

Abstract
Childhood HIV-1 associated nephropathy (HIVAN) is a clinical and renal histological disease characterized by heavy proteinuria associated with focal and segmental glomerular sclerosis and/or mesangial hyperplasia in combination with microcystic tubular dilatation. These lesions lead to renal enlargement and rapid progression to kidney failure. Children of African ancestry have a unique susceptibility to developing HIVAN. It is estimated that approximately 300,000 HIV-infected children living in the sub-Saharan Africa could develop HIVAN if they do not receive appropriate antiretroviral therapy. This article discusses recent developments and controversies related to the pathogenesis of childhood HIVAN. The role of host genetic factors, including the newly identified variants in the APOL1 gene, is discussed in the context of previous studies that established the pathological paradigm for HIVAN, and our current understanding of the functional genomics analysis. Hopefully, these advances will provide new research opportunities to generate better treatments for children with HIVAN.
Keywords: African–American, APOL1, autophagic cell death, autophagy, Duffy antigen, genetic susceptibility, HIVAN, kidney failure, nonsynonymous SNP
African–Americans represent approximately 65% of all children with HIV-1 infection or AIDS in the USA [1,2]. HIV-1 associated nephropathy (HIVAN) affects the clinical outcome, quality of life, and survival of HIV-infected children [1,2]. Unfortunately, despite the effective antiretroviral therapies (ART) available, we continue to see newly diagnosed children with HIVAN in the USA. In addition, an estimated 2.3 million children living with HIV in sub-Saharan Africa, are at high risk of developing HIVAN if they do not receive appropriate ART [2]. Moreover, young children represent a unique group in which to study the pathogenesis of HIVAN because they are usually infected through vertical transmission and do not develop many of the confounding comorbidities and renal diseases seen in adult patients [1,2]. For all these reasons, it is necessary to understand the basic mechanisms involved in the pathogenesis of childhood HIVAN, in order to develop better strategies to prevent or ameliorate the progression of this renal disease. This review will highlight the progress made during the last few years, and discuss how these findings may affect our understanding of HIVAN in children. Other pediatric renal diseases associated with HIV infection, including IgA nephropathy, immune complex glomerulonephritis and proliferative glomerulonephritis, are not part of HIVAN, and will not be discussed in this review.
Clinical & renal histological lesions
HIV-1 associated nephropathy is a clinical and renal histological disease characterized by the presence of heavy proteinuria and nephrotic syndrome leading to a rapid progression to chronic renal failure [1,3,4]. The classic renal pathological findings in HIVAN are focal and segmental glomerulosclerosis (FSGS) with hypertrophy and hyperplasia of glomerular epithelial cells, tubuloreticular inclusions in renal endothelial cells, and microcystic tubules filled with proteinaceous material and infiltrating mononuclear cells [1,3,4]. Immunofluorescence microscopic findings are negative or non-specific [1,3,4]. The microcystic tubular changes lead to renal enlargement, a finding that contrasts with the small fibrotic kidneys typically seen in patients with chronic renal disease of other etiologies. Another feature characteristic of HIVAN in adults is the presence of collapsing glomerulopathy. However, this lesion can be seen in HIV-negative patients, and children usually develop the classic type of FSGS [1,2]. In addition, at the onset of nephrotic proteinuria, children may only exhibit mesangial hyperplasia in combination with microcystic tubular dilatation (Figure 1) [1,2]. It is worth mentioning the possibility that in some cases, what is being seen as mesangial proliferation could in fact be collapsing FSGS disguised. However, children with mesangial hyperplasia progress at a slower rate when compared to those with classic or collapsing FSGS [1]. As previously discussed [5], it is unclear whether mesangial cells can be infected by HIV-1 in vivo, and more studies are needed to understand the pathogenesis of mesangial hyperplasia in HIV-infected children. In addition, it remains to be determined whether these changes could be the first step toward the development of FSGS, or whether they can evolve as an independent pathogenic process associated with the viral infection. In summary, HIVAN is a similar condition in adults and children, but children may respond to HIV-1 in a different manner because their tissues and immune system are undergoing growth and developmental changes. In addition, HIV-infected children are usually not exposed to many of the several comorbidities, drugs, or risk factors that affect adult patients.
Figure 1.

Representative hematoxylin and eosin staining of a renal biopsy from a child with HIV associated nephropathy.
Role of HIV-genes in HIVAN
The HIV-1 genome contains at least nine genes that encode viral proteins [6]. At least four of these genes, env, tat, nef and vpr, have been linked to the pathogenesis of the renal disease [5,7,8]. The env gene encodes for HIV-1 proteins gp120 and gp41, which constitute the viral envelope and form the surface unit of HIV-1 as it ‘buds’ out from cells [6,9]. gp120 binds to chemokine receptors and induces signaling pathways in many cell types [6]. Tat is a transcription factor that is essential for viral replication, but can also be released and taken up by uninfected cells, where it can bind to heparan sulfate proteoglycans (HSPGs) and activate several host genes [5,10]. Both gp120 and Tat are present in the circulation of HIV-infected patients and can activate the immune system and induce endothelial dysfunction [9,11]. The nef gene product helps HIV-1 escape host immunity by downregulating CD4 and other cell surface receptors [6]. Nef also activates several signaling pathways involved in cell growth and differentiation [12], and can induce the proliferation and de-differentiation of immortalized murine podocytes [13]. In addition, Nef can also affect the expression of the tumor suppression gene p53 [14], which normally inhibits the proliferation of podocytes [15]. However, to date, the mitogenic effect of Nef has not been demonstrated in cultured human podocytes. Vpr has multiple effects on host cells, including regulation of cell arrest, apoptosis, cytokine production, and can act as a transcriptional activator or repressor [6]. Vpr can facilitate the entry of viral DNA and proteins into the nucleus [16,17]. In summary, the exact mechanism by which HIV genes affect the outcome of HIVAN in children is not fully understood.
Host genetic variations in people with HIVAN
Host genetic factors appear to play an essential role in the pathogenesis of HIVAN [18–20]. People of African descent are at markedly, >18-fold-increased risk for developing HIVAN compared with people of European descent [18]. One of the first host genetic factors explored to explain the susceptibility of people from African ancestry to develop HIVAN, was the ‘Duffy negative phenotype’ [21,22]. This phenotype is determined by a mutation in the Duffy antigen receptor for chemokines (DARC), which disrupts the binding site for a transcription factor (GATA-1) that is required to express DARC on the surface of red blood cells [23]. Previous studies showed that DARC may promote the clearance of and/or neutralize the activity of inflammatory chemokines [24], and can bind HIV-1 particles [25–27]. Thus, people carrying the Duffy/DARC negative genotype might not be able to neutralize circulating chemokines and/or HIV-1 in an effective manner. Children with HIVAN show increased expression of DARC in glomerular endothelial and tubular epithelial cells [21], and these changes may facilitate the recruitment of HIV-infected cells, and increase its renal toxicity. However, one study done in HIV-infected adults found no association between the Duffy negative phenotype and HIVAN [22]. Alternatively, recent studies have explored the role of Duffy negative phenotype in the progression of HIV infection [27–31]. To date, the causative link between the Duffy/DARC negative phenotype and the potential outcome of HIVAN or HIV infection is unclear, and more studies are needed to determine how DARC may affect the clinical outcome of childhood HIVAN.
Host genetic factors in a region of chromosome 22q12 in HIVAN
Familial clustering of various kidney diseases including hypertension and diabetic-attributed end-stage kidney disease (ESKD), idiopathic FSGS, and HIVAN are often observed among populations of African descent. In the past few years, linkage analysis and mappingm by admixture linkage disequilibrium have localized a region in chromosome 22q12 that includes the MYH9 gene, and harbors risk variants associated with FSGS, ESKD, and HIVAN in people of African ancestry [18,20,32]. MYH9 encodes nonmuscle myosin IIA heavy chain, a cytoskeletal motor protein expressed in many cell types, including glomerular podocytes. Dense mapping of MYH9 has identified individual single nucleotide polymorphisms (SNPs) and groups of such SNPs as haplotypes that were found to be highly associated with ESKD risk phenotypes [33]. Previously, a number of mutations in the coding exons of MYH9 have been identified in patients with dominantly inherited giant platelet syndrome [34,35]. Two specific MYH9 variants (rs5750250 or S-haplotype and rs11912763 or F-haplotype), both in noncoding introns, were designated as most strongly predictive for ESKD on the basis of Receiver Operating Characteristic analysis. Despite intensive efforts including resequencing of the MYH9 gene, no functional/pathological causative mutations have been identified in people with HIVAN or idiopathic FSGS. Interestingly, a recent study showed that although C57BL/6 mice carrying a podocyte-specific deletion of the MYH9 gene did not develop proteinuria or renal insufficiency, they were predisposed to develop glomerulosclerosis when stressed with adriamycin [36]. These findings suggest that lack of expression of MYH9 in podocytes, together with a second (environmental or genetic) provocation/stress, could induce renal disease.
In 2010, using mappingm by admixture linkage disequilibrium and database mining in 1000 Genomes Project, three independent studies demonstrated that human APOL1 variants are associated with FSGS and ESKD [19,37,38]. In sum, two haplotypes, harboring three coding sequence mutations of APOL1, have been identified as risk variants. The first one, termed G1, is a two-nonsynonymous-SNP haplotype (rs73885319 (A→G; S342G) and rs60910145 (G→T; I384M). The second one, termed G2, is a two-codon-deletion haplotype (rs71785313 (6-bp in frame deletion; DN388Y389). All three of these mutations are located in the last/7th exon. In addition, it has been demonstrated that the distribution of the APOL1 G1 haplotype in African populations is consistent with the pattern of African ancestry risk of developing ESKD previously attributed to MYH9 [38]. Importantly, both G1 and G2 variants, located approximately 14 kb 3′ downstream from MYH9, are in linkage disequilibrium with common MYH9 SNPs known to confer a greater risk of FSGS and nondiabetic ESRD. Based on odds ratios and p values, APOL1 risk variants are more strongly associated with ESKD than the leading MYH9 risk variants (S-haplotype and F-haplotype). Furthermore, we have recently reported that apolipoprotein L1 (ApoL1) is a novel BH3-only, phospholipid-binding, pro-death protein that, when overexpressed intracellularly, induces autophagy and autophagic cell death in all cell types examined thus far, including those originating from normal kidney and cancerous tissues [39–41]. However, the normal function of APOL1, and/or the possible pathological consequences of APOL1 risk alleles in kidney cells have not been investigated. To explore this issue, we conducted a functional genomics analysis using NCBI RefSeqs Build 37.2, and showed that the human APOL1 gene encodes at least four putative human mRNA variants, from which three protein isoforms, ApoL1.a, ApoL1.b and ApoL1.c., can be made. ApoL1.a, a 398 amino-acid (aa) polypeptide, is the most abundant and relatively well-studied protein (Figure 2). We expect that the functional expression system we have established previously [39] will be valuable to further characterize the roles of the wild type, G1 and G2 alleles in kidney cells.
Figure 2. Sequences of three human ApoL1 isoforms and their naturally occurring nonsynonymous alleles.

ApoL1.a, a 398 amino-acid (aa) polypeptide, is shown in its complete sequence. Resulting from alternatively splicing, ApoL1.b, 414 aa, is 16 aa longer than ApoL1.a at the N-terminal (first line), while ApoL1.c, 380 aa, is missing a 18 aa motif (underlined) close to the N-terminal of ApoL1.a. Thus far, 14 naturally occurring, nonsynonymous alleles of APOL1 have been identified. According to residue numbers of ApoL1.a, they are N95K, G96R, K142fs, E150K, N176S, M218I, R255K, N264K, L266fs, G270D, D337N, S342G, I384M and ΔN388Y389 (aa changes are underwritten). The BH3 (aa 158–166) and leucine zipper (aa 365–392) domains are in bold and underlined. The two risk alleles of APOL1 in ESKD in African–Americans, S342G/I384M and ΔN388Y389, are in the C-terminal domain (aa 338–398), which interacts with trypanomosal serum resistance-associated protein. I384M mutation is also present in the leucine zipper domain.
The first report describing the cloning and characterization of human ApoL1 was published in 1997 [42]. This study showed that extracellular ApoL1 interacts with apolipoprotein A1 (ApoA1), as a component of circulating high density lipoprotein complexes in human plasma [42]. Subsequently, using immunoblot and immunohistochemical analyses, we and others have shown the intracellular expression and localization of ApoL1 in a number of cells and tissues [39,40,201]. In general, kidney tubular epithelial cells, pancreatic exocrine granular cells and smooth muscle cells have strong ApoL1 expression, whereas other cell types have weak to very low expression. As an illustration (Figure 3), a kidney section showed a remarkable expression of ApoL1 in tubular epithelial cells. In addition, expression of APOL1 can be induced by TNF-α, IFN-γ and p53 in various cell types [40]. Taken together, these findings suggest that cytokines released by HIV-infected cells may upregulate the expression of ApoL1 in renal tubular epithelial cells. Furthermore, as discussed above, it has been proposed that HIVAN arises due to HIV-1-induced dysregulation of glomerular parietal epithelial cells and podocytes. Interestingly, a recent report showed that podocytes have high levels of autophagic homeostasis for cellular integrity and survival [43]. Autophagy-deficient podocytes exhibited glomerulopathy in aging mice, suggesting that autophagy is a major protective means to maintain functional podocytes and therefore kidneys [43]. It is conceivable that if ApoL1 plays an important role in regulating autophagy in kidney cells, both gain-of-function and loss-of-function mutations of APOL1 may disrupt ApoL1-dependent cellular homeostasis, leading to apoptosis, and other forms of cell death. In this manner, in patients homozygous for APOL1 G1 or G2 alleles, HIV-1 may act as a second provocation stress to trigger apoptosis in renal epithelial cells and induce HIVAN. Moreover, our advanced genomic and SNP analysis identified additional nine nonsynonymous-SNPs and two frameshift deletion variants in the coding exons of the human APOL1 structural gene, N95K, G96R, K142fs, E150K, N176S, M218I, R255K, N264K, L266fs, G270D and D337N (Figure 2). It is logical to speculate that the protein products of some of these alleles of APOL1 may have altered activity, stability or subcellular localization, resulting in cytotoxicity.
Figure 3. Representative immunohistochemistry staining showing ApoL1 expression in human renal tubular epithelial cells.
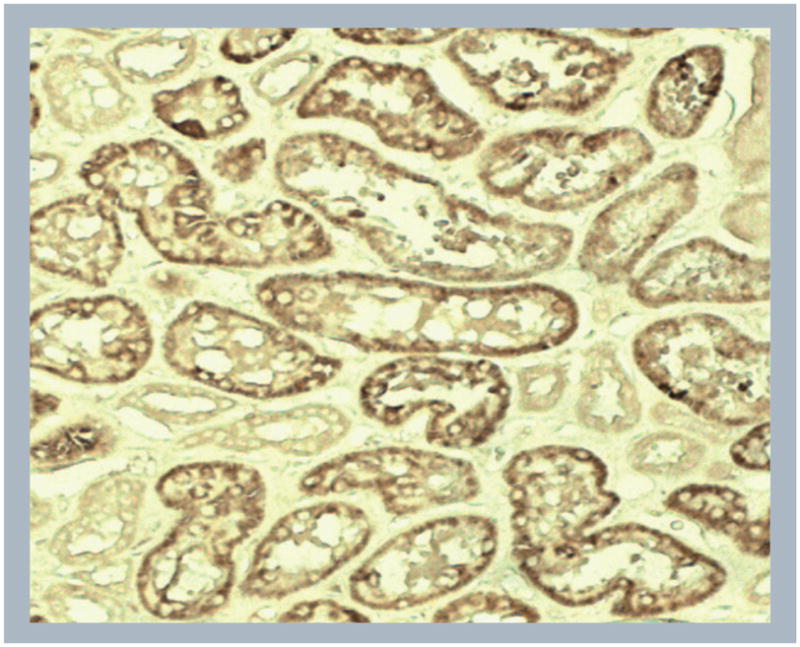
The ApoL1 antibody was generated and used as previously described [34].
With regard to other extracellular functions of ApoL1, intensive studies conducted by a number of parasite-focused groups demonstrated that ApoL1, functioning as the trypanolytic factor, kills the African trypanosome Trypanosoma brucei brucei, except two subspecies adapted to humans, T. b. rhodesiense, T. b. gambiense [44]. Resistance to ApoL1 is conferred by a trypanosomal protein known as serum resistance-associated protein (SRA), which is a lysosomal protein that interacts strongly with a carboxy-terminal α-helix of ApoL1 [45]. ApoL1 is taken up through the endocytic pathway into the lysosome of trypanosomes, and causes osmotic swelling of the lysosome until the trypanosome dies [46]. Interestingly, the two risk alleles of APOL1 in ESKD in African–Americans, S342G/I384M (G1) and ΔN388Y389 (G2), are in the C-terminal domain (Figure 2), which interacts with trypanosomal SRA. Consequently, these two mutant alleles lose binding affinity with SRA and are trypanolytic. Importantly, the I384M mutation is also present in the leucine zipper domain and therefore may disrupt the interaction of ApoL1 with other human proteins that lead to pathological consequences. However, whether extracellular or intracellular ApoL1 proteins produced by these two haplotypes are ‘toxic’ to kidney cells is currently unknown. It is worth noting that there are two other protein isoforms of ApoL1 that can be generated from alternative splicing, ApoL1.b (414 aa), 16 aa longer than ApoL1.a at the N-terminus, and ApoL1.c (380 aa) missing a 18 aa motif close to the N-terminus of ApoL1.a (Figure 2). The function of both isoforms is not clearly understood. Thus, future studies need to elucidate the normal function of ApoL1 isoforms in kidney cells, the crosstalk between ApoL1, HIV-1, and several host inflammatory proteins, as well as the clinicopathological consequences of G1, G2 and other nonsynoynmous mutations of APOL1.
Finally, APOL1 may affect other functions that are involved in the pathogenesis of kidney disease and hypertension, including lipid metabolism, chronic inflammation and endothelial dysfunction [40]. APOL1 is strongly induced by INF-γ and TNF-α in cultured endothelial cells [40]. To date, very little is known about the role of APOL1 in the pathogenesis of hypertension or chronic kidney failure in children. We speculate that the long-term metabolic consequences of ART in children, may cause changes in lipid metabolism, endothelial dysfunction, and renal injury per se, and therefore, patients carrying the APOL1 risk genotype may be a higher risk of developing hypertension and chronic renal disease in adulthood.
Host genetic factors in HIV-Tg26 mice
A few years after HIVAN was recognized as a new renal disease, three HIV-transgenic (Tg) founding lines were generated and used to support the notion that the expression of viral genes in renal epithelial cells can induce the full HIVAN phenotype in the absence of viral replication and immune dysregulation [47]. One of these mouse lines, named Tg26, was used to show that HIV-1 genes, in combination with endogenous host growth factors, can induce ‘uncontrolled’ hyperplasia of renal tubular epithelial cells, leading to renal enlargement [48]. Since the role of genetic factors in the pathogenesis of HIVAN was studied in HIV-Tg26 mice [49,50], we will review relevant features of this animal model.
The Tg26 mouse line carries a replication-deficient, Δ gag/pol, pNL4–3 HIV proviral genomic DNA driven by the endogenous HIV-1 promoter [47]. The mRNA expression of HIV is detected in a wide range of cells, including renal glomerular and tubular epithelial cells [47]. Viral proteins cannot however, be detected in the circulation or peripheral blood mononuclear cells. HIV-Tg26 mice are born with normal kidneys, and develop a progressive HIVAN-like renal disease. Homozygous mice are usually runted and die during the first month of life, sometimes without developing renal disease [51]. Many heterozygous Tg26 mice develop renal disease by approximately 40 days of life, however, not all mice develop renal disease [48]. During the initial stages of the renal disease, Tg26 mice show moderate proteinuria in association with glomerular and tubular epithelial injury [48]. Subsequently, these mice develop global or focal segmental glomerulosclerosis in combination with microcystic tubular dilatation, leading to renal enlargement. This stage is characterized by the presence of proliferating glomerular and tubular epithelial cells that are not well differentiated and express very low levels of HIV-1 transcripts [48]. Usually, mice died of uremia and/or ascites by approximately 60–80 days of life. Elegant studies done in different mouse strains carrying the Tg26 transgene, confirmed the notion that host genetic factors play a critical role in the pathogenesis of HIVAN [16,49,50,52]. For example, while Tg26 mice generated on the FVB/N mouse strain are susceptible to develop renal disease, Tg26 mice bred onto the mixed FBV/CAST background mouse strain do not developing renal disease [16]. Furthermore, these mice were used to identify several genetic loci responsible for the penetrance of the renal disease [16,50,52]. Taken together, these studies support the notion that in some mouse strains, the expression of HIV-1 transcripts in renal epithelial cells is not sufficient to induce the full HIVAN phenotype.
HIV-Tg rats
This model was generated in Sprague–Dawley rat carrying the Tg26 Δ gag/pol pNL4–3 HIV-transgene [53]. HIV-Tg rats develop a progressive AIDS-like disease, including immunological dysfunction, nephropathy, muscle wasting and skin lesions [53,54]. One advantage of the rat model, relative to HIV-Tg26 mice, is that the Tg26 transgene is more efficiently expressed in rat peripheral blood mononuclear cells, and that gp120 and Tat can be detected in the circulation. In the kidney, only glomerular and tubular epithelial cells express viral transcripts, and these rats develop microcystic tubular dilatation and FSGS. In addition, HIV-Tg rats develop two renal features frequently seen in HIV-infected children; prominent mesangial hyperplasia and renal vascular lesions [54]. Based on these findings, it appears that the pathogenesis of the renal disease in HIV-Tg rats is probably multifactorial, involving the expression of viral transcripts in renal epithelial cells, and the recruitment of circulating viral proteins, heparin binding growth factors, and mononuclear cells expressing HIV genes in the kidney [54]. More recently, HIV-Tg rats were used to show that HIV-1 proteins can induce oxidative stress in several tissues [55–57]. Free iron was also found accumulated in diseased kidneys [58]. Interestingly, HIV-infected children with renal disease also show elevated urinary levels of iron and iron-related proteins, including neutrophil gelatinase-associated lipocalin (NGAL) [58]. Thus, it is tempting to speculate that the accumulation of iron may accelerate the progression of the renal disease by causing oxidative stress. If this hypothesis is correct, both the urinary levels of iron and NGAL might become promising candidate biomarkers to identify HIV-infected children with renal disease and iron accumulation. In support of this notion, one study in HIV-Tg26 mice reveals renal iron accumulation in HIVAN, and upregulation of NGAL in renal tubular epithelial cells undergoing microcystic changes [59]. This study concluded that the urinary NGAL levels may mark the presence of renal microcysts in people with HIVAN, and could aid in the noninvasive diagnosis of HIVAN [59]. In summary, iron could become a therapeutic target against HIVAN, and more studies are needed to determine how much iron supplement should be given to HIV-infected children with renal disease. Alternatively, another study performed in HIV-infected children, showed that the urinary levels of EGF, FGF-2, and matrix metalloproteinase-2, could become a reliable biomarker profile to identify and follow the renal outcome of children with HIVAN [60].
Role of renal & circulating factors in childhood HIVAN
Several Tg mouse models demonstrate that the selective expression of HIV-1 genes in podocytes, including Nef and Vpr, play an important role in the pathogenesis of HIVAN [7,61]. These findings, however, do not exclude the possibility that circulating factors might play an additional role in the pathogenesis of childhood HIVAN, since the processes of viral infection, replication, and immunological response to the virus are all bypassed in HIV-Tg mice. For example, HIV-Tg mice do not develop tubuloreticular inclusion in renal endothelial cells, which is a typical renal feature caused by the host immune response to the virus in people with HIVAN. In addition, HIV-infected children develop progressive endothelial injury in response to a high viral load, circulating viral proteins, and cytokines release by the host immune response [5], and all these factors may affect the outcome of HIVAN. Moreover, the forced expression of high levels of HIV-genes in podocytes may cause renal injury by a different mechanism to that seen in HIV-infected children. Alternatively, Tg mice expressing the full HIV genome, or HIV-nef, under the control of the human CD4 promoter, which restricts the expression of HIV-1 genes mainly to circulating immune cells, can also develop renal microcysts and FSGS [44,62–67]. These findings challenge the notion that the expression of HIV-genes in renal epithelial cells is a necessary condition for developing HIVAN, and suggest that renal infiltrating mononuclear cells carrying HIV-1 genes, and/or cytokines released by these cells, can induce HIVAN without establishing a productive infection of renal epithelial cells. In summary, it is possible that both renal and systemic factors are needed to induce the full HIVAN phenotype in HIV-infected children.
Infection of renal epithelial cells
One critical question that remains to be answered to understand the pathogenesis of childhood HIVAN, is how HIV-1 infects renal epithelial cells in vivo. One study showed that cultured human renal tubular epithelial cells harvested from HIV-infected children can be infected via a CD4-independent process [68]. This event, however, results in the generation of very low levels of viral proteins [68]. Moreover, a high viral load was needed to infect these cells, and the mechanism mediating the process of viral entry remains a mystery [68]. Renal epithelial cells harvested from HIV-infected children do not express detectable protein levels of CD4, the primary HIV-1 receptor, or the coreceptors CCR-5 or CXCR-4. Nevertheless, cell–cell contact interactions with mononuclear cells, facilitate the transfer and entry of HIV-1 to renal epithelial cells [68]. Considering that children with HIVAN show a remarkable upregulation of renal HSPG [69], it is tempting to speculate that HSPGs facilitate the entry of HIV-1 into renal epithelial cells through endocytic pathways, as demonstrated in cultured endothelial cells [70]. Thus, HIV-1 may use HSPGs as low-affinity attachment receptors to concentrate viral particles close to specific entry receptors [70,71]. In support of this notion, a recent study showed that sulfated polysaccharides can block the transfer of viruses from T cells to cultured renal tubular epithelial cells by a mechanism that is not yet clearly understood [72]. Alternatively, macrophages harvested from HIV-infected children express CD4 and HIV coreceptors, and produce high levels of viral proteins [68]. These cells can transfer mature viral particles to renal epithelial cells by a Trojan-horse like mechanism [68], and may establish a clinically relevant renal reservoir in HIV-infected children [5].
In recent years, a variety of cellular binding proteins have been found to be associated with the entry of HIV-1 into human renal glomerular and tubular epithelial cell lines. Among these, the C-type lectin receptor DEC-205 can facilitate the entry of HIV-1 into the HK2 tubular epithelial cell line [73,74]. However, these cells were able to sustain a nonproductive silent infection for a short period of time. In a similar manner, the DC-specific ICAM-3-grabbing nonintegrin can mediate the internalization of HIV-1 into human podocytes [75], but these cells were also not productively infected. To date, it is unclear whether renal epithelial cells from HIV-infected children express significant protein levels of DEC-205 or DC-specific ICAM-3-grabbing nonintegrin. Alternatively, cholesterol and glycosphingolipids within lipid rafts can facilitate the entry of HIV-1 into several cell types, including cultured human podocytes [76]. Interestingly, recent work found that the HIV-1 envelope glycoprotein gp120 can bind with high affinity to the glycosphingolipid globotriaosylceramide (Gb3), which is expressed within ‘nonraft’ fractions in human renal tubular epithelial cells [77]. Renal sections harvested from HIV-Tg26 mice and HIV-infected children with renal disease show a high content of Gb3 in renal tubules [78,79]. Thus, it is tempting to speculate that Gb3 and other glycosphingolipids may modulate the binding and entry of HIV-1 into renal tubular epithelial cells [79]. In other cell types, viral infectivity can be affected by the reciprocal interactions between X4 and R5 viruses and Gb3 expression [80]. In addition, recent findings in mice and humans suggest that glycosphingolipids may play a role in the formation of renal cysts [81]. Thus, further experiments are needed to define whether pharmacological manipulation of the expression of Gb3 and other renal glycosphingolipids may provide a new approach to treat HIVAN. We also hope that future studies will elucidate the conditions and receptors needed to establish a productive infection of human podocytes and tubular epithelial cells in vivo.
Proliferating renal epithelial cells & childhood HIVAN
As discussed above, one distinctive feature of HIVAN is the presence of renal microcysts and proliferating glomerular epithelial cells [1,2]. The basic mechanisms responsible for these changes are not completely understood, and have been the subject of extensive studies and controversies. One current leading hypothesis proposes that HIV-1 can induce a productive infection of podocytes, leading to the expression of Nef and subsequent activation of several proliferative signaling pathways [12]. This hypothesis, however, is based on studies done in conditionally immortalized murine podocytes [13] and/or Tg mice [16]. Thus, they need to be validated in human epithelial cells and renal sections harvested from HIV-infected children. At the present time, we do not know whether mature human podocytes have the capacity to de-differentiate and proliferate when exposed to HIV-1 in vivo, and these events have not been reproduced in cultured human podocytes. Moreover, cultured human podocytes cannot be productively infected with HIV-1 [75,82–84], and it is unclear how these cells may produce Nef or other HIV proteins. Alternatively, podocytes exposed to HIV-1 may die, be shed in the urine, and be replaced by glomerular parietal epithelial cells [85,86] or renal stems cells [87–89]. This scenario could have implications for future therapies and is a promising field of research.
Previous studies in HIV-Tg26 mice and rats support the hypothesis that the renal epithelial proliferative changes characteristic of HIVAN, are a late event in the pathogenesis of the disease [48,54]. As discussed before, the expression of HIV-genes during the early stages of the renal disease in HIV-Tg26 mice induces renal epithelial injury [48]. Subsequently, these changes appear to trigger an exaggerated proliferative response involving undifferentiated renal epithelial cells that express almost undetectable levels of HIV-transcripts [48,54]. This response seems to be driven, at least partially, by the renal accumulation of heparin-binding growth factors, including FGF-2 [48,54] and VEGF-A [90]. HIV-Tat, acting from the extracellular space, can facilitate the release of FGF-2 from cultured human podocytes [91] and increase their proliferation and permeability, acting in synergy with VEGF-A [92]. FGF-2 increases the attachment of HIV-infected cells to renal tubular epithelial cells harvested from HIV-infected children [93], and can induce renal microcystic changes in young rodents [94]. Moreover, the serum levels of basic FGF-2 increase in correlation with the progression of AIDS [95], and the urinary levels of FGF-2 are elevated in children with HIVAN [60]. Renal sections from people with HIVAN and HIV-Tg26 mice show an up-regulated expression of VEGF-A in podocytes [90]. Experimental data suggest that too much or too little VEGF-A release by podocytes can cause proteinuria and renal disease [96,97]. For example, young mice overexpressing VEGF164 selectively in podocytes can develop collapsing glomerulopathy [96]. In addition, these heparin-binding growth factors stimulate signaling pathways that are known to be activated in patients with HIVAN [98,99]. Finally, cytokines released by HIV-infected mononuclear cells can also modulate the outcome of HIVAN [65,100,101]. Insight into the molecular and signaling mechanisms that are activated by these factors is rapidly expanding, and this information will facilitate our understanding of childhood HIVAN.
Conclusion
After more than 25 years of research, the scientific community has made significant progress to understand the pathogenesis of HIVAN. However, we need a better understanding of the mechanisms by which HIV-1 induces renal injury in order to prevent the development of cardiovascular complications and chronic renal failure in HIV-infected children. Recent studies show that host genetic factors play an essential role in HIVAN. Thus, we need to define how these factors modulate the infection and survival of renal cells in vivo, and determine the role of APOL1 in this process. Moreover, it is necessary to elucidate what intrinsic renal factors modulate the proliferation of renal epithelial cells, and why these cells continue to proliferate out of control. The role of circulating viral proteins, immune response, heparin-binding growth factors, and iron accumulation, should be also explored in more depth. Hopefully, all these studies will lead to better strategies to improve the quality of life of HIV-infected children.
Future perspective
During the early years of the AIDS epidemic, children with HIVAN progressed very rapidly to end stage renal disease and/or died before they developed chronic renal failure [5]. Subsequently, the early treatment with ART lead to the reduction of viral burden and has improved the outcome of all AIDS-defining conditions [5]. To date, ART is the best treatment available to prevent the development and/or block the progression of proteinuria and HIVAN [2,102]. Recent excellent articles have discussed the efficacy of ART to prevent and/or treat HIVAN [102–105], and therefore this topic will not be further discussed. We anticipate that the virological success of ART promise a longer lifespan for HIV-infected children, however, the long-term cardiovascular and renal consequences of ART are unknown. Angiotensin converting enzyme inhibitors or receptor blockers, may provide additional therapeutic benefits, but should be used with caution in young children with hypoalbuminemia, salt-wasting disorders, diarrhea and other gastrointestinal problems [2,102]. Measurements of renal function should be made in the patients on a serial basis to follow their progression. Overall, we anticipate that the medical community will continue to face several obstacles to improve the clinical outcome of HIV-infected children. In the USA, the prevalence of HIVAN in children may increase, reflecting the improved survival and increased number of HIV-infected teenagers that become noncompliant with ART. A very large number of children living in the sub-Saharan Africa may develop HIVAN if they do not receive appropriate ART. Among other challenges, the emergence of recombinant viruses that are resistant to ART must be considered. In addition, ART may induce renal injury per se [105], and HIV-infected children develop more severe cardiovascular complications during dialysis [106]. We expect that future studies will elucidate the role of host genetic factors, including the APOL1 polymorphisms, and improve our understanding of childhood HIVAN.
Executive summary.
Definition & clinical outcome of childhood HIV-1 associated nephropathy
Childhood HIV-1 associated nephropathy (HIVAN) is a renal disease characterized by the presence of heavy proteinuria and rapid progression to chronic renal failure.
Renal histological findings show mesangial hyperplasia and/or focal segmental glomerulosclerosis in combination with microcystic tubular dilatation.
Children of African ancestry show a unique susceptibility to developing HIVAN.
Role of HIV genes in HIVAN
The expression of viral genes in the kidney of HIV-transgenic (Tg) rodents can induce HIVAN in the absence of viral replication.
At least for HIV-1 genes, env, tat, nef and vpr, have been found to play a role in the pathogenesis of HIVAN.
nef and vpr appear to play a critical role in the pathogenesis of HIVAN in HIV-Tg mice, however, their role in childhood HIVAN is unclear at the present time.
Host genetic variations & HIVAN
Studies in HIV-Tg26 mice show that host genetic factors play a critical role in the pathogenesis of HIVAN.
The causative link between the Duffy/Duffy antigen receptor for chemokines negative phenotype and outcome of HIVAN or HIV infection is unclear at the present time.
There is a remarkable association between noncoding genetic variants in the MHY9 gene and the presence of focal and segmental glomerulosclerosis in African–Americans with HIVAN.
Role of APOL1 in HIVAN
Three coding-sequence mutations of the human APOL1 gene have been linked to HIVAN, idiopathic focal and segmental glomerulosclerosis, and end-stage kidney disease.
Recent linkage analysis using data generated by the 1000 Genome Project suggested that the risk alleles of APOL1 are more strongly associated with HIVAN than those of MYH9.
The functional consequences of those APOL1 risk alleles in kidney cells have not been elucidated.
The APOL1 gene encodes three protein isoforms and is highly expressed in kidney tubular epithelial cells.
Circulating factors & childhood HIVAN
HIV-Tg mouse models cannot exclude the possibility that circulating factors might play a role in childhood HIVAN, since the processes of viral infection, replication, and immunological response to HIV-1 are all bypassed in transgenic mice.
HIV-Tg mice carrying HIV genes under the control of the CD4 promoter, which restricts the expression of the transgene mainly to immune cells, can develop a HIVAN-like disease.
The HIV-Tg rat model suggests that circulating viral proteins and the accumulation of iron in diseased kidneys may play a pathological role by generating oxidative stress.
Infection of renal epithelial cells
Renal tubular epithelial cells cultured from children with HIVAN can be productively infected with primary isolates harvested from children with HIVAN by a CD4-independent mechanism.
Macrophages can transfer mature viral particles to cultured renal tubular epithelial cells by a Trojan horse-like mechanism, and may establish a clinically relevant renal HIV-1 reservoir.
Heparan sulfated proteoglycans, DEC-205, DC-specific ICAM-3-grabbing nonintegrin, lipid rafts and glycosphingolipids can modulate the process of HIV-1 entry into cultured renal epithelial cells.
Proliferation of renal epithelial cells
nef induces the proliferation of conditionally immortalized murine podocytes. These findings have not been confirmed in primary human renal epithelial cells harvested from HIV-infected children.
The heparin-binding growth factors, FGF-2 and VEGF-A, alone or in combination with HIV-Tat, can induce the proliferation of cultured human podocytes and tubular epithelial cells, and may play a role in the pathogenesis of childhood HIVAN.
The urinary levels of FGF-2 may become a promising biomarker to follow the outcome of childhood HIVAN.
Challenges facing the medical community to improve our understanding of childhood HIVAN
The role of genetic variations in APOL-1 and other candidate genes needs to be studied in depth.
The conditions and receptors that are required to establish a productive infection of human podocytes and tubular epithelial cells in vivo need to be defined.
A better understanding of the mechanism by which HIV-1 induces mesangial hyperplasia, renal epithelial injury and proliferation of human renal epithelial cells is needed.
The roles of circulating viral proteins, heparin-binding growth factors and immune response to the virus need further investigation in children.
New biomarkers are needed to follow the outcome of childhood HIVAN, and to define how much iron supplementation should be given to children with HIVAN.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
This manuscript was supported by the National Institutes of Health Grants: RO-1 HL55605, RO-1 DK-049419 and R0-1 CA106644. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Bibliography
- 1.Strauss J, Abitbol C, Zilleruelo G, et al. Renal disease in children with the acquired immunodeficiency syndrome. N Engl J Med. 1989;321(10):625–630. doi: 10.1056/NEJM198909073211001. [DOI] [PubMed] [Google Scholar]
- 2.McCulloch MI, Ray PE. Kidney disease in HIV-positive children. Semin Nephrol. 2008;28(6):585–594. doi: 10.1016/j.semnephrol.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao TK, Filippone EJ, Nicastri AD, et al. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. 1984;310(11):669–673. doi: 10.1056/NEJM198403153101101. [DOI] [PubMed] [Google Scholar]
- 4.Pardo V, Aldana M, Colton RM, et al. Glomerular lesions in the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101(4):429–434. doi: 10.7326/0003-4819-101-4-429. [DOI] [PubMed] [Google Scholar]
- 5.Ray PE. Taking a hard look at the pathogenesis of childhood HIV-associated nephropathy. Pediatr Nephrol. 2009;24(11):2109–2119. doi: 10.1007/s00467-009-1155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy JA. Discovery, structure, heterogeneity, and origins of HIV. In: Levy JA, editor. HIV and the Pathogenesis of AIDS. 3. ASM Press; Washington, DC, USA: 2007. pp. 1–26. [Google Scholar]
- 7.Zuo Y, Matsusaka T, Zhong J, et al. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. 2006;17(10):2832–2843. doi: 10.1681/ASN.2005080878. [DOI] [PubMed] [Google Scholar]
- 8.Rosenstiel PE, Gruosso T, Letourneau AM, et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. 2008;74(8):1049–1058. doi: 10.1038/ki.2008.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y. HIV-1 gp120 compromises blood–brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007;27(1):123–134. doi: 10.1038/sj.jcbfm.9600330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urbinati C, Nicoli S, Giacca M, et al. HIV-1 Tat and heparan sulfate proteoglycan interaction: a novel mechanism of lymphocyte adhesion and migration across the endothelium. Blood. 2009;114(15):3335–3342. doi: 10.1182/blood-2009-01-198945. [DOI] [PubMed] [Google Scholar]
- 11.Pu H, Hayashi K, Andras IE, Eum SY, Hennig B, Toborek M. Limited role of COX-2 in HIV Tat-induced alterations of tight junction protein expression and disruption of the blood–brain barrier. Brain Res. 2007;1184:333–344. doi: 10.1016/j.brainres.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 12.Feng X, Lu TC, Chuang PY, et al. Reduction of Stat3 activity attenuates HIV-induced kidney injury. J Am Soc Nephrol. 2009;20(10):2138–2146. doi: 10.1681/ASN.2008080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Husain M, Gusella GL, Klotman ME, et al. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol. 2002;13(7):1806–1815. doi: 10.1097/01.asn.0000019642.55998.69. [DOI] [PubMed] [Google Scholar]
- 14.Greenway AL, McPhee DA, Allen K, et al. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J Virol. 2002;76(6):2692–2702. doi: 10.1128/JVI.76.6.2692-2702.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wada T, Pippin JW, Marshall CB, Griffin SV, Shankland SJ. Dexamethasone prevents podocyte apoptosis induced by puromycin aminonucleoside: role of p53 and Bcl-2-related family proteins. J Am Soc Nephrol. 2005;16(9):2615–2625. doi: 10.1681/ASN.2005020142. [DOI] [PubMed] [Google Scholar]
- 16.Rosenstiel P, Gharavi A, D’Agati V, Klotman P. Transgenic and infectious animal models of HIV-associated nephropathy. J Am Soc Nephrol. 2009;20(11):2296–2304. doi: 10.1681/ASN.2008121230. [DOI] [PubMed] [Google Scholar]
- 17.Sakurai N, Kuroiwa T, Ikeuchi H, et al. Fluvastatin prevents podocyte injury in a murine model of HIV-associated nephropathy. Nephrol Dial Transplant. 2009;24(8):2378–2383. doi: 10.1093/ndt/gfp012. [DOI] [PubMed] [Google Scholar]
- 18.Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40(10):1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winkler CA, Nelson G, Oleksyk TK, Nava MB, Kopp JB. Genetics of focal segmental glomerulosclerosis and human immunodeficiency virus-associated collapsing glomerulopathy: the role of MYH9 genetic variation. Semin Nephrol. 2010;30(2):111–125. doi: 10.1016/j.semnephrol.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu XH, Hadley TJ, Xu L, Peiper SC, Ray PE. Up-regulation of Duffy antigen receptor expression in children with renal disease. Kidney Int. 1999;55(4):1491–1500. doi: 10.1046/j.1523-1755.1999.00385.x. [DOI] [PubMed] [Google Scholar]
- 22.Woolley IJ, Kalayjian R, Valdez H, et al. HIV nephropathy and the Duffy antigen/receptor for Chemokines in African Americans. J Nephrol. 2001;14(5):384–387. [PubMed] [Google Scholar]
- 23.Langhi DM, Jr, Bordin JO. Duffy blood group and malaria. Hematology. 2006;11(5):389–398. doi: 10.1080/10245330500469841. [DOI] [PubMed] [Google Scholar]
- 24.Lee JS, Wurfel MM, Matute-Bello G, et al. The Duffy antigen modifies systemic and local tissue chemokine responses following lipopolysaccharide stimulation. J Immunol. 2006;177(11):8086–8094. doi: 10.4049/jimmunol.177.11.8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lachgar A, Jaureguiberry G, Le Buenac H, et al. Binding of HIV-1 to RBCs involves the Duffy antigen receptors for chemokines (DARC) Biomed Pharmacother. 1998;52(10):436–439. doi: 10.1016/s0753-3322(99)80021-3. [DOI] [PubMed] [Google Scholar]
- 26.Beck Z, Brown BK, Wieczorek L, et al. Human erythrocytes selectively bind and enrich infectious HIV-1 virions. PLoS One. 2009;4(12):E8297. doi: 10.1371/journal.pone.0008297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walton RT, Rowland-Jones SL. HIV and chemokine binding to red blood cells – DARC matters. Cell Host Microbe. 2008;4(1):3–5. doi: 10.1016/j.chom.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 28.He W, Neil S, Kulkarni H, et al. Duffy antigen receptor for chemokines mediates trans-infection of HIV-1 from red blood cells to target cells and affects HIV-AIDS susceptibility. Cell Host Microbe. 2008;4(1):52–62. doi: 10.1016/j.chom.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horne KC, Li X, Jacobson LP, et al. Duffy antigen polymorphisms do not alter progression of HIV in African Americans in the MACS cohort. Cell Host Microbe. 2009;5(5):415–417. doi: 10.1016/j.chom.2009.04.013. author reply 418–419. [DOI] [PubMed] [Google Scholar]
- 30.Walley NM, Julg B, Dickson SP, et al. The Duffy antigen receptor for chemokines null promoter variant does not influence HIV-1 acquisition or disease progression. Cell Host Microbe. 2009;5(5):408–410. doi: 10.1016/j.chom.2009.04.011. author reply 418–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler CA, An P, Johnson R, Nelson GW, Kirk G. Expression of Duffy antigen receptor for chemokines (DARC) has no effect on HIV-1 acquisition or progression to AIDS in African Americans. Cell Host Microbe. 2009;5(5):411–413. doi: 10.1016/j.chom.2009.04.010. author reply 418–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40(10):1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bostrom MA, Freedman BI. The spectrum of MYH9-associated nephropathy. Clin J Am Soc Nephrol. 2010;5(6):1107–1113. doi: 10.2215/CJN.08721209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopp JB. Glomerular pathology in autosomal dominant MYH9 spectrum disorders: what are the clues telling us about disease mechanism? Kidney Int. 2010;78(2):130–133. doi: 10.1038/ki.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekine T, Konno M, Sasaki S, et al. Patients with Epstein–Fechtner syndromes owing to MYH9 R702 mutations develop progressive proteinuric renal disease. Kidney Int. 2010;78(2):207–214. doi: 10.1038/ki.2010.21. [DOI] [PubMed] [Google Scholar]
- 36.Johnstone DB, Zhang J, George B, et al. Podocyte-specific deletion of Myh9 encoding nonmuscle myosin heavy chain 2A predisposes mice to glomerulopathy. Mol Cell Biol. 2011;31(10):2162–2170. doi: 10.1128/MCB.05234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genovese G, Tonna SJ, Knob AU, et al. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. 2010;78(7):698–704. doi: 10.1038/ki.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. 2010;128(3):345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283(31):21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4(8):1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19(5):850–858. doi: 10.1101/gr.085647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duchateau PN, Pullinger CR, Orellana RE, et al. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272(41):25576–25582. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 43.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wheeler RJ. The trypanolytic factor-mechanism, impacts and applications. Trends Parasitol. 2010;26(9):457–464. doi: 10.1016/j.pt.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 45.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422(6927):83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 47.Dickie P, Felser J, Eckhaus M, et al. HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology. 1991;185(1):109–119. doi: 10.1016/0042-6822(91)90759-5. [DOI] [PubMed] [Google Scholar]
- 48.Ray PE, Bruggeman LA, Weeks BS, et al. bFGF and its low affinity receptors in the pathogenesis of HIV-associated nephropathy in transgenic mice. Kidney Int. 1994;46(3):759–772. doi: 10.1038/ki.1994.331. [DOI] [PubMed] [Google Scholar]
- 49.Kiryluk K, Martino J, Gharavi AG. Genetic susceptibility, HIV infection, and the kidney. Clin J Am Soc Nephrol. 2007;2(Suppl 1):S25–S35. doi: 10.2215/CJN.00320107. [DOI] [PubMed] [Google Scholar]
- 50.Chan KT, Papeta N, Martino J, et al. Accelerated development of collapsing glomerulopathy in mice congenic for the HIVAN1 locus. Kidney Int. 2009;75(4):366–372. doi: 10.1038/ki.2008.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franks RR, Ray PE, Babbott CC, et al. Maternal-fetal interactions affect growth of human immunodeficiency virus type 1 transgenic mice. Pediatr Res. 1995;37(1):56–63. doi: 10.1203/00006450-199501000-00012. [DOI] [PubMed] [Google Scholar]
- 52.Papeta N, Chan KT, Prakash S, et al. Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. J Clin Invest. 2009;119(5):1178–1188. doi: 10.1172/JCI37131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reid W, Sadowska M, Denaro F, et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci USA. 2001;98(16):9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray PE, Liu XH, Robinson LR, et al. A novel HIV-1 transgenic rat model of childhood HIV-1-associated nephropathy. Kidney Int. 2003;63(6):2242–2253. doi: 10.1046/j.1523-1755.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 55.Kline ER, Kleinhenz DJ, Liang B, et al. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. Am J Physiol Heart Circ Physiol. 2008;294(6):H2792–H2804. doi: 10.1152/ajpheart.91447.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lassiter C, Fan X, Joshi PC, et al. HIV-1 transgene expression in rats causes oxidant stress and alveolar epithelial barrier dysfunction. AIDS Res Ther. 2009;6:1. doi: 10.1186/1742-6405-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otis JS, Ashikhmin YI, Brown LA, Guidot DM. Effect of HIV-1-related protein expression on cardiac and skeletal muscles from transgenic rats. AIDS Res Ther. 2008;5:8. doi: 10.1186/1742-6405-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soler-Garcia AA, Johnson D, Hathout Y, Ray PE. Iron-related proteins: candidate urine biomarkers in childhood HIV-associated renal diseases. Clin J Am Soc Nephrol. 2009;4(4):763–771. doi: 10.2215/CJN.0200608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paragas N, Nickolas TL, Wyatt C, et al. Urinary NGAL marks cystic disease in HIV-associated nephropathy. J Am Soc Nephrol. 2009;20(8):1687–1692. doi: 10.1681/ASN.2009010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soler-Garcia AA, Rakhmanina NY, Mattison PC, Ray PE. A urinary biomarker profile for children with HIV-associated renal diseases. Kidney Int. 2009;76(2):207–214. doi: 10.1038/ki.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhong J, Zuo Y, Ma J, et al. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1-associated nephropathy. Kidney Int. 2005;68(3):1048–1060. doi: 10.1111/j.1523-1755.2005.00497.x. [DOI] [PubMed] [Google Scholar]
- 62.Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21(9):1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell. 1998;95(2):163–175. doi: 10.1016/s0092-8674(00)81748-1. [DOI] [PubMed] [Google Scholar]
- 64.Hanna Z, Priceputu E, Hu C, Vincent P, Jolicoeur P. HIV-1 Nef mutations abrogating downregulation of CD4 affect other Nef functions and show reduced pathogenicity in transgenic mice. Virology. 2006;346(1):40–52. doi: 10.1016/j.virol.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 65.Heckmann A, Waltzinger C, Jolicoeur P, Dreano M, Kosco-Vilbois MH, Sagot Y. IKK2 inhibitor alleviates kidney and wasting diseases in a murine model of human AIDS. Am J Pathol. 2004;164(4):1253–1262. doi: 10.1016/S0002-9440(10)63213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanna Z, Priceputu E, Chrobak P, et al. Selective expression of human immunodeficiency virus Nef in specific immune cell populations of transgenic mice is associated with distinct AIDS-like phenotypes. J Virol. 2009;83(19):9743–9758. doi: 10.1128/JVI.00125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rahim MM, Chrobak P, Hu C, Hanna Z, Jolicoeur P. Adult AIDS-like disease in a novel inducible human immunodeficiency virus type 1 Nef transgenic mouse model: CD4+ T-cell activation is Nef dependent and can occur in the absence of lymphopenia. J Virol. 2009;83(22):11830–11846. doi: 10.1128/JVI.01466-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ray PE, Liu XH, Henry D, et al. Infection of human primary renal epithelial cells with HIV-1 from children with HIV-associated nephropathy. Kidney Int. 1998;53(5):1217–1229. doi: 10.1046/j.1523-1755.1998.00900.x. [DOI] [PubMed] [Google Scholar]
- 69.Ray PE, Tassi E, Liu XH, Wellstein A. Role of fibroblast growth factor-binding protein in the pathogenesis of HIV-associated hemolytic uremic syndrome. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R105–R113. doi: 10.1152/ajpregu.00492.2005. [DOI] [PubMed] [Google Scholar]
- 70.Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol. 2003;77(22):12140–12151. doi: 10.1128/JVI.77.22.12140-12151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bobardt MD, Saphire AC, Hung HC, et al. Syndecan captures, protects, and transmits HIV to T lymphocytes. Immunity. 2003;18(1):27–39. doi: 10.1016/s1074-7613(02)00504-6. [DOI] [PubMed] [Google Scholar]
- 72.Chen P, Chen BK, Mosoian A, et al. Virological synapses allow HIV-1 uptake and gene expression in renal tubular epithelial cells. J Am Soc Nephrol. 2011;22(3):496–507. doi: 10.1681/ASN.2010040379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hatsukari I, Singh P, Hitosugi N, et al. DEC-205-mediated internalization of HIV-1 results in the establishment of silent infection in renal tubular cells. J Am Soc Nephrol. 2007;18(3):780–787. doi: 10.1681/ASN.2006121307. [DOI] [PubMed] [Google Scholar]
- 74.Mikulak J, Teichberg S, Faust T, Schmidtmayerova H, Singhal PC. HIV-1 harboring renal tubular epithelial cell interaction with T cells results in T cell trans-infection. Virology. 2009;385(1):105–114. doi: 10.1016/j.virol.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 75.Mikulak J, Teichberg S, Arora S, et al. DC-specific ICAM-3-grabbing nonintegrin mediates internalization of HIV-1 into human podocytes. Am J Physiol Renal Physiol. 2010;299(3):F664–F673. doi: 10.1152/ajprenal.00629.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mikulak J, Singhal PC. HIV-1 entry into human podocytes is mediated through lipid rafts. Kidney Int. 2009;77(1):72–73. doi: 10.1038/ki.2009.366. author reply 73–74. [DOI] [PubMed] [Google Scholar]
- 77.Khan F, Proulx F, Lingwood CA. Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int. 2009;75(11):1209–1216. doi: 10.1038/ki.2009.7. [DOI] [PubMed] [Google Scholar]
- 78.Liu XH, Lingwood CA, Ray PE. Recruitment of renal tubular epithelial cells expressing verotoxin-1 (Stx1) receptors in HIV-1 transgenic mice with renal disease. Kidney Int. 1999;55(2):554–561. doi: 10.1046/j.1523-1755.1999.00278.x. [DOI] [PubMed] [Google Scholar]
- 79.Ray PE. Shiga-like toxins and HIV-1 ‘go through’ glycosphingolipids and lipid rafts in renal cells. Kidney Int. 2009;75(11):1135–1137. doi: 10.1038/ki.2009.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lund N, Branch DR, Mylvaganam M, et al. A novel soluble mimic of the glycolipid, globotriaosyl ceramide inhibits HIV infection. AIDS. 2006;20(3):333–343. doi: 10.1097/01.aids.0000206499.78664.58. [DOI] [PubMed] [Google Scholar]
- 81.Natoli TA, Smith LA, Rogers KA, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med. 2010;16(7):788–792. doi: 10.1038/nm.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Green DF, Resnick L, Bourgoignie JJ. HIV infects glomerular endothelial and mesangial but not epithelial cells in vitro. Kidney Int. 1992;41(4):956–960. doi: 10.1038/ki.1992.146. [DOI] [PubMed] [Google Scholar]
- 83.Conaldi PG, Bottelli A, Wade-Evans A, et al. HIV-persistent infection and cytokine induction in mesangial cells: a potential mechanism for HIV-associated glomerulosclerosis. AIDS. 2000;14(13):2045–2047. doi: 10.1097/00002030-200009080-00021. [DOI] [PubMed] [Google Scholar]
- 84.Khatua AK, Taylor HE, Hildreth JE, Popik W. Non-productive HIV-1 infection of human glomerular and urinary podocytes. Virology. 2010;408(1):119–127. doi: 10.1016/j.virol.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poulsom R, Little MH. Parietal epithelial cells regenerate podocytes. J Am Soc Nephrol. 2009;20(2):231–233. doi: 10.1681/ASN.2008121279. [DOI] [PubMed] [Google Scholar]
- 86.Suzuki T, Matsusaka T, Nakayama M, et al. Genetic podocyte lineage reveals progressive podocytopenia with parietal cell hyperplasia in a murine model of cellular/collapsing focal segmental glomerulosclerosis. Am J Pathol. 2009;174(5):1675–1682. doi: 10.2353/ajpath.2009.080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Becker JU, Hoerning A, Schmid KW, Hoyer PF. Immigrating progenitor cells contribute to human podocyte turnover. Kidney Int. 2007;72(12):1468–1473. doi: 10.1038/sj.ki.5002524. [DOI] [PubMed] [Google Scholar]
- 88.Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol. 2009;20(12):2593–2603. doi: 10.1681/ASN.2009020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ronconi E, Sagrinati C, Angelotti ML, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol. 2009;20(2):322–332. doi: 10.1681/ASN.2008070709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korgaonkar SN, Feng X, Ross MD, et al. HIV-1 upregulates VEGF in podocytes. J Am Soc Nephrol. 2008;19(5):877–883. doi: 10.1681/ASN.2007050629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Conaldi PG, Bottelli A, Baj A, et al. Human immunodeficiency virus-1 tat induces hyperproliferation and dysregulation of renal glomerular epithelial cells. Am J Pathol. 2002;161(1):53–61. doi: 10.1016/S0002-9440(10)64156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doublier S, Zennaro C, Spatola T, et al. HIV-1 Tat reduces nephrin in human podocytes: a potential mechanism for enhanced glomerular permeability in HIV-associated nephropathy. AIDS. 2007;21(4):423–432. doi: 10.1097/QAD.0b013e328012c522. [DOI] [PubMed] [Google Scholar]
- 93.Tang P, Jerebtsova M, Przygodzki R, Ray PE. Fibroblast growth factor-2 increases the renal recruitment and attachment of HIV-infected mononuclear cells to renal tubular epithelial cells. Pediatr Nephrol. 2005;20(12):1708–1716. doi: 10.1007/s00467-005-2018-2. [DOI] [PubMed] [Google Scholar]
- 94.Li Z, Jerebtsova M, Liu XH, Tang P, Ray PE. Novel cystogenic role of basic fibroblast growth factor in developing rodent kidneys. Am J Physiol Renal Physiol. 2006;291(2):F289–F296. doi: 10.1152/ajprenal.00382.2005. [DOI] [PubMed] [Google Scholar]
- 95.Ascherl G, Sgadari C, Bugarini R, et al. Serum concentrations of fibroblast growth factor 2 are increased in HIV type 1-infected patients and inversely related to survival probability. AIDS Res Hum Retroviruses. 2001;17(11):1035–1039. doi: 10.1089/088922201300343717. [DOI] [PubMed] [Google Scholar]
- 96.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kumar D, Konkimalla S, Yadav A, et al. HIV-associated nephropathy: role of mammalian target of rapamycin pathway. Am J Pathol. 2010;177(2):813–821. doi: 10.2353/ajpath.2010.100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sharma M, Callen S, Zhang D, Singhal PC, Vanden Heuvel GB, Buch S. Activation of Notch signaling pathway in HIV-associated nephropathy. AIDS. 2010;24(14):2161–2170. doi: 10.1097/QAD.0b013e32833dbc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martinka S, Bruggeman LA. Persistent NF-κB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy. Am J Physiol Renal Physiol. 2006;290(3):F657–F665. doi: 10.1152/ajprenal.00208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bruggeman LA, Nelson PJ. Controversies in the pathogenesis of HIV-associated renal diseases. Nat Rev Nephrol. 2009;5(10):574–581. doi: 10.1038/nrneph.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chaparro AI, Mitchell CD, Abitbol CL, et al. Proteinuria in children infected with the human immunodeficiency virus. J Pediatr. 2008;152(6):844–849. doi: 10.1016/j.jpeds.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 103.Longenecker CT, Scherzer R, Bacchetti P, Lewis CE, Grunfeld C, Shlipak MG. HIV viremia and changes in kidney function. AIDS. 2009;23(9):1089–1096. doi: 10.1097/QAD.0b013e32832a3f24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Post FA, Campbell LJ, Hamzah L, et al. Predictors of renal outcome in HIV-associated nephropathy. Clin Infect Dis. 2008;46(8):1282–1289. doi: 10.1086/529385. [DOI] [PubMed] [Google Scholar]
- 105.Judd A, Boyd KL, Stohr W, et al. Effect of tenofovir disoproxil fumarate on risk of renal abnormality in HIV-1-infected children on antiretroviral therapy: a nested case-control study. AIDS. 2010;24(4):525–534. doi: 10.1097/QAD.0b013e3283333680. [DOI] [PubMed] [Google Scholar]
- 106.Gordillo R, Kumar J, Del Rio M, Flynn JT, Woroniecki RP. Outcome of dialysis in children with human immunodeficiency virus infection. Pediatr Nephrol. 2009;24(1):171–175. doi: 10.1007/s00467-008-0976-x. [DOI] [PubMed] [Google Scholar]
- Website
- 201.The Human Protein Atlas. www.proteinatlas.org/ENSG00000100342.