

Abstract
The schweinfurthins are an intriguing group of anti-proliferative agents that display low nanomolar activities against several cell types, including the human-derived glioblastoma cell line SF-295, but have little impact on other cell lines even at micromolar concentrations. This activity has inspired the synthesis of seven of the natural schweinfurthins, all with the correct absolute stereochemistry, and a variety of analogues designed to probe different facets of the pharmacophore. Reported herein is the synthesis of several new schweinfurthin analogues varied at the C-5 position along with data on their biological activity in the NCI 60 cell-line assay.
Keywords: schweinfurthins, apoptosis, cytotoxic, anti-proliferative, bioassay
1. Introduction
In 1998 the structures of schweinfurthins A and B (1 and 2, Figure 1) were reported as part of the National Cancer Institute's (NCI) search for anti-proliferative agents with new mechanisms of action. These compounds were isolated from an extract of Macaranga schweinfurthii through bioassay guided fractionation,1 and subsequently were subjected to the NCI 60 cell-line screen. This assay revealed a unique pattern of activity which does not correlate to any known mechanism of cell growth inhibition. Several cell lines were particularly sensitive to the schweinfurthins, including the human glioblastoma line SF-295, while other cell lines (e.g. the lung cancer line A549) were only marginally affected even at elevated doses. These observations led to the hypothesis that further exploration of the schweinfurthins might uncover a new target for treatment of specific malignancies, especially for conditions with poor clinical prognoses such as glioblastoma multiforme.
Figure 1.
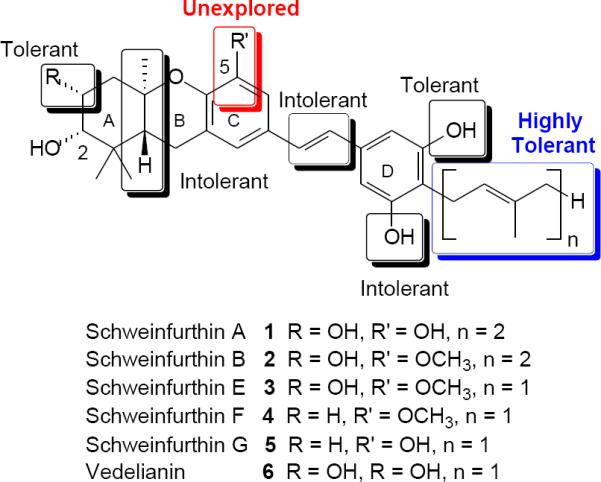
Key schweinfurthins and current understanding of the pharmacophore
At this time seven of the eleven natural schweinfurthins1–4 have been synthesized, including specifically schweinfurthins A (1),5 B (2),6 C,7 E (3),6 F (4),8 and G (5),8 as well as vedelianin9 (6). Our group also has synthesized numerous schweinfurthin analogues and in the process has gained considerable knowledge of the schweinfurthin pharmacophore. Several portions of the schweinfurthin structure have shown tolerance to modification, including the presence or absence of a hydroxyl group at C-3,10 methylation of one D-ring phenol11 (Figure 1, “tolerant”), and especially incorporation of substituents at the para position of the D-ring, including hydrogen or an alkyl, allyl, prenyl, or geranyl group (“highly tolerant”).12–15 Conversely, modification of other regions resulted in a significant loss of activity, including a cis-fused A/B system,16 a cis-stilbene or a saturated linkage,13 and simultaneous methylation of both D-ring phenols (Figure 1, “intolerant”).11
Investigations of the schweinfurthin mechanism17 have revealed that schweinfurthin A can phenocopy the tumor suppressor NF1 in CNS and peripheral nervous system tumor cell lines,17a although a proximate target has not yet been identified conclusively. Recent work by the Shair group has implicated oxysterol binding proteins in the action of schweinfurthins and other apparently similar natural products, but the evidence for schweinfurthins is less compelling than for OSW-1 and cephalostatin 1.17b A recent study more specific to the schweinfurthins found that the synthetic analogue 3-deoxyschweinfurthin B had a pronounced impact on isoprenoid homeostasis,17c but more work is clearly needed before the mechanism(s) will be understood in any detail.
An increased understanding of the pharmacophore has allowed synthesis of schweinfurthins for use as fluorescent12,14 or biotinylated15 probes that may allow more specific determination of the basis of schweinfurthins' cellular activity. However, one region of the schweinfurthin structure that, until now, has remained relatively unexplored is the C-5 position. Within the natural family, this position has been found only as a free phenol or a methoxy group, with the free phenol typically 2–4 fold more potent. Given this difference in potency, it appeared to be worthwhile to explore the effect that various substituents at the C-5 position might have on activity. Reported herein is the synthesis of a small set of C-5 modified schweinfurthins with an evaluation of their relative activity in the NCI's 60 cell-line assay.
2. Synthesis
Our strategy for schweinfurthin synthesis is based on a late stage Horner-Wadsworth-Emmons (HWE) condensation between an aryl aldehyde, usually representing the hexahydroxanthene side of the target, and a benzylic phosphonate.10 To use this approach to obtain C-5 analogues required preparation of several key aldehydes (Scheme 1). The first aldehyde prepared carried only a hydrogen substituent at C-5. To pursue this compound, protection of the previously reported arene 718 as the TBS ether afforded compound 8, which was epoxidized under Shi conditions19 to obtain the nonracemic compound 9. Removal of the silyl group in epoxide 9 afforded the phenol 10, and subsequent treatment with BF3·OEt2 brought about cyclization to afford the hexahydroxanthene 11 in very good yield. After reduction of this methyl ester to the benzylic alcohol 12, oxidation with MnO2 gave the parent aldehyde 13. While aldehyde 13 underwent direct bromination at the C-5 position in modest yield (23%), a longer sequence provided the MOM-protected bromide 18 in much better yield. Thus, bromination of the methyl ester 11 in methylene chloride and acetic acid provided a mixture of bromides 14 and 15, but cleavage of the acetate could be accomplished in near-quantitative yield. After protection of the hydroxyl group of compound 14 through reaction with MOMCl, reduction provided the benzylic alcohol 17 and a final oxidation with MnO2 gave the desired aldehyde 18. This aldehyde was condensed with a “right-half” phosphonate (vide infra) that was also MOM-protected, so a separate step was not required for the eventual removal of the C-2 MOM group.
Scheme 1.

Synthesis of four intermediate aldehydes for the “left half” of the schweinfurthins.
A substantial number of important drugs bear a fluorine substituent, at least in part because fluorine's electronic properties are significantly different from hydrogen but its size is sufficiently similar that there is minimal potential for introduction of unfavorable steric interactions in a monofluoro compound.20 To prepare a C-5 fluorinated schweinfurthin analogue, synthesis of the requisite aldehyde began with ester 19, obtained in three steps from commercial 3-fluoro-4-hydroxybenzoic acid (esterification, bromination and protection as the MOM acetal).21 After this ester was reduced to alcohol 20 it was converted to the methyl ether 21 via a standard Williamson synthesis. Halogen metal exchange and reaction of the resulting aryl lithium intermediate with geranyl bromide provided compound 22 in low yield. Application of a Shi epoxidation furnished epoxide 23 in moderate yield and good enantiomeric excess. Cyclization of this epoxide afforded the hexahydroxanthene 24. The benzyl methyl ether has been utilized as a latent benzaldehyde in other schweinfurthin syntheses,5,8 and in this specific case DDQ oxidation provided aldehyde 25 cleanly in a single step from the methyl ether 24.
A slightly different approach based on sequential and selective directed ortho metallation was employed to gain access to the C-5 methylthio schweinfurthin analogue. In this case, after alcohol 26 was converted to the methyl ether 27, deprotonation with n-BuLi followed by treatment with (CH3S)2 provided the thiomethyl ether 28. A second ortho metallation and copper-mediated coupling with the known22 epoxide of geranyl bromide provided epoxide 29. Cyclization to compound 30 proceeded in reasonable yield, and DDQ oxidation of the benzyl methyl ether provided aldehyde 31 smoothly without detectable oxidation of the thiomethyl group.
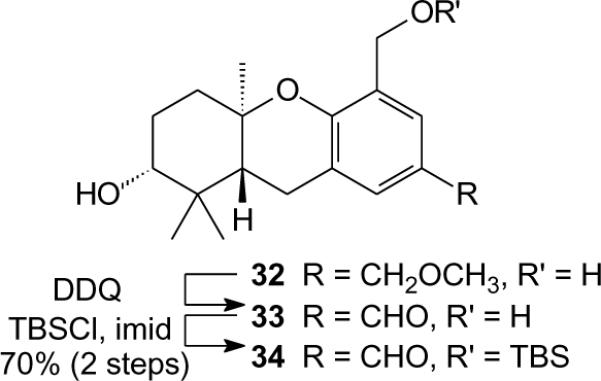
We also prepared compounds with carbon-containing substituents at C-5 (Scheme 2). During the course of these studies, the benzyl alcohol 32 became available through a cascade cyclization terminated by electrophilic aromatic substitution.5 Treatment of compound 32 with DDQ preferentially oxidized the benzyl methyl ether in the presence of the free benzyl alcohol to provide aldehyde 33, corroborating previous observations on the regioselectivity of this oxidation.5 Protection of the benzyl alcohol as a TBS ether afforded aldehyde 34 for use as a partner in HWE reactions.
Scheme 2.
Synthesis of C-5 alkyl compounds
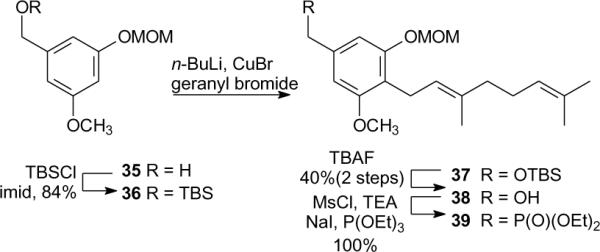
The right half of the new schweinfurthin analogues was prepared as shown in Scheme 3. Directed ortho metallation of alcohol 3523 occurred upon exposure to multiple equivalents of n-BuLi, and subsequent reaction with geranyl bromide afforded the expected geranylated arene 38 in 47% yield.24 This approach presumably involves formation of an intermediate dianion, and was more efficient than the 34% overall yield if alcohol 35 was protected as the TBS ether 36, alkylated to obtain compound 37, and then deprotected to afford alcohol 38. Conversion of benzyl alcohol 38 to the phosphonate 39 was conducted by a classical Arbuzov reaction25 after formation of the mesylate and iodide. This provided phosphonate 39 which was used as an HWE coupling partner throughout this series.
Scheme 3.
Synthesis of phosphonate 39
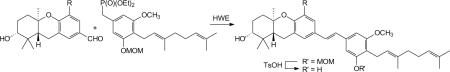
With phosphonate 39 and a set of aldehydes now available, stilbene formation was accomplished by standard HWE olefination (Table 1). Treatment of the new aldehydes 13, 18, 25, 31, and 34, as well as the known aldehydes 40,541,8 and 42,10 with phosphonate 39 and base provided stilbenes 43–50 in moderate to high yield. Hydrolysis of the MOM acetals 43–49 was accomplished by exposure to methanolic p-TsOH to provide schweinfurthin analogues 51–57.
Table 1.
HWE Condensations and subsequent hydrolysis.
| Entry | RCHO | R = | HWE (% Yield) | Stilbene | Hydrolysis (% Yield) | Schwein. | R = |
|---|---|---|---|---|---|---|---|
| 1 | 13 | H | 66% | 43 | 53% | 51 | H |
| 2 | 18 a | Br | 54% | 44 a | 92% | 52 | Br |
| 3 | 25 | F | 53% | 45 | 77% | 53 | F |
| 4 | 31 | SCH3 | 74% | 46 | 40% | 54 | SCH3 |
| 5 | 40 | CH2OCH3 | 50% | 47 | 41% | 55 | CH2OCH3 |
| 6 | 41 | OMOM | 77% | 48 | 61% | 56 | OH |
| 7 | 42 | OCH3 | 90% | 49 | 92% | 57 | OCH3 |
| 8 | 34 | CH2OTBS | 36% | 50 | |||
In this series, the aldehyde and stilbene bear a C-2 MOM group that is cleaved in the final hydrolysis.
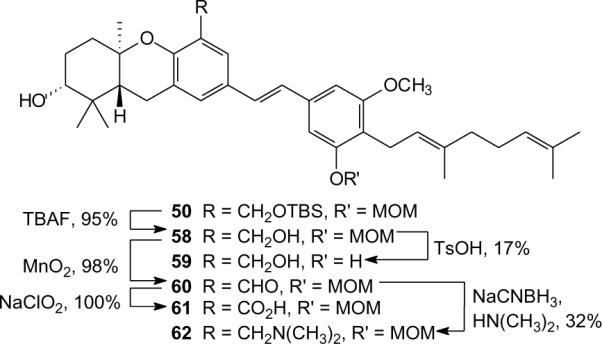
The benzylic C-5 position of compound 50 allowed the possibility of further elaboration and this intermediate was exploited to obtain several additional analogues. After cleavage of the silyl ether through treatment with TBAF, several analogues were obtained through straightforward reactions of the resulting benzyl alcohol 58 (Scheme 4). Exposure of alcohol 58 to MnO2 resulted in aldehyde 60. Further oxidation of this intermediate with sodium chlorite26 provided the carboxylic acid 61, while reductive amination of aldehyde 60 with NaCNBH3 and dimethylamine resulted in the tertiary amine 62. The D-ring MOM group of compound 58 could be removed via hydrolysis to afford schweinfurthin 59, but this reaction proceeded in low yield. Given this disappointing yield, and because MOM groups can sometimes be cleaved in vivo during cellular assays, hydrolysis of the other MOM acetals in this small set was not pursued pending the results of bioassays on two representative members of this group, the alcohol 58 and the corresponding aldehyde 60.
Scheme 4.
3. Biological results and discussion
Nine of these new schweinfurthin analogues have been submitted to the NCI's 60 cell-line screen,27 compounds 51–58, and 60. The current protocol for this assay requires initial testing at a single dose, and then the more active compounds are subjected to the full 5-dose assay. Of these nine schweinfurthins, compounds 52 and 55 did not pass the single dose assay with a level of activity sufficient to justify the full screen. Given the substantial size of the bromide substituent, it may not be surprising that compound 52 showed little activity, but the limited activity of the fluoride 55 was disappointing. This may suggest that a C-5 substituent capable of hydrogen bond donation is important to activity, or that the limited ability of fluorine28 to serve as hydrogen bond acceptor diminishes activity.
Five of the compounds that did pass this test carried one phenol and one methoxy substituent in the D-ring, and these five compounds displayed a range of activity (Table 2). Compound 56 was the most potent, with a mean GI50 of 0.47 μM, and this compound also showed the greatest difference in activity between the most and least sensitive cell lines (2.84 log units). Reflecting the same trend as the natural products schweinfurthin A and B, introduction of a methyl ether at the C-5 position (i.e. compound 57) resulted in about a 3-fold loss of potency as measured by the mean GI50 values. The difference in activity is even more striking if one considers just the sensitive SF-295 and insensitive A549 cell lines. For these two cell lines, compound 56 was more than 30-fold more potent against SF-295 cells than against A549 cells, while compound 57 showed only about 13-fold greater activity in the SF-295 assay. A lesser differential activity also is observed in the full 60 cell-line data for compound 57 versus compound 56 (1.58 versus 2.84).
Table 2.
Activity of Schweinfurthin Analogues in the NCI 60 Cell Line Screen.27
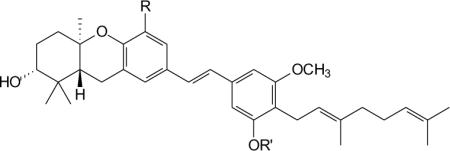
| Entry | Schweinfurthin | R = | R' = | Mean GI50 (μM) | Differential (log units) | SF-295 GI50 (μM) | A-549 GI50 (μM) |
|---|---|---|---|---|---|---|---|
| 1 | 51 a | H | H | 10.15 | 1.56 | 1.66 | 13.2 |
| 2 | 53 a | F | H | 6.23 | 1.40 | 5.01 | 2.14 |
| 3 | 54 a | SCH3 | H | 7.95 | 1.58 | 3.80 | 6.03 |
| 4 | 56 b | OH | H | 0.47 | 2.84 | 0.022 | 0.72 |
| 5 | 57 a | OCH3 | H | 1.67 | 1.58 | 0.19 | 2.45 |
| 6 | 58 b | CH2OH | MOM | 1.66 | 2.56 | 0.15 | 1.41 |
| 7 | 60 a | CHO | MOM | 6.30 | 2.46 | 1.20 | 7.8 |
Duplicate wells were run for each of these compounds in one assay.
Duplicate wells were run for each of these compounds in each of two independent assays and the average value is given in this table.
By some measures, compound 57 also is an interesting compound: it has a relatively low mean GI50 (1.67 μM) and shows a 13-fold difference in activity between the SF-295 and A549 cells. However, perhaps the most surprising compound is the benzyl alcohol 58. As noted above, because attempted hydrolysis of the D-ring MOM group in compound 58 was accompanied by extensive decomposition, both compounds 58 and 60 were submitted for this assay with the D-ring MOM group still in place. Thus there are two points of difference between compounds 58 and 57 which complicates any direct comparison of their biological activity. Nevertheless, the fact that compound 58 displays potency comparable to compound 57 and high differential activity (2.56 log units in the 60 cell assay) is intriguing. If one assumes that the D-ring MOM group is lost after cell uptake, the resulting compound would be simply isomeric to compound 56 at C-5 and it is nearly equivalent in mean GI50. However compound 58 may be significantly more stable to metabolism. Because ortho quinone formation in the C-ring would require extensive metabolism with any carbon substituent at C-5, it may be worthwhile to explore other schweinfurthin analogues that include a C-5 hydroxymethyl group.
4. Conclusions
In conclusion, several C-5 modified schweinfurthin analogues have been prepared through new variations on the strategies that have been used to prepare the natural products in this family. These syntheses required sequences of varied length and gave varied yields, but averaged 11 linear steps and proceeded in an average yield of ~5%. Of the nine schweinfurthin analogues synthesized for this study and tested in the NCI 60 cell assay, only compounds 56 and 58 have in vitro potency comparable to schweinfurthin A (1),1 although most of them show selectivity for inhibition of CNS tumor cell growth. From these studies, it appears that the phenol group at C-5 is one structural feature that preserves both good potency and a high differential activity. Perhaps surprisingly, incorporation of a hydroxymethyl group at this position also led to an analogue with good activity. Thus the contribution of the C-5 substituent may be more reliant on its ability to undergo hydrogen bonding than on its electronic effect on the extended π system. It is now apparent that preservation of a C-5 substituent capable of H-bonding will be important during studies that probe other aspects of the schweinfurthin pharmacophore. Furthermore, the activity observed for compound 58, despite the presence of a D-ring MOM acetal, suggests that this group is biodegradable upon cell uptake and/or that there is more flexibility to substituents at this position than previously recognized. Thus it is reasonable to conclude that there is still more to be learned about the activity of synthetic compounds modeled upon the natural schweinfurthins, and further studies in this vein will be forthcoming.
5. Experimental procedures and methods
5.1 General Experimental Conditions
Tetrahydrofuran was freshly distilled from sodium/benzophenone, while methylene chloride was distilled from calcium hydride prior to use. All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions in nonaqueous solvents were conducted in flame-dried glassware under a positive pressure of argon and with magnetic stirring. All NMR spectra were obtained at 300 MHz for 1H, and 75 MHz for 13C with CDCl3 as solvent, and (CH3)4Si (1H, 0.00 ppm) or CDCl3 (13C, 77.0 ppm) as internal standards unless otherwise noted. The 31P chemical shifts were reported in ppm relative to 85% H3PO4 (external standard). High resolution mass spectra were obtained at the University of Iowa Mass Spectrometry Facility. Silica gel (60 Å, 0.040–0.063 mm) was used for flash chromatography.
5.2 Silyl ether 8
To a solution of phenol 7 (1.86 g, 6.4 mmol) in CH2Cl2 (200 mL) was added imidazole (2.19 g, 32.2 mmol), followed by TBSCl (1.15 g, 7.7 mmol) in one portion at 0 °C. The solution was allowed to stir for 12 h, and then the reaction was quenched by addition of water. The resulting solution was extracted with CH2Cl2, dried (MgSO4), and concentrated in vacuo. Purification by column chromatography (1–5% EtOAc in hexanes) produced ester 8 as an oil (2.46 g, 95%): 1H NMR δ 7.83 (d, J = 2.2 Hz, 1H), 7.78 (dd, J = 8.4, 2.2 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 5.34–5.29 (m, 1H), 5.14–5.09 (m, 1H), 3.86 (s, 3H), 3.32 (d, J = 6.8 Hz, 2H), 2.12–2.01 (m, 4H), 1.69 (s, 3H), 1.67 (s, 3H), 1.60 (s, 3H), 1.02 (s, 9H), 0.26 (s, 6H); 13C NMR δ 167.2, 157.8, 136.7, 132.5, 131.4, 129.6, 128.8, 124.3, 122.9, 122.0, 117.9, 51.8, 39.8, 28.5, 26.7, 25.7 (3C), 25.7, 18.3, 17.7, 16.3, −3.7 (2C). Anal. Calcd for C24H38O3Si: C, 71.59; H, 9.51. Found C, 71.44; H, 9.58.
5.3 Epoxide 9
To a solution of ester 8 (879 mg, 2.2 mmol), CH2Cl2 (6 mL), CH3CN (3 mL), EtOH (3 mL), and aqueous buffer (2 M K2CO3, 4 × 10−3 M EDTA, 12 mL), and the Shi catalyst (140 mg, 0.5 mmol) was added H2O2 (30% wt. in H2O, 1.2 mL, 10.6 mmol) via syringe pump (0.14 mL/h) at –10 °C. After 17 h, the reaction was quenched by addition of Na2SO3. The resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (2–5% EtOAc in hexanes) provided recovered ester 8 (318 mg, 36%) and epoxide 9 (459 mg, 50%) as oils: 1H NMR δ 7.81 (d, J = 2.1 Hz, 1H), 7.77 (dd, J = 8.4, 2.4 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 5.38–5.33 (m, 1H), 3.86 (s, 3H), 3.33 (d, J = 6.9 Hz, 2H), 2.72 (t, J = 6.4 Hz, 1H), 2.28–2.09 (m, 2H), 1.77–1.56 (m, 2H), 1.71 (s, 3H), 1.27 (s, 3H), 1.25 (s, 3H), 1.01 (s, 9H), 0.26 (s, 6H); 13C NMR δ 167.1, 157.8, 135.8, 132.2, 131.4, 128.9, 122.8, 122.5, 117.9, 64.2, 58.4, 51.9, 36.3, 28.5, 27.4, 25.7 (3C), 24.9, 18.7, 18.3, 16.3, −3.7 (2C). Anal. Calcd for C24H38O4Si: C, 68.86; H, 9.15. Found C, 69.09; H, 9.19.
5.4 Phenol 10
To a solution of epoxide 9 (459 mg, 1.1 mmol) in THF (20 mL) at rt was added TBAF (1 M in THF, 2.2 mL, 2.2 mmol). After 3 h, the reaction was quenched by addition of water. The resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (10–15% EtOAc in hexanes) afforded phenol 10 (333 mg, 100%) as an oil: 1H NMR δ 7.81–7.77 (m, 2H), 6.84–6.81 (m, 1H), 5.36 (td, J = 7.1, 1.4 Hz, 1H), 3.87 (s, 3H), 3.38 (d, J = 6.9 Hz, 2H), 2.74 (t, J = 6.2 Hz, 1H), 2.29–2.12 (m, 2H), 1.77 (s, 3H), 1.71–1.64 (m, 2H), 1.29 (s, 3H), 1.27 (s, 3H); 13C NMR δ 167.3, 158.7, 137.0, 131.8, 129.6, 127.2, 122.2, 122.0, 115.4, 64.4, 58.9, 51.9, 36.4, 29.1, 27.2, 24.8, 18.7, 16.2. Anal. Calcd for C18H24O4: C, 71.03; H, 7.95. Found C, 70.73; H, 7.96.
5.5 Hexahydroxanthene 11
To a solution of epoxide 10 (353 mg, 1.16 mmol) in CH2Cl2 (125 mL) at −78 °C was added BF3·OEt2 (0.73 mL, 5.76 mmol). After 6 min, the reaction was quenched by addition of triethylamine (TEA, 1.5 mL). Water was added, the resulting solution was extracted with CH2Cl2, dried (MgSO4), and concentrated in vacuo. Purification by column chromatography (25% EtOAc in hexanes) afforded compound 11 (332 mg, 94%) as a white solid: [α]26.4D = +66.8 (c 7.3, CHCl3, 93% ee by HPLC); 1H NMR δ 7.82 (d, J = 1.8 Hz, 1H), 7.77 (dd, J = 8.4, 2.1 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 3.87 (s, 3H), 3.44 (dd, J = 11.3, 4.2 Hz, 1H), 2.82–2.66 (m, 2H), 2.03 (dt, J = 12.4, 3.3 Hz, 1H), 1.92–1.54 (m, 5H), 1.23 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR (CDCl3) δ 167.1, 157.3, 131.9, 129.2, 121.7, 117.0, 77.9, 76.7, 51.8, 46.7, 38.4, 37.6, 28.2, 27.3, 22.9, 20.0, 14.3; 13C NMR (75.5 MHz, acetone-d6) δ 167.4, 158.7, 132.9, 129.9, 123.6, 122.8, 118.0, 78.8, 78.0, 52.2, 48.0, 39.5, 38.9, 29.4, 28.1, 23.9, 20.6, 15.2. Anal. Calcd for C18H24O4: C, 71.03; H, 7.95. Found C, 70.82; H, 8.10.
5.6 Hexahydroxanthene 12
To a solution of ester 11 (108 mg, 0.4 mmol) in THF (20 mL) at 0 °C was added LiAlH4 (74 mg, 2.0 mmol). After 2 h, the reaction was quenched by addition of H2O, acidified (pH 2), extracted into ethyl acetate, dried (MgSO4), and concentrated in vacuo to afford alcohol 12 (98 mg, 100%) as a white solid: 1H NMR δ 7.10–7.06 (m, 2H), 6.75 (d, J = 8.1 Hz, 1H), 4.58 (s, 2H), 3.41 (dd, J = 11.5, 4.4 Hz, 1H), 2.72 (bd, 1H), 2.69 (d, J = 3.9 Hz, 1H), 2.00 (dt, J = 12.3, 3.2 Hz, 1H), 1.88–1.82 (m, 1H), 1.80–1.50 (m, 4H), 1.25 (br s, 1H), 1.21 (s, 3H), 1.09 (s, 3H), 0.88 (s, 3H); 13C NMR δ 152.8, 132.3, 129.0, 126.6, 122.0, 117.1, 78.0, 76.5, 65.3, 46.9, 38.4, 37.8, 28.3, 27.4, 23.1, 19.9, 14.3. Anal. Calcd for C17H24O3: C, 69.84; H, 8.27. Found C, 70.16; H, 7.92.
5.7 Aldehyde 13
To a solution of diol 12 (98 mg, 0.35 mmol) in CH2Cl2 (25 mL) at rt was added MnO2 (812 mg, 8.22 mmol). After 2 h, the reaction was diluted, filtered through a pad of celite, and the filtrate was concentrated in vacuo. Final purification by crystalization (hexanes) afforded aldehyde 13 (78 mg, 90%) as white needles: 1H NMR δ 9.83 (s, 1H), 7.64–7.61 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 3.45 (d, J = 12.0 Hz, 1H), 2.79–2.75 (m, 2H), 2.07–2.02 (m, 1H), 1.92–1.60 (m, 5H), 1.25 (s, 3H), 1.12 (s, 3H), 0.90 (s, 3H); 13C NMR δ 191.0, 158.9, 132.2, 129.7, 129.2, 122.5, 117.7, 78.0, 77.8, 46.6, 38.4, 37.6, 28.2, 27.3, 22.9, 20.1, 14.3; HRMS (EI) m/z calcd for C17H22O3 (M+) 274.1570, found 274.1579.
5.8 C5-Bromo hexahydroxanthene 14
Method A
To a flask containing unsubstituted hexahydroxanthene 11 (107 mg, 0.4 mmol), glacial acetic acid (1 mL), and CH2Cl2 (35 mL) was added Br2 (0.02 mL, 0.4 mmol) in CH2Cl2 (1 mL) dropwise at room temperature. The solution was allowed to stir for 24 h, and then the reaction was quenched by addition of Na2SO3. Water was added and the product was extracted into CH2Cl2, dried (MgSO4), and concentrated under reduced pressure. Final purification by flash chromatography produced bromide 14 as a white solid (48 mg, 36%): 1H NMR δ 8.03 (d, J = 2.1 Hz, 1H), 7.76 (d, J = 2.2 Hz, 1H), 3.88 (s, 3H), 3.44 (dd, J = 11.4, 4.1 Hz, 1H), 2.78–2.75 (m, 2H), 2.17–2.10 (m, 1H), 1.94–1.55 (m, 5H), 1.24 (s, 3H), 1.11 (s, 3H), 0.88 (s, 3H); 13C NMR δ 166.0, 154.0, 132.5, 130.7, 123.1, 122.4, 111.2, 79.0, 77.7, 52.1, 46.5, 38.4, 37.3, 28.1, 27.3, 23.3, 20.2, 14.3; HRMS (EI) m/z calcd for C18H23O4Br (M+) 382.0780, found 382.0777. In addition the acetate-protected compound 15 was isolated as a white solid (78 mg, 52%): 1H NMR δ 8.04 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 2.1 Hz, 1H), 4.66 (dd, J = 11.5, 3.9 Hz, 1H), 3.88 (s, 3H), 2.78–2.75 (m, 2H), 2.18–1.37 (m, 5H), 2.09 (s, 3H), 1.37 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H); 13C NMR δ 170.6, 165.9, 153.9, 132.5, 130.6, 122.8, 122.5, 111.3, 79.0, 78.5, 52.0, 46.6, 37.4, 37.0, 27.2, 24.6, 23.1, 21.2, 20.2, 15.4; HRMS (EI) m/z calcd for C20H25O5Br (M+) 424.0886, found 424.0889.
Method B
To a flask containing acetate 15 (76 mg, 0.2 mmol), and CH3OH (10 mL) was added potassium carbonate (137 mg, 1.0 mmol) at room temperature. The solution was allowed to stir for 6 h, the CH3OH was removed in vacuo and then the reaction was quenched by addition of NH4Cl. The product was extracted into CH2Cl2, dried (MgSO4), and concentrated under reduced pressure. Final purification by flash chromatography produced the bromide 14 as a white solid (66 mg, 96%).
5.9 MOM-protected bromide 16
To a flask containing alcohol 14 (174 mg, 0.5 mmol), and CH2Cl2 (10 mL) was added DIPEA (0.5 mL, 2.9 mmol) followed by MOMCl (0.08 mL, 1.1 mmol) at room temperature. The solution was allowed to stir for 24 h, and then the reaction was quenched by addition of water. The product was extracted into CH2Cl2, dried (MgSO4), and concentrated under reduced pressure. Final purification by flash chromatography produced 16 as a yellow oil (185 mg, 82%): 1H NMR δ 8.03 (d, J = 1.8 Hz, 1H), 7.76–7.75 (m, 1H), 4.78 (d, J = 6.9 Hz, 1H), 4.65 (d, J = 7.1 Hz, 1H), 3.88 (s, 3H), 3.41 (s, 3H), 3.28 (dd, J = 11.7, 4.1 Hz, 1H), 2.80–2.68 (m, 2H), 2.12 (dt, J = 12.8, 3.3 Hz, 1H), 2.05–1.97 (m, 1H), 1.85–1.51 (m, 3H), 1.25 (s, 3H), 1.09 (s, 3H), 0.90 (s, 3H); 13C NMR δ 166.0, 154.0, 132.5, 130.7, 123.2, 122.4, 111.2, 96.2, 83.7, 78.9, 55.7, 52.0, 46.8, 38.2, 37.3, 27.4, 25.2, 23.3, 20.2, 15.1; HRMS (EI) m/z calcd for C20H27O5Br (M+) 426.1042, found 426.1051.
5.10 Benzyl alcohol 17
To a flask containing ester 16 (185 mg, 0.4 mmol) and THF (10 mL) was added DIBAL-H (2.2 mL, 5.2 mmol) at 0 °C. The solution was allowed to stir for 3 hours, and then the reaction was quenched by addition of saturated NH4Cl. The product was extracted into ether, dried (MgSO4), and concentrated under reduced pressure. Final purification by flash chromatography produced alcohol 17 (158 mg, 92%): 1H NMR δ 7.36 (d, J = 1.8 Hz, 1H), 7.04 (d, J = 1.8 Hz, 1H), 4.77 (d, J = 7.1 Hz, 1H), 4.65 (d, J = 6.6 Hz, 1H), 4.56 (s, 2H), 3.41 (s, 3H), 3.28 (dd, J = 11.4, 4.2 Hz, 1H), 2.72 (d, J = 9.5 Hz, 2H), 2.09 (dt, J = 12.6, 3.0 Hz, 1H), 1.95–1.43 (m, 5H), 1.22 (s, 3H), 1.08 (s, 3H), 0.89 (s, 3H); 13C NMR δ 149.5, 133.2, 129.9, 127.9, 123.7, 111.3, 96.2, 84.0, 77.7, 64.6, 55.7, 47.1, 38.2, 37.4, 27.4, 25.2, 23.4, 20.0, 15.1; HRMS (EI) m/z calcd for C19H27O4Br (M+) 398.1093, found 398.1093.
5.11 Brominated aldehyde 18
To a flask containing benzyl alcohol 17 (76 mg, 0.2 mmol) in methylene chloride (52 mL) was added MnO2 (88% precipitated active, 315 mg, 3.2 mmol). The mixture was allowed to stir for 3.25 h and then the reaction was quenched by filtration through celite. Final purification by flash chromatography produced aldehyde 18 (61 mg, 81%) as a white solid: [α]26.4D = +75.9 (c 0.61, CHCl3, 96% ee by HPLC); 1H NMR δ 9.71, (s, 1H), 7.81 (d, J = 1.7 Hz, 1H), 7.51 (d, J = 1.6 Hz, 1H), 4.71 (d, J = 7.1 Hz, 1H), 4.58 (d, J = 7.1 Hz, 1H), 3.34 (s, 3H), 3.22 (dd, J = 11.7, 4.3 Hz, 1H), 2.77–2.65 (m, 2H), 2.07 (dt, J = 12.7, 3.2 Hz, 1H), 2.01–1.45 (m, 4H), 1.20 (s, 3H), 1.03 (s, 3H), 0.84 (s, 3H); 13C NMR δ 189.8, 155.5, 132.8, 130.8, 129.7, 123.9, 112.3, 96.2, 83.7, 79.4, 55.7, 46.8, 38.3, 37.2, 27.4, 25.2, 23.3, 20.3, 15.1. Anal. Calcd for C19H25O4Br: C, 57.44; H, 6.34. Found C, 57.54; H, 6.37.
5.12 Benzyl alcohol 20
To a solution of ester 19 (254 mg, 0.9 mmol) in THF (3 mL) at 0 °C was added LiAlH4 (35 mg, 0.9 mmol). After 20 min, the reaction was quenched by slow addition of saturated NH4Cl. The resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography afforded alcohol 20 (208 mg, 90%) as a colorless oil: 1H NMR δ 7.26–7.23 (m, 1H), 6.97 (dd, JHF = 11.3 Hz, J = 2.0 Hz, 1H), 5.09 (s, 2H), 4.50 (s, 2H), 3.56 (s, 3H), 2.68 (br s, 1H); 13C NMR δ 156.0 (d, JCF = 254 Hz), 141.1 (d, JCF = 14.0 Hz), 138.7 (d, JCF = 7.0 Hz), 126.5 (d, JCF = 2.3 Hz), 117.8 (d, JCF = 3.4 Hz), 114.3 (d, JCF = 21.2 Hz), 98.9 (d, JCF = 5.6 Hz), 63.5, 57.8; 19F NMR (280 MHz, CDCl3) δ −125.8. Anal. Calcd for C9H10O3BrF: C, 40.93; H, 3.82. Found C, 41.17; H, 3.85.
5.13 Benzyl ether 21
To a solution of benzyl alcohol 20 (198 mg, 0.7 mmol) in THF (5 mL) at 0 °C was added NaH (60% wt. in mineral oil, 35 mg, 0.9 mmol) followed by iodomethane (0.06 mL, 1.0 mmol). After 10 h, the reaction was quenched by addition of water, the resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (8% EtOAc in hexanes) afforded ether 21 (158 mg, 76%) as an oil: 1H NMR δ 7.25–7.24 (m, 1H), 6.98 (dd, JHF = 11.1 Hz, J = 2.0 Hz, 1H), 5.11 (s, 2H), 4.29 (s, 2H), 3.56 (s, 3H), 3.31 (s, 3H); 13C NMR δ 155.9 (d, JCF = 250 Hz), 141.4 (d, JCF = 13.4 Hz), 136.1 (d, JCF = 6.7 Hz), 127.3 (d, JCF = 3.5 Hz), 117.8 (d, JCF = 3.5 Hz), 115.1 (d, JCF = 20.9 Hz), 99.0 (d, JCF = 6.1 Hz), 72.9 (d, JCF = 1.8 Hz), 58.3, 57.8 (d, JCF = 1.7 Hz); 19F NMR (280 MHz, CDCl3) δ −126.1. Anal. Calcd for C10H12BrF O3: C, 43.03; H, 4.33. Found C, 42.87; H, 4.33
5.14 Geranylated Arene 22
To a solution of ether 21 (153 mg, 0.6 mmol) in THF (10 mL) at −78 °C was added n-BuLi (0.27 mL, 0.6 mmol). After 10 min, CuBr·DMS (136 mg, 0.7 mmol) was added. After an additional 30 min, geranyl bromide (0.11 mL, 0.6 mmol) was added and after an additional 1 h, the reaction was quenched by addition of water. The resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography afforded arene 22 (31 mg, 17%) as an oil: 1H NMR δ 6.94–6.79 (m, 2H), 5.21 (t, J = 7.2 Hz, 1H), 5.04 (s, 2H), 5.04–5.00 (m, 1H), 4.28 (s, 2H), 3.51 (s, 3H), 3.34–3.27 (m, 2H), 3.30 (s, 3H), 2.09–1.84 (m, 4H), 1.63 (s, 3H), 1.60 (s, 3H), 1.52 (s, 3H); 13C NMR δ 155.3 (d, JCF = 247 Hz), 141.7 (d, JCF = 10.8 Hz), 136.8, 136.7, 134.4 (d, JCF = 6.9 Hz), 131.5, 124.2, 124.0 (d, JCF = 2.9 Hz), 122.0, 113.6 (d, JCF = 20.6 Hz), 99.1 (d, JCF = 6.0 Hz), 73.9 (d, JCF = 1.1 Hz), 58.2, 57.4 (d, JCF = 1.0 Hz), 39.7, 28.3 (d, JCF = 2.5 Hz), 27.6, 25.7, 17.7, 16.2; 19F NMR (280 MHz, CDCl3): δ −130.6; HRMS (EI) m/z calcd for C20H29O3F (M+) 336.2102, found 336.2106.
5.15 (R)-Epoxide 23
To a solution of benzyl ether 22 (30 mg, 0.1 mmol) in CH2Cl2 (1 mL), CH3CN (0.5 mL), EtOH (0.5 mL), aqueous buffer (2 M K2CO3, 4 × 10−3 M EDTA, 2 mL), and Shi catalyst (24 mg, 0.1 mmol) at 0 °C, H2O2 (30% wt. in H2O, 0.05 mL, 0.44 mmol) was added over 2 h. After an additional 45 min the reaction was quenched by addition of Na2SO3. The resulting solution was extracted with CH2Cl2, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography afforded epoxide 23 (17 mg, 54%) as an oil: 1H NMR δ 6.91–6.82 (m, 2H), 5.26 (t, J = 7.1 Hz, 1H), 5.04 (s, 2H), 4.23 (s, 2H), 3.51 (s, 3H), 3.34 (d, J = 7.1 Hz, 2H), 3.30 (s, 3H), 2.64 (t, J = 6.3 Hz, 1H), 2.29–1.86 (m, 4H), 1.66 (s, 3H), 1.21 (s, 3H), 1.19 (s, 3H); 13C NMR δ 155.3 (d, JCF = 246 Hz), 141.7 (d, JCF = 12.4 Hz), 136.5 (d, JCF = 1.8 Hz), 135.8, 134.5 (d, JCF = 7.4 Hz), 124.0 (d, JCF = 2.8 Hz), 122.6, 113.6 (d, JCF = 20.4 Hz), 99.1 (d, JCF = 6.5 Hz), 73.9 (d, JCF = 1.9 Hz), 64.1, 58.4, 58.2, 57.5, 36.4, 28.4, 27.4, 24.9, 18.8, 16.2; 19F NMR (280 MHz, CDCl3) δ −130.6; HRMS (EI) m/z calcd for C20H29O3F (M+) 352.2051, found 352.2048.
5.16 5-Fluoro-hexahydroxanthene 24
To a solution of epoxide 23 (16 mg, 0.05 mmol) in CH2Cl2 (6 mL) at −78 °C was added BF3·OEt2 (0.03 mL, 0.2 mmol). After 6 min, the reaction was quenched by addition of TEA. Water was added and the product was extracted into CH2Cl2, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (20% EtOAc in hexanes) produced hexahydroxanthene 24 (8 mg, 57%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 6.91–6.85 (m, 2H), 4.33 (s, 2H), 3.44 (dd, J = 11.3, 6.9 Hz, 1H), 3.38 (s, 3H), 2.79–2.66 (m, 2H), 2.12–1.58 (m, 5H), 1.25 (s, 3H), 1.11 (s, 3H), 0.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 151.8 (d, JCF = 246 Hz), 141.0 (d, JCF = 11.3 Hz), 129.4 (d, JCF = 5.6 Hz), 124.4 (d, JCF = 2.8 Hz), 124.0 (d, JCF = 2.8 Hz), 113.4 (d, JCF = 18.3 Hz), 77.9, 74.1 (d, JCF = 1.5 Hz), 58.1, 46.7, 38.4, 37.5, 28.3, 27.3, 23.0, 22.9, 19.8, 14.3; 19F NMR (CDCl3) δ −136.9; HRMS (EI) m/z calcd for C18H25O3F (M+) 308.1789, found 308.1782.
5.17 5-Fluorinated aldehyde 25
To a solution of methyl ether 24 (7 mg, 0.02 mmol) in CH2Cl2 (1.0 mL) and water (0.1 mL) at rt was added DDQ (12 mg, 0.05 mmol). After 4 h, the reaction was quenched by addition of saturated NaHCO3. The resulting solution was extracted with CH2Cl2, dried (MgSO4), and concentrated in vacuo. Final purification by radial chromatography (40% EtOAc in hexanes) produced aldehyde 25 (6 mg, 90%) as a film: 1H NMR (400 MHz, CDCl3) δ 9.81 (d, JHF = 1.9 Hz, 1H), 7.45–7.42 (m, 2H), 3.47 (dd, J = 11.7, 4.0 Hz, 1H), 2.88–2.75 (m, 2H), 2.17–1.58 (m, 5H), 1.30 (s, 3H), 1.14 (s, 3H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.9 (d, JCF = 2.0 Hz), 152.0 (d, JCF = 249 Hz), 147.3 (d, JCF = 11.0 Hz), 128.4 (d, JCF = 5.2 Hz), 127.8 (d, JCF = 2.6 Hz), 124.6 (d, JCF = 2.6 Hz), 113.7 (d, JCF = 19.5 Hz), 78.8, 77.5, 46.3, 38.3, 37.1, 28.0, 27.1, 22.7, 19.9, 14.1; 19F NMR (CDCl3) δ −134.7. HRMS (EI) calcd for C17H21O3F (M+) 292.1477, found 292.1478.
5.18 Ether 27
To a solution of alcohol 2629 (2.66 g, 15.8 mmol) in THF (50 mL) at 0 °C was added NaH (750 mg, 18.8 mmol, 60% dispersion oil). After 30 min, MeI (1.09 mL, 17.5 mmol) was added dropwise and the reaction mixture was allowed to stir overnight. The reaction was quenched by the addition of NH4Cl (sat), and extracted with EtOAc. The combined organic extracts were washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. Final purification by flash column (15% to 40% ethyl acetate in hexanes) afforded arene 27 (2.34 g, 81%) as a colorless oil: 1H NMR δ 7.25 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 5.15 (s, 2H), 4.38 (s, 2H), 3.46 (s, 3H), 3.35 (s, 3H); 13C NMR δ 156.7, 131.5, 129.1 (2C), 116.0 (2C), 94.3, 74.1, 57.7, 55.8; HRMS (ESI+) calcd for C10H14O (M+) 182.0943, found 182.0949.
5.19 Thiol Ether 28
To a solution of arene 27 (2.34 g, 12.8 mmol) in THF (60 mL) at 0°C was added n–BuLi (6.0 mL, 2.2 M in hexanes) and after 5 minutes dimethyldisulfide (1.23 mL, 14 mmol) was added dropwise. After 5 h the reaction mixture was quenched by addition of NH4Cl (sat), extracted with EtOAc, washed with brine, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification by flash column chromatography (13% EtOAc in hexanes) afforded 28 (1.29 g, 44%) as an oil: 1H NMR δ 7.13 (s, 1H), 7.04 (m, 2H), 5.22 (s, 2H), 4.38 (s, 2H), 3.49 (s, 3H), 3.36 (s, 3H), 2.43 (s, 3H); 13C NMR δ 153.2, 132.2, 128.2, 125.2, 125.2, 113.9, 94.7, 74.1, 57.5, 56.0, 14.4; HRMS (EI+) calcd for C11H16O3S (M+) 228.0820, found 228.0822.
5.20 Epoxide 29
To a solution of arene 28 (829 mg, 3.63 mmol), in THF (15 mL) at to 0 °C was added n–BuLi (1.6 mL, 3.68 mmol). After 1 h, the solution was cooled to –20 °C and CuBr·DMS (784 mg, 3.81 mmol) was added. After an additional 1 h, epoxygeranyl bromide22 (887 mg, 3.93 mmol) was added dropwise as a THF solution (2 mL). After 2 h, the reaction mixture was quenched by addition of NH4Cl (sat), diluted with water and extracted with EtOAc. The combined organic extracts were washed with water, brine, dried (MgSO4), filtered, and then concentrated in vacuo. Final purification by column chromatography (15% EtOAc in hexanes) afforded epoxide 29 (522 mg, 38%) as a light yellow oil: 1H NMR δ 6.99 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 1.6 Hz, 1H), 5.36 (m, 1H), 5.03 (s, 2H), 4.37 (s, 2H), 3.63 (s, 3H), 3.43 (d, J = 7.2 Hz, 2H), 3.38 (s, 3H), 2.71 (t, J = 6.2 Hz, 1H), 2.43 (s, 3H), 2.04 (m, 2H), 1.73 (s, 3H), 1.69–1.62 (m, 2H), 1.28 (s, 3H), 1.25 (s, 3H); 13C NMR δ 151.6, 135.4, 134.8, 134.8, 132.6, 125.9, 123.1, 123.0, 99.2, 74.3, 64.0, 58.2, 58.0, 57.6, 36.3, 28.4, 27.3, 24.7, 18.6, 16.1, 14.8; HRMS (EI) calcd for C21H32O4S (M+) 380.2120, found 380.2127.
5.21 Hexahydroxanthene 30
To a solution of epoxide 29 (207 mg, 0.54 mmol) in CH2Cl2 (136 mL) at −78 °C was added BF3·OEt2 (0.40 mL, 3.3 mmol). After 10 minutes, the reaction was quenched by addition of TEA (0.3 mL), allowed to warm to room temperature, and the solvent was removed in vacuo. Final purification by flash column chromatography (25–30% EtOAc in hexanes) gave hexahydroxanthene 30 (93 mg, 51%) as an oil: 1H NMR δ 6.93 (d, J = 1.6 Hz, 1H), 6.87 (d, J = 1.6 Hz, 1H), 4.34 (s, 2H), 3.40–3.35 (m, 1H), 3.74 (s, 3H), 2.71–2.68 (m, 2H), 2.40 (s, 3H), 2.09–2.04 (m, 1H), 1.87–1.77 (m, 2H), 1.71–1.57 (m, 3H), 1.21 (s, 3H), 1.07 (s, 3H), 0.86 (s, 3H); 13C NMR δ 149.6, 129.6, 126.3, 126.2, 123.6, 121.2, 77.9, 77.2, 74.6, 57.9, 46.8, 38.3, 37.5, 28.2, 27.2, 23.1, 19.9, 14.6, 14.2; HRMS (EI) calcd for C19H28O3S (M+) 336.1759, found 336,1750.
5.22 Aldehyde 31
To a solution of methyl ether 30 (84 mg, 0.25 mmol) in a 9:1 mixture of CH2Cl2 and H2O (10 mL) at rt was added DDQ (79 mg, 0.35 mmol). After 20 min, the reaction was quenched by the addition of NaHCO3 (sat.), diluted with water and extracted with CH2Cl2. The combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated in vacuo to afford aldehyde 31 (61 mg, 76%) as a white solid: 1H NMR δ 9.82 (s, 1H), 7.48 (d, J = 1.6 Hz, 1H), 7.42 (d, J = 1.1 Hz, 1H), 3.45 (dd, J = 11.2, 3.6 Hz, 1H), 2.80–2.76 (m, 2H), 2.45 (s, 3H), 2.16–2.10 (m, 1H), 1.94–1.81 (m, 2H), 1.76–1.56 (m, 3H), 1.27 (s, 3H), 1.12 (s, 3H), 0.90 (s, 3H); 13C NMR δ 190.9, 155.2, 130.0, 129.5, 128.9, 123.6, 121.5, 79.1, 77.9, 46.7, 38.5, 37.5, 28.3, 27.4, 23.1, 20.4, 14.4, 14.4; HRMS (EI) calcd for C18H24O3S (M+) 320.1446, found 320.1447.
5.23 Aldehyde 33
To a solution of methyl ether 325 (350 mg, 1.1 mmol), in CH2Cl2/water (10:1) at rt was added DDQ (320 mg, 1.4 mmol). After 15 min, the reaction was quenched by addition of brine and NaHCO3. The resulting solution was extracted with CH2Cl2, and the combined organic extracts were washed with a small amount of water followed by brine. After the organic phase was dried (MgSO4) and concentrated in vacuo, aldehyde 33 was obtained as a faintly yellow wax that was used without further purification: 1H NMR δ 9.76 (s, 1H), 7.65 (d, J = 1.6 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 5.26 (s, 1H), 4.64 (d, J = 13.2 Hz, 1H), 4.60 (d, J = 13.6 Hz, 1H), 3.38 (dd, J = 11.4, 4.2 Hz, 1H), 2.79–2.67 (m, 2H), 2.34 (br, 1H), 2.04–1.99 (m, 1H), 1.87–1.57 (m, 4H), 1.20 (s, 3H), 1.07 (s, 3H), 0.85 (s, 3H); 13C NMR δ 191.2, 156.2, 131.3, 129.6, 128.6, 127.7, 122.2, 78.4, 77.5, 60.7, 46.4, 38.3, 37.5, 27.9, 27.1, 22.7, 20.2, 14.2; HRMS (EI) calcd for C18H24O4 (M+) 304.1675, found 304.1668.
5.24 Silyl Ether 34
To a solution of alcohol 33, in CH2Cl2 at rt was added TBSCl (485 mg, 3.2 mmol) followed by imidazole (394 mg, 5.8 mmol). After 45 min, the reaction was quenched by addition of water. The resulting solution was extracted with CH2Cl2, and the combined organic extracts were washed with a small amount of water followed by brine. After which the organic phase was dried (MgSO4) and concentrated in vacuo. Final purification by column chromatography (30% EtOAc in hexanes) afforded aldehyde 34 (321 mg, 70% over 2-steps) as a colorless oil: 1H NMR (CDCl3) δ 9.82 (s, 1H), 7.78 (d, J = 1.2 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 4.70 (d, J = 14.4 Hz, 1H), 4.62 (d, J = 14.1 Hz, 1H), 3.40 (dd, J = 11.4, 3.9 Hz, 1H), 2.77–2.72 (m, 2H), 2.07–2.00 (m, 1H), 1.89–1.62 (m, 4H), 1.20 (s, 3H), 1.09 (s, 3H), 0.94 (s, 9H), 0.87 (s, 3H), −0.11 (s, 6H); 13C NMR δ 191.5, 155.3, 130.1, 130.0, 128.6, 127.4, 121.6, 77.9, 77.6, 59.7, 46.4, 38.3, 37.5, 28.0, 27.1, 25.9 (3C), 22.8, 20.2, 18.4, 14.2, −5.4 (2C); HRMS (EI) calcd for C24H38O Si (M+–tBu) 362.1869, found 362.1861.
5.25 Silyl ether 36
To a flask containing benzyl alcohol 35 (1.81 g, 9.2 mmol) and CH2Cl2 (150 mL) was added imidazole (3.19 g, 46.9 mmol), followed by TBSCl (1.61 g, 10.7 mmol) at room temperature. The solution was allowed to stir for 9 h, and then the reaction was quenched by addition of saturated NH4Cl. The product was extracted into CH2Cl2, dried (MgSO4), and concentrated under reduced pressure. Final purification by flash chromatography produced the silyl ether 36 as an oil (2.40 g, 84%): 1H NMR δ 6.62–6.57 (m, 2H), 6.49–6.48 (m, 1H), 5.15 (s, 2H), 4.68 (s, 2H), 3.78 (s, 3H), 3.47 (s, 3H), 0.94 (s, 9H), 0.10 (s, 6H); 13C NMR δ 160.9, 158.5, 144.4, 106.2, 105.2, 101.2, 94.7, 65.0, 56.2, 55.5, 26.2 (3C), 18.6, −5.0 (2C); HRMS (EI) calcd for C16H28O4Si (M+) 312.1757, found 312.1753.
5.26 Geranylated benzyl alcohol 38
To a flamed dried Schlenk flask under argon, ether (30 mL) was added via syringe followed by TMEDA (3.8 mL, 25 mmol) and n-BuLi (12 mL, 29 mmol, 2.4 M solution in hexanes) and this solution was cooled to 0 °C. Compound 35 (2.97 g, 12.6 mmol) was dissolved in ether (20 mL) and transferred via cannula to the reaction vessel, which gave a white precipitate. After 20 min, solid CuI (2.64 g, 13.8 mmol) was added in one portion leading to an immediate color change to black. The resulting mixture was allowed to stir for 20 min and then geranyl bromide (3.3 mL, 16.3 mmol) was added dropwise over a 10 min period. The reaction mixture was allowed to stir for 4 h, and then was quenched by addition of water. The product was extracted into diethyl ether, dried (MgSO4), and concentrated under reduced pressure. Final purification by column chromatography (30% EtOAc in hexanes) produced alcohol 38 (1.98 g, 47%) as an oil: 1H NMR δ 6.72 (s, 1H), 6.61 (s, 1H), 5.20–5.06 (m, 1H), 5.18 (s, 2H), 5.06–5.04 (m, 1H), 4.63 (s, 2H), 3.82 (s, 3H), 3.46 (s, 3H), 3.36 (d, J = 6.9 Hz, 2H), 2.05–2.00 (m, 2H), 1.96–1.91 (m, 2H), 1.77 (s, 3H), 1.64 (s, 3H), 1.57 (s, 3H); 13C NMR δ 158.3, 155.6, 139.9, 134.6, 131.1, 124.4, 122.6, 118.6, 105.6, 103.3, 94.4, 65.6, 55.9, 55.7, 39.8, 26.7, 25.6, 22.3, 17.6, 16.0; HRMS (EI) calcd for C20H30O4 (M+) 334.2144, found 334.2141.
5.27 Phosphonate 39
To a solution of alcohol 38 (1.00 g, 3.0 mmol) in CH2Cl2 (200 mL) at 0 °C was added TEA (1.70 mL, 12.2 mmol), followed by MsCl (0.93 mL, 12.0 mmol). After 20 h, the reaction was quenched by addition of saturated NH4Cl. The resulting solution was extracted with CH2Cl2, dried (MgSO4), and concentrated in vacuo to yield the intermediate mesylate as an oil which was used without further purification.
To a solution of the crude intermediate mesylate in acetone (11 mL) in a foil-wrapped flask at rt was added sodium iodide (1.81 g, 12.1 mmol). After 30 min, the reaction was concentrated in vacuo. The residue was diluted with water and the resulting solution was extracted with ether, dried (MgSO4), and concentrated in vacuo to yield the intermediate iodide as a yellow oil, which was used without further purification.
To a flask containing the intermediate iodide, triethylphosphite (1.1 mL, 12.1 mmol) was added at rt. The solution was heated to 62 °C, and after 16 h the reaction was quenched by addition of water. The resulting solution was extracted with ethyl acetate, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography afforded phosphonate 39 (1.36 g, 100% for 3 steps) as a colorless oil: 1H NMR δ 6.65–6.64 (m, 1H), 6.55–6.54 (m, 1H), 5.19–5.14 (m, 1H), 5.16 (s, 2H), 5.09–5.04 (m, 1H), 4.08–4.00 (m, 4H), 3.81 (s, 3H), 3.45 (s, 3H), 3.33 (d, J = 7.1 Hz, 2H), 3.10 (d, JHP = 21.5 Hz, 2H), 2.07–1.91 (m, 4H), 1.75 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.26 (t, J = 7.0 Hz, 6H); 13C NMR δ 158.0, 155.5, 134.6, 131.2, 130.0, 124.5, 122.7, 118.0 (d, JCP = 3.9 Hz), 108.7 (d, JCP = 6.6 Hz), 106.4 (d, JCP = 6.1 Hz), 94.4, 62.1 (d, JCP = 7.4 Hz, 2C), 55.9, 55.8, 39.8, 34.0 (d, JCP = 138.2 Hz), 26.7, 25.7, 22.2, 17.7, 16.4 (d, JCP = 6.1 Hz, 2C), 16.0; 31P NMR δ 27.1; HRMS (EI) m/z calcd for C24H39O6P (M+) 454.2486, found 454.2471.
5.28 Stilbene 43
Under the general conditions for HWE condensations (vide infra), the reaction of aldehyde 13 (36 mg, 0.1 mmol), phosphonate 39 (31 mg, 0.1 mmol), and NaH (60% wt. in mineral oil, 34 mg, 0.9 mmol) provided stilbene 43 as a white solid (49 mg, 66%): 1H NMR (400 MHz, CDCl3) δ 7.26–7.25 (m, 1H), 6.99–6.86 (m, 4H), 6.74 (d, J = 8.3 Hz, 1H), 6.70 (s, 1H), 5.22 (s, 2H), 5.20–5.18 (m, 1H), 5.07 (t, J = 6.7 Hz, 1H), 3.86 (s, 3H), 3.49 (s, 3H), 3.43 (dd, J = 11.8, 4.6 Hz, 1H), 3.37 (d, J = 7.0 Hz, 2H), 2.76–2.66 (m, 2H), 2.07–1.48 (m, 9H), 1.78 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.23 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.1, 155.6, 152.7, 136.5, 134.4, 131.0, 129.2, 127.7, 127.2, 126.3, 125.4, 124.3, 122.5, 121.8, 118.6, 117.1, 105.2, 102.4, 94.4, 77.9, 76.5, 55.8, 55.6, 46.7, 39.7, 38.2, 37.6, 28.1, 27.2, 26.6, 25.5, 22.9, 22.3, 19.8, 17.5, 15.9, 14.1; HRMS (EI) m/z calcd for C37H50O5 (M+) 574.3660, found 574.3651.
5.29 Schweinfurthin analogue 51
Under general conditions for the removal of MOM-ethers from protected stilbenes (vide infra), stilbene 53 (33 mg, 0.1 mmol) was treated with methanol (0.3 mL) and p-TsOH·H2O (56 mg, 0.3 mmol) for 2.5 h to provide analogue 51 as a clear oil (16 mg, 53%): 1H NMR (400 MHz, CDCl3) δ 7.25–6.61 (m, 7H), 5.33 (br s, 1H), 5.25 (t, J = 7.0 Hz, 1H), 5.07 (t, J = 6.7 Hz, 1H), 3.87 (s, 3H), 3.47–3.42 (m, 3H), 2.80–2.68 (m, 2H), 2.18–1.55 (m, 10H), 1.82 (s, 3H), 1.69 (s, 3H), 1.60 (s, 3H), 1.24 (s, 3H), 1.13 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.8, 155.5, 152.8, 138.1, 136.9, 131.7, 129.1, 128.0, 127.7, 126.0, 125.4, 123.7, 121.8, 121.6, 117.1, 114.3, 106.7, 101.1, 77.9, 77.0, 55.6, 46.7, 39.5, 38.2, 37.6, 28.1, 27.2, 26.3, 25.5, 22.9, 22.1, 19.8, 17.5, 16.0, 14.1; HRMS (EI) m/z calcd for C35H46O4 (M+) 530.3398, found 530.3399.
5.30 C-5 Bromostilbene 44
Under the general conditions for HWE condensations, the reaction of aldehyde 18 (60 mg, 0.2 mmol), phosphonate 39 (72 mg, 0.2 mmol), and NaH (60% wt. in mineral oil, 33 mg, 0.8 mmol) provided stilbene 44 as a white solid (57 mg, 54%): [α]26.4D = +64.2 (c 0.25, CHCl3, 96% ee by HPLC); 1H NMR δ 7.46 (d, J = 1.9 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 6.82–6.79 (m, 3H), 6.62 (s, 1H), 5.15 (s, 2H), 5.12 (t, J = 7.0 Hz, 1H), 5.00 (t, J = 6.4 Hz, 1H), 4.71 (d, J = 6.9 Hz, 1H), 4.58 (d, J = 7.0 Hz, 1H), 3.79 (s, 3H), 3.42 (s, 3H), 3.35 (s, 3H), 3.30 (d, J = 7.0 Hz, 1H), 3.22 (dd, J = 11.6, 4.1 Hz, 1H), 2.81 (d, J = 9.5 Hz, 2H), 2.06–1.36 (m, 9H), 1.70 (s, 3H), 1.57 (s, 3H), 1.50 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 0.83 (s, 3H); 13C NMR δ 158.2, 155.8, 149.5, 136.2, 134.7, 131.2, 130.3, 128.8, 127.6, 127.0, 126.5, 124.5, 123.6, 122.6, 119.1, 111.7, 105.4, 102.7, 96.2, 94.5, 83.9, 77.9, 56.0, 55.7, 55.7, 47.1, 38.8, 38.2, 37.3, 27.4, 26.7, 25.7, 25.2, 23.4, 22.5, 20.1, 17.6, 16.1, 15.1; HRMS (EI) m/z calcd for C39H53O6Br (M+) 696.3027, found 696.3098.
5.31 5-Bromoschweinfurthin analogue 52
Under the general conditions for removal of MOM-acetals, the reaction of stilbene 44 (10 mg, 0.01 mmol), methanol (1.5 mL), and p-TsOH·H2O (11 mg, 0.06 mmol) for 96 h provided analogue 52 as a white solid (8 mg, 92%): [α]26.4D = +29.2 (c 0.27, CHCl3, 96% ee by HPLC); 1H NMR (400 MHz, CDCl3) δ 7.53–6.60 (m, 6H), 5.33 (br s, 1H), 5.25 (t, J = 6.6 Hz, 1H), 5.07 (t, J = 6.6 Hz, 1H), 3.87 (s, 3H), 3.48–3.38 (m, 3H), 2.79–2.70 (m, 2H), 2.14–1.35 (m, 10H), 1.82 (s, 3H), 1.69 (s, 3H), 1.60 (s, 3H), 1.25 (s, 3H), 1.12 (s, 3H), 0.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.8, 155.6, 149.4, 138.2, 136.5, 131.7, 130.1, 128.7, 127.1, 126.8, 126.6, 123.7, 123.3, 121.5, 114.6, 111.6, 106.9, 101.2, 77.8, 77.7, 55.6, 46.6, 39.5, 38.2, 37.3, 28.1, 27.1, 26.2, 25.5, 23.2, 22.1, 19.9, 17.5, 16.0, 14.0; HRMS (EI) m/z calcd for C35H45O4Br (M+ 608.2503, found 608.2498.
5.32 5-Fluoro stilbene 45
Under the general conditions for HWE reactions, aldehyde 25 (6 mg, 0.02 mmol), phosphonate 39 (10 mg, 0.02 mmol), and NaH (60% wt. in mineral oil, 5 mg, 0.1 mmol), and purification by column chromatography (25% EtOAc in hexanes) provided stilbene 45 as a white solid (6 mg, 53%): 1H NMR (400 MHz, CDCl3) δ 7.11–6.70 (m, 6H), 5.23 (s, 2H), 5.23–5.19 (m, 1H), 5.08 (t, J = 6.7 Hz, 1H), 3.88 (s, 3H), 3.51 (s, 3H), 3.46 (dd, J = 11.1, 3.6 Hz, 1H), 3.38 (d, J = 6.9 Hz, 2H), 2.81–2.68 (m, 2H), 2.13–1.60 (m, 9H), 1.78 (s, 3H), 1.66 (s, 3H), 1.58 (s, 3H), 1.28 (s, 3H), 1.13 (s, 3H), 0.91 (s, 3H); 13C NMR (CDCl3) δ 158.1, 155.7, 151.9 (d, JCF = 244 Hz), 140.9 (d, JCF = 10.3 Hz), 136.0, 134.5, 131.0, 129.0 (d, JCF = 8.3 Hz), 127.5, 126.8 (d, JCF = 2.3 Hz), 125.3, 124.3, 122.8 (d, JCF = 2.3 Hz), 122.4, 119.0, 111.0 (d, JCF = 19.0 Hz), 105.3, 102.6, 94.3, 77.7, 77.3, 55.8, 55.6, 46.6, 39.6, 38.2, 37.3, 28.1, 27.1, 26.6, 25.5, 22.8, 22.3, 19.7, 17.5, 15.9, 14.1; 19F NMR (CDCl3) δ −136.8; HRMS (EI) m/z calcd for C37H49O5F (M+) 592.3566, found 592.3574.
5.33 5-Fluoro schweinfurthin analogue 53
Under the general conditions for MOM hydrolysis, stilbene 45 (6 mg, 0.01 mmol), methanol (0.6 mL), and p-TsOH·H2O (12 mg, 0.1 mmol) were allowed to react for 24 h. Final purification by column chromatography provided analogue 53 as a white solid (4 mg, 72%): 1H NMR (400 MHz, CDCl3) δ 7.09 (dd, J = 12.1, 1.7 Hz, 1H), 6.99 (s, 1H), 6.91 (d, J = 16.3 Hz, 1H), 6.84 (d, J = 16.3 Hz, 1H), 6.64 (d, J = 1.3 Hz, 1H), 6.60 (d, J = 1.3 Hz, 1H), 5.34 (br s, 1H), 5.27–5.23 (m, 1H), 5.09–5.05 (m, 1H), 3.87 (s, 3H), 3.48–3.42 (m, 3H), 2.82–1.84 (m, 11H), 1.82 (s, 3H), 1.69 (s, 3H), 1.60 (s, 3H), 1.27 (s, 3H), 1.13 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 157.8, 155.6, 151.9 (d, JCF = 243 Hz), 141.0 (d, JCF = 11.3 Hz), 138.2, 136.5, 131.7, 129.0 (d, JCF = 7.5 Hz), 127.1, 127.1 (d, JCF = 2.3 Hz), 124.3 (d, JCF = 1.6 Hz), 123.7, 122.9 (d, JCF = 2.1 Hz), 121.5, 114.6, 111.0 (d, JCF = 19.2 Hz), 106.8, 101.2, 77.7, 77.3, 55.6, 46.6, 39.5, 38.2, 37.3, 28.1, 27.1, 26.3, 25.5, 22.8, 22.1, 19.7, 17.5, 15.9, 14.1; 19F NMR (CDCl3) δ −136.8; HRMS (EI) m/z calcd for C35H45O4F + 35 45O4F (M+) 548.3304, found 548.3299.
5.34 Stilbene 46
Under the general conditions for HWE condensations, aldehyde 31 (23 mg, 0.07 mmol), phosphonate 39 (40 mg, 0.09 mmol), THF (0.7 mL), and NaH (50 mg, 1.25 mmol, 60% dispersion oil) were allowed to react for 18 h. Final purification by flash column chromatography (25% EtOAc in hexanes) afforded stilbene 46 (33 mg, 74%) as a colorless oil; 1H NMR δ 7.11 (d, J = 1.7 Hz, 1H), 7.07 (d, J = 1.6 Hz, 1H), 6.96 (d, J = 16.3 Hz, 1H), 6.92–6.88 (m, 2H), 6.71 (s, 1H), 5.23 (s, 2H), 5.23–5.18 (m, 1H), 5.09–5.05 (m, 1H), 3.87 (s, 3H), 3.50 (s, 3H), 3.43 (dd, J = 11.5, 3.8 Hz, 1H), 3.37 (d, J = 7.0 Hz, 2H), 2.74–2.70 (m, 2H), 2.45 (s, 3H), 2.11–1.57 (m, 10H), 1.78 (s, 3H), 1.64 (s, 3H), 1.57 (s, 3H), 1.24 (s, 3H), 1.10 (s, 3H), 0.88 (s, 3H); 13C NMR δ 158.3, 155.8, 149.8, 136.5, 134.6, 131.1, 129.5, 127.7, 126.8, 126.6, 124.7, 124.4, 122.6, 122.0, 121.4, 118.9, 105.4, 102.6, 94.5, 77.8, 77.5, 55.9, 55.7, 46.8, 39.8, 38.4, 37.5, 28.2, 27.3, 26.7, 25.6, 23.1, 22.4, 20.0, 17.6, 16.0, 14.8, 14.2 HRMS (EI) calcd for C38H52O5S (M+) 620.3535, found 620.3536.
5.35 Schweinfurthin analogue 54
Under general conditions for MOM hydrolysis, stilbene 46 (16.5 mg, 0.03 mmol), methanol (1.5 mL), and TsOH (25 mg, 0.13 mmol) were allowed to react for 18 h. Final purification by column chromatography (25% EtOAc in hexanes) afforded analogue 54 (6 mg, 40%) as an off-white solid: 1H NMR δ 7.11 (d, J = 1.5 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 6.95 (d, J = 16.2 Hz, 1H), 6.86 (d, J = 16.2 Hz, 1H), 6.64 (d, J = 1.1 Hz, 1H), 6.60 (d, J = 1.0 Hz, 1H), 5.26–5.23 (m, 1H), 5.08–5.04 (m, 1H), 3.86 (s, 3H), 3.47–3.41 (m, 3H), 2.74–2.71 (m, 2H), 2.45 (s, 3H), 2.10–2.03 (m, 4H), 1.90–1.63 (m, 5H), 1.81 (s, 3H), 1.68 (s, 3H), 1.59 (s, 3H), 1.24 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR (CDCl3) δ 158.0, 155.8, 153.0, 138.3, 137.2, 131.9, 129.3, 128.2, 127.9, 126.2, 125.6, 123.9, 122.0, 121.8, 117.4, 114.5, 106.9, 101.3, 78.1, 77.2, 55.8, 46.9, 39.8, 38.4, 37.8, 30.3, 28.3, 27.4, 26.5, 25.7, 23.1, 22.3, 20.0, 17.7, 16.2, 14.3; HRMS (EI) calcd for C36H48O4S (M+) 576.3273, found 576.3279.
5.36 5-Methoxymethyl stilbene 47
Under the general conditions for HWE condensations, aldehyde 40 (26 mg, 0.1 mmol), phosphonate 39 (42 mg, 0.1 mmol), and NaH (60% wt. in mineral oil, 48 mg, 1.2 mmol) were allowed to react. Final purification by column chromatography provided stilbene 47 (26 mg, 50%) as a solid: [α]26.4D = +31.1 (c 0.30, CHCl3, 81% ee by HPLC); 1H NMR δ 7.30 (d, J = 1.6 Hz, 1H), 7.11 (d, J = 1.6 Hz, 1H), 6.89–6.64 (m, 4H), 5.15 (s, 2H), 5.19–5.10 (m, 1H), 5.02–4.97 (m, 1H), 4.38 (s, 2H), 3.79 (s, 3H), 3.42 (s, 3H), 3.37 (s, 3H), 3.35–3.38 (m, 3H), 2.67–2.63 (m, 2H), 2.02–1.75 (m, 5H), 1.70 (s, 3H), 1.57 (s, 3H), 1.50 (s, 3H), 1.14–1.13 (m, 4H), 1.13 (s, 3H), 1.03 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.2, 155.8, 150.4, 136.7, 134.6, 131.2, 128.9, 128.0, 127.0, 126.5 (2C), 124.5, 124.5, 122.7, 121.7, 118.7, 105.4, 102.6, 94.5, 78.1, 76.7, 69.2, 58.5, 56.0, 55.7, 46.8, 39.9, 38.7, 37.7, 28.3, 27.3, 26.8, 25.7, 23.1, 22.7, 20.2, 17.7, 16.1, 14.3; HRMS (EI) m/z calcd for C39H54O6 (M+) 618.3922, found 618.3922.
5.37 5-Methoxymethyl schweinfurthin analogue 55
Under the general conditions for MOM hydrolysis, stilbene 47 (34 mg, 0.1 mmol), methanol (3.0 mL), and p-TsOH·H2O (59 mg, 0.3 mmol) were allowed to react for 14 h. Purification by radial chromatography (20% EtOAc in hexanes) afforded analogue 55 (13 mg, 41%) as an oil: [α]26.4D = +40.0 (c 0.68, CHCl3, 81% ee by HPLC); 1H NMR (CDCl3) δ 7.31–7.01 (m, 2H), 6.90 (d, J = 16.3 Hz, 1H), 6.80 (d, J = 16.3 Hz, 1H), 6.58–6.46 (m, 2H), 5.29 (br s, 1H), 5.21–5.11 (m, 1H), 5.00–4.97 (m, 1H), 4.38 (s, 2H), 3.78 (s, 3H), 3.37 (s, 3H), 3.34 (d, J = 6.7 Hz, 2H), 2.73–2.58 (m, 2H), 2.09–1.40 (m, 11H), 1.73 (s, 3H), 1.60 (s, 3H), 1.52 (s, 3H), 1.13 (s, 3H), 1.03 (s, 3H), 0.81 (s, 3H); 13C NMR (CDCl3) δ 157.8, 155.5, 150.3, 138.1, 137.0, 131.7, 128.7, 128.1, 126.9, 126.3, 125.9, 124.3, 123.7, 121.6, 121.5, 114.2, 106.7, 101.1, 77.9, 77.0, 69.0, 58.3, 55.6, 46.6, 39.5, 38.2, 37.5, 28.1, 27.1, 26.3, 25.5, 22.9, 22.1, 20.0, 17.5, 16.0, 14.1; HRMS (EI) m/e calcd for C37H49O5 (M-H)− 573.3580, found 573.3560.
5.38 Stilbene 48. General Procedure for HWE Condensations
To a suspension of NaH (85 mg, 60% oil dispersion, 2.1 mmol) and 15-crown-5 (0.01 mL, 0.05 mmol) in THF (10 mL) at 0 °C was added a solution of phosphonate 39 (65 mg, 0.14 mmol) in THF (1.5 mL). The resulting mixture was stirred for 0.5 h. and aldehyde 41 (39 mg, 0.12 mmol) in THF (0.5 mL) was then added to the cooled solution. After the reaction was allowed to warm to room temperature and stirred for 16 h, it was quenched by addition of water and extracted with EtOAc. The combined organic extracts were washed with water and brine, dried (MgSO4), concentrated in vacuo to a yellow liquid, and purified by flash column chromatography (2:1 hexanes/EtOAc) to afford stilbene 48 (57 mg, 77%) as a colorless oil: [α]26.4D = +38.4 (c 3.74, CHCl3); 1H NMR δ 7.13 (d, J = 1.8 Hz, 1H), 6.95 (d, J = 1.8 Hz, 1H), 6.94 (d, J = 15.9 Hz, 1H), 6.90–6.86 (m, 1H), 6.86 (s, 1H), 6.71 (s, 1H), 5.25–5.18 (m, 5H), 5.07 (t, J = 6.6 Hz,1H), 3.86 (s, 3H), 3.54 (s, 3H), 3.49 (s, 3H), 3.42 (dd, J = 11.4, 3.9 Hz, 1H), 3.39 (d, J = 6.9 Hz, 2H), 2.74 (s, 1H), 2.71 (d, J = 2.7 Hz, 1H), 2.15–2.05 (m, 1H), 2.00–1.80 (m, 5H), 1.78 (s, 3H), 1.75–1.70 (m, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.24 (s, 3H), 1.10 (s, 3H), 0.88 (s, 3H); 13C NMR δ 158.4, 155.9, 146.3, 143.9, 136.6, 134.7, 131.3, 129.2, 127.9, 126.9, 124.6, 123.3, 122.8, 122.1, 118.9, 113.6, 105.5, 102.7, 96.0, 94.6, 78.1, 77.1, 56.3, 56.1, 55.8, 46.9, 39.9, 38.5, 37.9, 28.4, 27.4, 26.8, 25.8, 23.3, 22.6, 20.0, 17.8, 16.2, 14.4; HRMS (EI+) m/z calcd for C39H54O7 (M+) 634.3870, found 634.3876.
5.39 3-Deoxy-5'-O-Methylschweinfurthin A (56). General Procedure for MOM hydrolysis
To a solution of stilbene 48 (54 mg, 0.09 mmol) in MeOH (10 mL) was added TsOH (80 mg, 0.47 mmol) at room temperature and the solution was stirred for 23 h. The reaction was quenched by addition of NaHCO3 (sat.) and extracted with EtOAc. The organic extracts were washed with H2O and brine, dried (MgSO4), and concentrated in vacuo to afford a yellow oil. Final purification by flash column chromatography (2:1 hexanes/EtOAc) gave compound 56 (29 mg, 61%) as a white solid: [α]26.4D = +44.7 (c 1.84, CH3OH); 1H NMR (CD3OD) δ 6.89 (d, J = 16.2, 1H), 6.83 (d, J = Hz, 1H), 6.79 (d, J = 16.2, 1H), 6.73 (s, 1H), 6.58 (s, 2H), 5.19 (t, J = 7.2 Hz, 1H), 5.05 (t, J = 6.9 Hz, 1H), 3.81 (s, 3H), 3.37-3.33 (m, 1H), 3.31–3.28 (m, 2H), 2.70–2.67 (m, 2H), 2.05–2.00 (m, 2H), 1.95–1.90 (m, 2H), 1.82–1.78 (m, 2H), 1.78 (s, 3H), 1.70–1.65 (m, 3H), 1.61 (s, 3H), 1.55 (s, 3H), 1.21 (s, 3H), 1.08 (s, 3H), 0.86 (s, 3H); 13C NMR (CD3OD) δ 159.9, 156.9, 147.0, 142.2, 137.8, 134.8, 131.9, 130.9, 128.9, 127.6, 125.5, 124.5, 123.9, 120.6, 117.1, 111.1, 107.1, 101.6, 78.7, 78.2, 56.1, 48.6, 41.0, 39.5, 38.9, 29.0, 27.9, 27.8, 25.9, 24.0, 23.1, 20.3, 17.7, 16.2, 14.9; HRMS (EI+) m/z calcd for C35H46O5 (M+) 546.3345, found 546.3340.
5.40 Stilbene 49
Under the general conditions for HWE condensations, aldehyde 42 (98 mg, 0.32 mmol), phosphonate 39 (172 mg, 0.38 mmol), and NaH (130 mg, 3.2 mmol, 60% in oil) were allowed to react in THF (6.2 mL) for 15 h. Final purification by column chromatography (1:1 hexanes/ethyl acetate) afforded stilbene 49 (175 mg, 90%) as a yellow oil: 1H NMR (CDCl3) δ 7.00–6.72 (m, 6H), 5.23 (s, 2H), 5.23–5.21 (m, 1H), 5.08 (t, J = 8.5 Hz, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 3.50 (s, 3H), 3.46–3.37 (m, 3H), 2.73 (d, J = 9.2 Hz, 2H), 2.17–1.51 (m, 10H), 1.78 (s, 3H), 1.65 (s, 3H), 1.58 (s, 3H), 1.26, (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H);13C NMR (CDCl3) δ 158.2, 155.8, 148.9, 142.5, 136.5, 134.6, 131.1, 128.9, 128.0, 126.6, 124.4, 122.59, 122.56, 120.5, 118.7, 106.8, 105.3, 102.5, 94.4, 77.9, 77.1, 55.94, 55.92, 55.7, 46.7, 39.8, 38.3, 37.6, 28.2, 27.3, 26.7, 25.6, 23.1, 22.4, 19.8, 17.6, 16.0, 14.2; HRMS (ESI) m/z calcd for C38H52O6 (M+) 604.3764, found 604.3754.
5.41 3-Deoxy-schweinfurthin B Analogue 57
Under the general conditions for MOM hydrolysis, stilbene 49 (80 mg, 0.13 mmol), MeOH (35 mL), and p-TsOH (75 mg, 0.42 mmol) were allowed to react for 4 days. Final purification by column chromatography (1:1 hexanes/ethyl acetate) afforded compound 57 (68 mg, 92%) as a yellow oil: 1H NMR (CDCl3) δ 7.00–6.60 (m, 6H), 5.28–5.24 (m, 1H), 5.07–5.05 (m, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.43–3.37 (m, 3H), 2.73 (d, J = 9.2 Hz, 2H), 2.17–1.51 (m, 10H), 1.80 (s, 3H), 1.68 (s, 3H), 1.59 (s, 3H), 1.26, (s, 3H), 1.11 (s, 3H), 0.88 (s, 3H); 13C NMR (CDCl3) δ 158.0, 155.6, 148.8, 142.5, 137.7, 136.8, 131.7, 128.9, 128.2, 126.3, 123.9, 122.6, 121.9, 120.5, 114.7, 106.79, 106.77, 101.2, 77.9, 77.0, 55.94, 55.7, 46.7, 39.7, 38.3, 37.6, 28.2, 27.3, 26.4, 25.6, 23.1, 22.2, 19.8, 17.6, 16.1, 14.2; HRMS (ESI) m/z calcd for C36H48O5 (M+) 560.3502, found 560.3481.
5.42 Stilbene 50
Under the general conditions for HWE condensations, aldehyde 34 (320 mg, 0.76 mmol) phosphonate 39 (560 mg, 1.23 mmol), and KHMDS (0.5 M in toluene, 5 mL, 2.5 mmol) were allowed to react in THF (10 mL) for 10 min. Final purification by column chromatography (30% EtOAc in hexanes) afforded stilbene 50 (195 mg, 36%) as a colorless oil: 1H NMR (CDCl3) δ 7.44 (d, J = 0.8 Hz, 1H), 7.14 (d, J = 1.6 Hz, 1H), 6.96 (d, J = 16.0 Hz, 1H), 6.90 (J = 16.0 Hz, 1H), 6.86 (d, J = 0.8 Hz, 1H), 6.71 (d, J = 0.8 Hz, 1H), 5.23 (s, 2H), 5.20 (t, J = 6.8 Hz, 1H), 5.07 (t, J = 6.8 Hz, 1H), 4.70 (d, J = 13.6 Hz, 1H), 4.63 (d, J = 13.6 Hz, 1H), 3.87 (s, 3H), 3.50 (s, 3H), 3.43 (dd, J = 11.6, 4.4 Hz, 1H), 3.37 (d, J = 6.8 Hz, 2H), 2.73–2.70 (m, 2H), 2.06–1.84 (m, 8H), 1.78 (s, 3H), 1.72–1.68 (m, 2H), 1.65 (s, 3H), 1.57 (s, 3H), 1.20 (s, 3H), 1.10 (s, 3H), 0.98 (s, 9H), 0.88 (s, 3H), – 0.13 (s, 6H); 13C NMR δ 158.2, 155.8, 149.6, 136.8, 134.6, 131.2, 129.4, 128.8, 128.4, 126.3, 126.3, 124.4, 123.4, 122.7, 121.1, 118.6, 105.3, 102.6, 94.5, 78.0, 76.5, 60.2, 56.0, 55.7, 46.8, 39.8, 38.3, 37.8, 28.2, 27.3, 26.7, 26.0, 25.7 (3C), 23.0, 22.4, 20.1, 18.5, 17.6, 16.0, 14.2, −5.2 (2C); HRMS (EI) calcd for C44H66O6Si (M+) 718.4629, found 718.4631.
5.43 Alcohol 58
To a solution of silyl ether 50 (195 mg, 0.27 mmol) in THF at rt was added TBAF (0.5 mL, 1 M in THF, 0.5 mmol). After 4 h, the reaction was quenched by addition of water, the resulting solution was extracted with ethyl acetate, and the combined organic phases were washed with brine. The organic phase was dried (MgSO4) and concentrated in vacuo, which provided nonracemic alcohol 58 (193 mg, 100% yield, 89% ee by HPLC) as a colorless oil: 1H NMR (CDCl3) δ 7.27 (d, J = 1.2 Hz, 1H), 7.18 (d, J = 1.2 Hz, 1H), 6.97 (d, J = 16.0 Hz, 1H), 6.92 (d, J = 16.0 Hz, 1H), 6.84 (s, 1H), 6.71 (s, 1H), 5.23 (s, 2H), 5.22 (m, 1H), 5.08 (t, J = 6.2 Hz, 1H), 4.66 (d, J = 13.2 Hz, 1H), 4.59 (d, J = 13.2 Hz, 1H), 3.87 (s, 3H), 3.50 (s, 3H), 3.40–3.37 (m, 3H), 2.75–2.64 (m, 2H), 2.08–1.95 (m, 5H), 1.86–1.80 (m, 2H), 1.79 (s, 3H), 1.75–1.67 (m, 3H), 1.66 (s, 3H), 1.58 (s, 3H), 1.22 (s, 3H), 1.10 (s, 3H), 0.88 (s, 3H); 13C NMR (CDCl3) δ 158.1, 155.7, 150.5, 136.5, 134.4, 130.9, 129.0, 128.7, 127.7, 127.2, 126.6, 124.3, 124.2, 122.6, 121.8, 118.7, 105.3, 102.6, 94.4, 77.6, 77.1, 61.8, 55.8, 55.6, 46.7, 39.7, 38.2, 37.7, 28.0, 27.2, 26.6, 25.6, 22.8, 22.4, 20.1, 17.5, 15.9, 14.2; HRMS (EI) m/z calcd for C38H52O6 (M+) 604.3764, found 604.3751.
5.44 Schweinfurthin analogue 59
Under the general conditions for MOM hydrolysis, stilbene 58 (14 mg, 0.025 mmol), methanol (1 mL), and p-TsOH·H2O (68 mg, 0.37 mmol) were allowed to react for 24 h to provide analogue 59 (2 mg, 17%) as a white solid after purification by thin layer chromatography (50% EtOAc in hexanes): 1H NMR (CDCl3) δ 7.26 (s, 1H), 7.18 (s, 1H), 6.95 (d, J = 16.0 Hz, 1H), 6.86 (d, J = 16.0 Hz, 1H), 6.63 (s, 1H), 6.60 (s, 1H), 5.23 (m, 1H), 5.06 (m, 1H), 4.67 (d, J = 12.4 Hz, 1H), 4.60 (d, J = 12.4 Hz, 1H), 3.85 (s, 3H), 3.46–3.40 (m, 3H), 2.76 (m, 2H), 2.09–1.88 (m, 8H), 1.80 (s, 3H), 1.75–1.70 (m, 2H), 1.66 (s, 3H), 1.59 (s, 3H), 1.25 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); HRMS (EI) m/z calcd for C36H48O5 (M+) 560.3502, found 560.3508.
5.45 Aldehyde 60
To a solution of alcohol 58 (50 mg, 0.10 mmol) in CH2Cl2 at rt was added activated MnO2 (250 mg, 2.3 mmol). After 22 h at rt, the solution was diluted with ethyl acetate, filtered through celite, and concentrated in vacuo which afforded aldehyde 60 (49 mg, 98%) as a yellow oil: 1H NMR (CDCl3) δ 10.41 (s, 1H), 7.78 (s, 1H), 7.47 (s, 1H), 6.96 (s, 2H), 6.87 (s, 1H), 6.70 (s, 1H), 5.20 (s, 2H), 5.19 (m, 1H), 5.06 (m, 1H), 3.86 (s, 3H), 3.49 (s, 3H), 3.44 (dd, J = 11.4, 3.4 Hz, 1H), 3.37 (d, J = 6.8 Hz, 2H), 2.77–2.73 (m, 2H), 2.08–1.82 (m, 8H), 1.77 (s, 3H), 1.74–1.70 (m, 2H), 1.64 (s, 3H), 1.50 (s, 3H), 1.28 (s, 3H), 1.12 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 189.9, 158.2, 155.8, 155.6, 136.0, 134.6, 133.6, 131.1, 129.2, 128.0, 126.6, 124.4, 124.4, 123.8, 123.7, 122.5, 119.2, 105.4, 102.7, 94.5, 78.0, 77.7, 55.9, 55.7, 46.5, 39.7, 38.4, 37.4, 28.1, 27.2, 26.7, 25.6, 22.9, 22.4, 20.2, 17.6, 16.0, 14.2; HRMS (EI) m/z calcd for C38H50O6 (M+) 602.3607, found 602.3616.
5.46 Acid 61
To a solution of aldehyde 60 (17 mg, 0.028 mmol) in (CH3)3COH (1 mL) at rt was added 2-methyl-2-butene (0.3 mL). Dropwise addition of NaH2PO4 (40 mg) and NaClO2 (34 mg, 0.38 mmol) as an aqueous solution (0.3 mL) resulted in a darkening of the reaction solution. After 45 min, the reaction was quenched by addition of 1N HCl. The resulting solution was extracted with ethyl acetate, and the combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo to afford acid 61 (18 mg, 100%) as a yellow oil: 1H NMR (CDCl3) δ 8.23 (d, J = 2.4 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.09 (d, J = 16.8 Hz, 1H), 7.05 (d, J = 16.6 Hz, 1H), 6.97 (d, J = 0.8 Hz, 1H), 6.79 (d, J = 1.2 Hz, 1H), 5.30 (s, 2H), 5.27 (m, 1H), 5.15 (t, J = 5.1 Hz, 1H), 3.95 (s, 3H), 3.58 (s, 3H), 3.56 (m, 1H), 3.45 (d, J = 7.2 Hz, 2H), 2.92–2.87 (m, 2H), 2.20–1.88 (m, 8H), 1.85 (s, 3H), 1.81–1.74 (m, 2H), 1.72 (s, 3H), 1.65 (s, 3H), 1.44 (s, 3H), 1.23 (s, 3H), 1.00 (s, 3H); 13C NMR (CDCl3) δ 165.7, 158.3, 155.9, 151.1, 135.8, 134.7, 132.8, 131.2, 130.9, 129.5, 129.1, 126.0, 124.4, 123.4, 122.5, 119.5, 117.4, 105.6, 102.9, 94.6, 81.3, 77.2, 56.0, 55.7, 46.4, 39.8, 38.5, 37.5, 28.0, 27.1, 26.7, 25.6, 22.9, 22.4, 20.3, 17.6, 16.0, 14.2; HRMS (EI) m/z calcd for C38H50O7 (M+ 618.3557, found 618.3560.
5.47 Amine 62
Aldehyde 60 (7.5 mg, 0.012 mmol) was dissolved in dimethylamine (2 M solution in THF, 1 mL, 2 mmol) at rt and molecular sieves were added. After 2 h, additional dimethylamine (1 mL, 2 mmol) was added along with AcOH (0.05 mL). After an additional 5 h, NaBH(OAc)3 (58 mg, 0.4 mmol) was added in one portion. After 15 h, the reaction was quenched by addition of 1N NaOH. The resulting solution was extracted with ethyl acetate, and the combined organic phases were washed with brine, dried (Na2SO4), and concentrated in vacuo. Final purification by preparative thin layer chromatography on a base-washed plate (75% EtOAc and 5% TEA in hexanes) afforded amine 62 (2.5 mg, 33%) as a colorless oil: 1H NMR (CDCl3) δ 7.34 (s, 1H), 7.30 (s, 1H), 6.95 (s, 2H), 6.86 (s, 1H), 6.70 (s, 1H), 5.22 (s, 2H), 5.19 (m, 1H), 5.05 (m, 1H), 3.88 (s, 3H), 3.64 (s, 2H), 3.50 (s, 3H), 3.44 (dd, J = 11.4, 7.6 Hz, 1H), 3.36 (d, J = 7.6 Hz, 2H), 2.76–2.73 (m, 2H), 2.59 (s, 6H), 2.06–2.03 (m, 6H), 1.97–1.88 (m, 2H), 1.81 (s, 3H), 1.73–1.68 (m, 2H), 1.67 (s, 3H), 1.59 (s, 3H), 1.24 (s, 3H), 1.11 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 159.4, 156.9, 152.6, 137.3, 135.7, 132.2, 130.6, 130.3, 129.9, 129.7, 128.7, 128.6, 128.0, 125.5, 123.7, 120.2, 106.6, 103.8, 95.6, 78.9, 78.2, 71.6, 57.0, 56.8, 48.0, 43.9, 40.9 (2C), 39.5, 38.9, 29.3, 28.4, 27.8, 26.7, 24.3, 23.5, 21.3, 18.7, 17.1, 15.4; HRMS (EI) m/z calcd for C40H57NO5 (M+) 631.4237, found 631.4232.
Supplementary Material
Acknowledgements
We thank the ACS Division of Medicinal Chemistry for support in the form of a predoctoral fellowship (to J.J.T.), the University of Iowa Department of Chemistry for a Shriner Fellowship (to J.J.T.), the UI Graduate College for a Presidential Fellowship (to J.G.K.), and the Iowa Center for Research by Undergraduates for a summer fellowship (to J.D.I.). Financial support from the Roy J. Carver Charitable Trust as a Research Program of Excellence is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Supplementary data associated with this article, including NMR spectra and complete bioassay data, can be found in the online version, at
References and notes
- 1).Beutler JA, Shoemaker RH, Johnson T, Boyd MR. J. Nat. Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 2).Beutler JA, Jato J, Cragg GM, Boyd MR. Nat. Prod. Lett. 2000;14:399–404. [Google Scholar]
- 3).Yoder BJ, Cao S, Norris A, Miller JS, Ratovoson F, Razafitsalama J, Andriantsiferana R, Rasamison VE, Kingston DGI. J. Nat. Prod. 2007;70:342–346. doi: 10.1021/np060484y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Thoison O, Hnawia E, Gueritte-Voegelein F, Sevenet T. Phytochemistry. 1992;31:1439–1442. [Google Scholar]
- 5).Topczewski JJ, Kodet JG, Wiemer DF. J. Org. Chem. 2011;76:909–919. doi: 10.1021/jo1022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Topczewski JJ, Neighbors JD, Wiemer DF. J. Org. Chem. 2009;74:6965–6972. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 7).Treadwell EM, Cermak SC, Wiemer DF. J. Org. Chem. 1999;64:8718–8723. [Google Scholar]
- 8).Mente NR, Neighbors JD, Wiemer DF. J. Org. Chem. 2008;73:7963–7970. doi: 10.1021/jo800951q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Topczewski JJ, Wiemer DF. Tetrahedron Lett. 2011;52:1628–1630. doi: 10.1016/j.tetlet.2011.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Neighbors JD, Beutler JA, Wiemer DF. J. Org. Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 11).Neighbors JD, Salnikova MS, Beutler JA, Wiemer DF. Bioorg. Med. Chem. 2006;14:1771–1784. doi: 10.1016/j.bmc.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 12).Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2009;17:4718–4723. doi: 10.1016/j.bmc.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Ulrich NC, Kodet JG, Mente NR, Kuder CH, Beutler JA, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2010;18:1676–1683. doi: 10.1016/j.bmc.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Topczewski JJ, Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2010;18:6734–6741. doi: 10.1016/j.bmc.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Ulrich NC, Kuder CH, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. Lett. 2010;20:6716–6720. doi: 10.1016/j.bmcl.2010.08.143. [DOI] [PubMed] [Google Scholar]
- 16).Neighbors JD, Topczewski JJ, Swenson DC, Wiemer DF. Tetrahedron Lett. 2009;50:3881–3884. [Google Scholar]
- 17).a) Turbyville TJ, Gürsel D, Tuskan RG, Walrath JC, Lipshultz CA, Lockett S, Wiemer DF, Beutler JA, Reilly KM. Mol. Cancer Ther. 2010;9:1234–1243. doi: 10.1158/1535-7163.MCT-09-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Burgett AWG, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Mimaki Y, Kuroda M, Murphy JP, Schwalb DJ, Petrella EC, Cornella-Taracido I, Schirle M, Tallarico JA, Shair MD. Nature: Chemical Biology. 2011;7:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Holstein SA, Kuder CH, Tong H, Hohl RJ. Lipids. 2011;46:907–921. doi: 10.1007/s11745-011-3572-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Blunt SB, Chen T-B, Wiemer DF. J. Nat. Prod. 1998;61:1400–1403. doi: 10.1021/np980098j. [DOI] [PubMed] [Google Scholar]
- 19).Shi Y. Acc. Chem. Res. 2004;37:488–496. doi: 10.1021/ar030063x. [DOI] [PubMed] [Google Scholar]
- 20).O'Hagan D. J. Fluorine Chem. 2010;131:1071–1081. [Google Scholar]
- 21).Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, Mendelsohn B, Mdluli K, Raetz CRH, Stover CK, Witte PR, Yabannavar A, Zhu S. J. Med. Chem. 2002;45:3112–3129. doi: 10.1021/jm010579r. [DOI] [PubMed] [Google Scholar]; b) Hagiwara A, Ikenogami T, Kurihara K, Taniguchi T, Takahashi M, Ida A. PCT Int. Appl. 2006 WO 2006043510 A1 20060427. [Google Scholar]
- 22).Neighbors JD, Mente NR, Boss KD, Zehnder DW, II, Wiemer DF. Tetrahedron Lett. 2008;49:516–519. [Google Scholar]
- 23).Mente NR, Wiemer AJ, Neighbors JD, Beutler JA, Hohl RJ, Wiemer DF. Bioorg Med. Chem. Letters. 2007;17:911–915. doi: 10.1016/j.bmcl.2006.11.096. [DOI] [PubMed] [Google Scholar]
- 24).Neighbors JD, Salnikova MS, Wiemer DF. Tetrahedron Lett. 2005;46:1321–1324. [Google Scholar]
- 25).Battacharya AK, Thyagarajan G. Chem. Rev. 1981;81:415–431. [Google Scholar]
- 26).a) Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175–1176. [Google Scholar]; b) Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 27).a) Shoemaker RH. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]; b) Additional details about this screen can be found at http://dtp.nci.nih.gov/screening.html.
- 28).Carosati E, Sciabola S, Cruciani G. J. Med. Chem. 2004;47:5114–5125. doi: 10.1021/jm0498349. [DOI] [PubMed] [Google Scholar]
- 29).Takatori K, Nishihara M, Nishiyama Y, Kajiwara M. Tetrahedron. 1998;54:15861–15869. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.