008;13:3215–3238. doi: 10.2741/2922. [DOI] [PMC free article] [PubMed] [Google Scholar]