

Abstract
The retinal network increases its sensitivity in low-light conditions to detect small visual inputs and decreases its sensitivity in bright-light conditions to prevent saturation. However, the cellular mechanisms that adjust visual signaling in the retinal network are not known. Here, we show that voltage-gated sodium channels in bipolar cells dynamically control retinal light sensitivity. In dim conditions, sodium channels amplified light-evoked synaptic responses mediated by cone pathways. Conversely, in bright conditions, sodium channels were inactivated by dopamine released from amacrine cells, and they did not amplify synaptic inputs, minimizing signal saturation. Our findings demonstrate that bipolar cell sodium channels mediate light adaptation by controlling retinal signaling gain.
Keywords: patch clamp, sodium channel, ganglion cell, bipolar cell, dopamine, retinal network adaptation
Introduction
The retina processes visual information over a wide range of ambient light conditions from dim starlight to bright sunlight. To achieve this, the retina adjusts its sensitivity to the mean intensity and the variance (contrast) of ambient light. Adaptation to the mean intensity of ambient light occurs in rod and cone photoreceptors (Pugh et al., 1999; Fain et al., 2001) and at sites in the retinal network postsynaptic to photoreceptors (Fig. 1) (Green et al., 1975; Naka et al., 1979; Green and Powers, 1982; Shapley and Enroth-Cugell, 1984; Page-McCaw et al., 2004; Dunn and Rieke, 2006). Contrast adaptation occurs in the retinal network but not in photoreceptors (Shapley and Enroth-Cugell, 1984; Smirnakis et al., 1997; Dunn and Rieke, 2006). Adaptation mechanisms increase retinal sensitivity in low-light settings but decrease retinal sensitivity in bright-light settings to prevent saturation. However, the cellular mechanisms that mediate network adaptation remain unknown.
Figure 1.

Schematic of retinal neural network and sites of adaptation. Voltage-gated sodium channels in bipolar cells contribute to network adaptation in dim (left) but not in bright (right) light conditions. C, Cone photoreceptors; R, rod photoreceptors; B, bipolar cells; G, ganglion cells; H, horizontal cells; A, amacrine cells.
Voltage-gated sodium channels boost EPSPs in cerebral cortex pyramidal neurons and hippocampal cells (Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Lipowsky et al., 1996; Gonzalez-Burgos and Barrionuevo, 2001). In the retina, sodium channels in a subset of bipolar cells boost light-evoked EPSPs (L-EPSPs) in dim-light conditions and amplify the excitatory drive to ganglion cells (Ichinose et al., 2005). Because bipolar cells have been implicated as sites of network adaptation (Green et al., 1975; Naka et al., 1979; Shapley and Enroth-Cugell, 1984; Smirnakis et al., 1997; Rieke, 2001; Baccus and Meister, 2002; Page-McCaw et al., 2004), the voltage-gated sodium channels in bipolar cells may regulate network adaptation by increasing retinal sensitivity in dim-light conditions (Fig. 1, left). However, if sodium channels contribute to network adaptation, then in bright-light conditions, the amplification of L-EPSPs by sodium channels must be reduced to decrease retinal sensitivity and avoid response saturation (Fig. 1, right).
To elucidate their role in retinal network adaptation, we examined how bipolar cell sodium channels affected synaptic inputs in different ambient light conditions. In dim-light-adapted conditions, sodium channels enhanced cone-mediated L-EPSPs in bipolar cells, which increased ganglion cell sensitivity to the mean intensity and the contrast of the visual scene. However, in bright-light-adapted conditions, these sodium channels were inactivated by the light-evoked release of dopamine and no longer boosted cone-mediated L-EPSPs, avoiding signal saturation. Our findings suggest that the voltage-gated sodium channels in bipolar cells regulate network adaptation by dynamically controlling retinal signal gain.
Materials and Methods
Retinal slice preparation.
Larval tiger salamanders were obtained from Charles Sullivan (Nashville, TN) and were kept in aquaria at 5°C on a 12 h light/dark cycle. Procedures for preparing and recording from salamander retinal slices were described previously (Ichinose et al., 2005). Light- and voltage-evoked responses were recorded from bipolar cells with axon terminals and ganglion cells with dendrites that ramified in the mid-inner plexiform layer (IPL) (Ichinose et al., 2005). Using a gravity-fed system, the preparation was continually superfused at 2 ml/min with a Ringer's solution containing the following (in mm): 112 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 5 HEPES, adjusted to pH 7.75 with NaOH. For voltage-gated sodium current recordings from bipolar cells, we used a Ringer's solution containing the following (in mm): 100 NaCl, 2 KCl, 10 tetraethylammonium (TEA)-Cl, 3 MgCl2, 5 glucose, 5 HEPES, and 2 CoCl2.
Whole-cell recording.
Electrodes were pulled from borosilicate glass (IB150F-4; World Precision Instruments, Sarasota, FL) with a P-97 Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA). Whole-cell recordings were made from bipolar cells with electrodes containing (in mm) 85 K-gluconate, 10 KCl, 10 HEPES, 10 BAPTA, 4 Mg-ATP, and 1 Na-GTP, adjusted to pH 7.4 with KOH for recordings of light-evoked responses. For voltage-gated sodium current recordings from bipolar cells, the electrode solution contained (in mm) 85 K-gluconate, 10 TEA-Cl, 10 HEPES, 10 BAPTA, 4 Mg-ATP, and 1 Na-GTP. For ganglion cell recordings, pipettes were filled with solution containing (in mm) 95.25 Cs-gluconate, 8 TEA-Cl, 0.4 MgCl2, 1 EGTA, and 10 Na-HEPES, adjusted to pH 7.5 with HCl. Sulforhodamine B (90 μm) was included in the pipette solution to identify bipolar and ganglion cell morphologies. Ganglion cells were only analyzed when their dendrites ramified mid-IPL, where the depths of the IPL were 20–80% (Ichinose et al., 2005). The axonal processes of transient and sustained ON bipolar cells ramified 50–80% and 80–100%, respectively (Ichinose et al., 2005).
The voltage-clamp or current-clamp recordings were made with an Axopatch 200A Patch Clamp Amplifier (Axon Instruments, Foster City, CA). Data were digitized and stored with a personal computer using a TL-1 data acquisition system (Axon Instruments). Patchit software (White Perch Software, Somerville, MA) was used to generate voltage command outputs, acquire data, control the light stimuli, and operate the drug perfusion system. Data were filtered at 1 kHz with a four-pole Bessel filter and were sampled at 1–10 kHz.
Light stimulation.
The bipolar or ganglion cell receptive field center was activated by a small spot of light (180 μm diameter) from a light-emitting diode (LED) light source [peak, 630 nm; unattenuated light intensity (0 log), 1.33 × 1010 photons · μm−2 · s−1]. The background and spot stimuli were projected through a 40× objective lens. For scotopic conditions, animals were dark adapted for ≥12 h, and dissections and recordings were performed under infrared (IR) illumination. For dim conditions, animals were dark adapted for 1 h, and retinas were either dissected in dim red light and recorded with no background illumination or dissected in IR and recorded with dim background illumination (−4 to −5 log). For bright conditions, animals were dark adapted for 1 h, and retinas were dissected in IR and recorded with bright background (−2 to −3 log) illumination. For some experiments, dissections and sodium current recordings were performed in “room light” (1.6 × 109 photons · μm−2 · s−1).
To obtain light-evoked responses (L-EPSPs or L-EPSCs), the light stimulus was presented for 1–3 s every 30–60 s. To adapt the preparation, background illumination was presented for >5 min continuously after whole-cell configuration was achieved, followed by a perfusion of receptor blockers. Background illumination was maintained through the experiment. The retina was stimulated with an increment of luminance above the background illumination. Light intensity was varied by changing the LED current.
For sodium current recordings in bipolar cells, the slices were prepared in dim conditions. Background illumination was present for >5 min after whole-cell recordings were obtained, followed by a perfusion of receptor and channel blockers. The voltage dependence of the sodium channel was determined either in dim conditions or in room light, which was equivalent to unattenuated background illumination.
Pharmacology.
Bipolar cells were pharmacologically isolated to avoid indirect, network effects when bath applying tetrodotoxin (TTX) (0.5 μm). For L-EPSP recordings from bipolar cells, GABAA, GABAC, glycine, NMDA, and non-NMDA receptors were blocked with a mixture of strychnine (5 μm), bicuculline (200 μm), imidazole-4-acetic acid (I4AA; 20 μm), picrotoxin (200 μm), CNQX (30 μm), and d-AP5 (50 μm). For sodium current recordings from bipolar cells, a calcium channel blocker, CoCl2 (2 mm), a potassium channel blocker, TEA (10 mm both for intracellular and extracellular solutions), and a metabotropic glutamate receptor 6 (mGluR6) agonist, l-AP4 (2 μm), were used in addition to the mixture of receptor blockers for L-EPSP recordings. When investigating the effects of light adaptation on bipolar cell L-EPSPs or sodium current, the retina was first adapted to background illumination, followed by application of the antagonist mixture to measure the L-EPSPs or sodium currents, because some of the blockers directly affect the release of dopamine. For the dopamine receptor experiments, dopamine receptor antagonist was applied before bright-light background illumination, and dopamine agonist was applied in dim-light conditions, and then, as above, the antagonist mixture was applied just before response recording. For L-EPSC recordings from ganglion cells, only strychnine (5 μm) was used for blocking glycine receptors to isolate the steady surround from the change-sensitive surround (Cook et al., 1998).
Voltage-gated bipolar cell currents.
To activate voltage-gated currents, bipolar cells were voltage clamped at −60 mV and then prepulsed at −80 mV for 500 ms to remove inactivation, followed by a test pulse to −10 mV for 50 ms every 15 s, which fully activated sodium channels. The TTX-sensitive, inward current was obtained by subtracting the averaged TTX current from the averaged control current. To determine the activation and inactivation properties of the sodium current, we varied either test pulse or prepulse (500 ms), for the activation and inactivation experiments, respectively.
Data analysis.
The voltage dependence of sodium channel inactivation and activation was analyzed as described previously (Ichinose et al., 2005). Normalized sodium currents (I) were plotted for inactivation, whereas normalized conductance (G) was plotted for activation, and data points were fit by Boltzmann equation: I = Imax/(1 + exp[(V − Vh)/k]) for inactivation, and G = Gmax/(1 + exp[(V − Vh)/k]) for activation, where Vh is the half-inactivation or activation voltage, and k is the slope factor.
Light intensity–response relationships were obtained from individual ganglion cells. We plotted the light-evoked charge transfer (R) versus stimulus light intensity (L). The Naka–Rushton equation was used to fit the relationship: R = aI/(L50 + L), where a is the maximum charge transfer, and L50 is the light intensity at the half-maximum response.
The sensitivity of mean intensity and contrast was calculated for individual ganglion cells (Shapley and Enroth-Cugell, 1984). The sensitivity of mean intensity (Sm) is defined as “the ratio of the magnitude of the physiological response to the stimulus magnitude” (Shapley and Enroth-Cugell, 1984) and was calculated from the following equation: Sm = dR/dL = aL50/(L50 + L)2. The ratio of Sm-control to Sm-TTX was plotted in Figure 6E.
Figure 6.
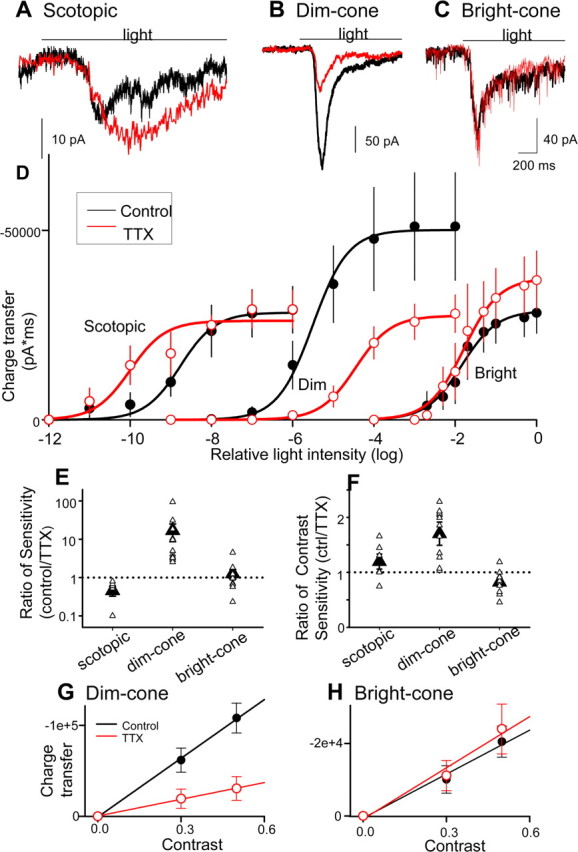
Bipolar cell (BC) sodium currents enhanced ganglion cell light sensitivity. A–C, Ganglion cell (GC) L-EPSCs (black lines) were reduced by TTX (red lines) in dim (B) but not bright (C) or scotopic (A) conditions. D, Intensity–response curves in dim light but not in scotopic or bright conditions were shifted (p < 0.05) and compressed (p < 0.01) by TTX, suggesting that BC sodium currents only boosted GC responses in dim conditions. E–H, The sensitivities for mean intensity (E) and contrast (F–H) were enhanced by bipolar cell sodium currents in dim but not scotopic or bright conditions [mean sensitivity, p < 0.01 (E); contrast sensitivity, p < 0.05 (F–H); scotopic, n = 6; dim, n = 11; bright, n = 11]. ctrl, Control.
The contrast sensitivity was calculated in two ways. Charge transfer of L-EPSCs in dim- and bright-light conditions was plotted as a function of contrast (see Fig. 6G,H), and contrast sensitivity was calculated from the slopes of low-contrast linear portions of the curves, in control and TTX solutions. Also, contrast sensitivity ΔR(ΔL/LB)−1 (where LB is background illumination) was obtained from the intensity–response curves using the following equation: Scon = dR/dlogL = aL50L/(L50 + L)2 ≈ ΔR(ΔL/LB)−1. Scon at the half-maximum EPSC was calculated for each cell in the absence and in the presence of TTX, and the ratio (control/TTX) was plotted in Figure 6F.
Most statistical comparisons were made using a paired t test. Sodium current data were compared using the unpaired t test. Spearman's rank test was used to evaluate correlation between sodium current amplitude or charge transfer and background light intensity. Differences were considered significant if p < 0.05. Data are presented as mean ± SE.
Results
Sodium channels in bipolar cells differentially modulated L-EPSPs in dim and bright conditions
We investigated whether sodium channels in bipolar cells affect network adaptation by assessing the sodium channel contributions to light-evoked synaptic responses in different background light conditions. L-EPSPs were recorded in whole-cell configuration from transient ON bipolar cells in retinal slices. When retinal slices were adapted to dim light, the sodium channel blocker TTX (0.5 μm) reduced L-EPSPs (Fig. 2A). Because these bipolar cells are cone dominant (Wu et al., 2000), this result suggested that bipolar cell sodium channels amplified cone-mediated synaptic inputs. TTX shifted and compressed the bipolar cell intensity–response curve (Fig. 2C), suggesting that sodium channels enhanced the light sensitivity and the maximal response in dim light. TTX did not alter the bipolar cell resting membrane potential (control, −59.0 ± 0.7 mV; TTX, −59.3 ± 0.9 mV; p > 0.1; n = 7), indicating that the reduction of L-EPSPs was not attributable to changes in the resting membrane potential.
Figure 2.

Voltage-gated sodium channels amplified light-evoked synaptic responses only in dim-light conditions. A, B, L-EPSPs recorded from ON bipolar cells in dim conditions (A) were reduced by TTX, but those recorded in bright conditions (B) were not affected by TTX. C, TTX shifted and compressed the intensity–response relationship in dim (p < 0.01; n = 5) but not in bright conditions (p > 0.1; n = 5). D, The intensity–response relationship in ON sustained bipolar cells, which do not possess sodium channels, were shifted to the right compared with the curve in transient bipolar cells in dim-light conditions (curves for the transient cells are the same as C). The shift of the sustained bipolar cell curves from dim to bright light is attributable to cone and OPL adaptation.
When the retina was adapted to bright light, TTX did not reduce the L-EPSPs (Fig. 2B) or affect the intensity–response curve (Fig. 2C), although the resting membrane potential was adjusted with current injection to ensure that voltage-gated channels were similarly activated (dim, −57.7 ± 1.8 mV; bright, −56.0 ± 2.7 mV; p = 0.30). Because bright background light can depolarize salamander ON bipolar cells (3–5 mV) (Werblin, 1974; Fahey and Burkhardt, 2001), the sodium current contributions in bright-light conditions were probably overestimated because of sodium channel inactivation at more positive potentials (Ichinose et al., 2005). These findings indicate that sodium channels did not boost L-EPSPs in bright conditions (Fig. 1) and suggest that the ambient light regulates the ability of the sodium channels to boost synaptic responses.
The rightward shift of the intensity–response curve by bright light was only partially attributable to the suppression of bipolar cell sodium channels, as illustrated by comparing the control and TTX curves in dim conditions (Fig. 2C). Comparisons of the TTX curves in dim and bright conditions indicate that the remainder of the shift by bright light was independent of bipolar sodium channels and was likely attributable to adaptation occurring in the cone photoreceptor outer segments and in the outer plexiform layer (OPL) (Normann and Werblin, 1974; Werblin, 1974). The intensity–response curves for ON sustained bipolar cells, which do not possess sodium channels (Ichinose et al., 2005), were also shifted to right by increasing background (Fig. 2D), supporting the idea that the TTX-insensitive shift of the intensity–response curves was attributable to adaptation in cones and the OPL.
Bright background illumination reduced the activity of voltage-gated sodium channels
We recorded voltage-gated sodium currents from bipolar cells (evoked by voltage steps from −80 to −10 mV) in dim- and bright-light conditions to determine how ambient illumination affected the TTX sensitivity of bipolar cell L-EPSPs. We found that sodium current magnitudes varied with background illumination levels. The sodium current was the largest in dim conditions (Fig. 3A, top). When retinal slices were exposed to brighter background illumination, the sodium currents were reduced (Fig. 3A). Sodium current peak amplitude (Fig. 3B) and charge transfer (Fig. 3C) were reduced by background illumination in an intensity-dependent manner (p < 0.01).
Figure 3.

Bright background illumination attenuated bipolar cell sodium currents by changing the channel voltage dependence. A, TTX-sensitive sodium currents recorded from bipolar cells at different background illuminations were obtained by subtracting the current responses to voltage steps in TTX from those obtained in control conditions. Each record was obtained from a different cell. B, C, The peak amplitude (B) and the charge transfer (measured at 0–50 ms; C) of sodium currents were decreased with background light in an intensity-dependent manner (p < 0.01; no background, n = 7; −4 log, n = 2; −2 log, n = 3; 0 log, n = 4). D, Sodium channel inactivation curves in dim (solid circles) and bright (open circles) conditions. Bright light shifted the curve to the left (half-maximum inactivation voltage, bright, −51.6 mV, n = 6; dim, −36.4 mV, n = 5; p < 0.05). E, Conductances in each condition was normalized to the maximum dim condition value and plotted. Bright light (open triangles) also shifted and compressed the activation curves for sodium currents in bipolar cells compared with dim light (solid triangles; half-maximum activation voltage, bright, −2.95 ± 4.7 mV; dim, −19.2 ± 5.9 mV; p < 0.05). Dotted line represents scaled bright-light curve.
To investigate how background illumination affected sodium currents, we determined the voltage dependence of activation and inactivation of the sodium channel in dim- and bright-light-adapted conditions. In dim light (Fig. 3D, closed circles), sodium channels inactivated with a half-inactivation voltage of −36.4 mV (Ichinose et al., 2005) (Fig. 3D, solid line, filled circles). However, in bright light, the sodium channels inactivated at more negative potentials, with a half-inactivation voltage of −51.6 mV (Fig. 3D, dashed line, open circles) (n = 6). Thus, bright light reduced sodium currents by shifting the inactivation curve to negative potentials (p < 0.05), resulting in a decreased “availability” of the sodium channels.
The voltage dependence of sodium channel activation was also examined. Figure 3E shows the voltage dependence of the sodium channel conductance when normalized to the maximum conductance of sodium channels in dim light (closed triangles). Compared with dim-light conditions, the maximum conductance was reduced in bright conditions (Fig. 3E, open triangles) (0.38 ± 0.08 vs 0.84 nS in dim light; p < 0.01; n = 4). In addition, the half-maximal activation voltage was shifted to right in bright- compared with dim-light conditions (p < 0.05). Thus, the reduction in sodium channel activation may also contribute to the decrease in sodium current observed in bright-light conditions.
We previously estimated the magnitude of the Na+ conductance in dim-light conditions during the L-EPSPs (depolarization from −62 to −54 mV) to be 0.04 nS (Ichinose et al., 2005). This conductance enhances the L-EPSPs by 7.4 mV, in agreement with the observed enhancement of 7.1 mV. In bright-light conditions, the activation curve was shifted to the right (Fig. 3), and the estimated Na+ conductance during a similar magnitude of L-EPSP is 0 nS, which produces no enhancement. However, if bright-light conditions depolarize the bipolar cell to a resting potential of −50 mV (Werblin, 1974; Fahey and Burkhardt, 2001), then the estimated magnitude of the Na+ conductance during an L-EPSP (depolarization from −50 to −40 mV) is 0.0025 nS, attributable to more activation and inactivation of the Na+ channels (Fig. 3). This small conductance only enhances the L-EPSP, at most, by 0.4 mV (assuming a 1 GΩ input resistance). These findings explain why we observed little or no amplification of bipolar cell L-EPSPs in bright conditions (Fig. 2B).
Dopamine mimicked the effects of light on voltage-gated sodium currents and L-EPSPs
Dopamine is a known mediator of light adaptation (Witkovsky, 2004) that is released from amacrine cells by cone-dependent signals (Myhr et al., 1994). Extracellular dopamine levels are low in dim conditions and are high in bright conditions (Boatright et al., 1989). Because the activity of voltage-gated sodium channels in other parts of the CNS is modulated by dopamine (Cantrell et al., 1997; Hayashida and Ishida, 2004), and dopamine receptors are present on bipolar cells (Veruki and Wassle, 1996; Nguyen-Legros et al., 1997; Mora-Ferrer et al., 1999), we investigated whether dopamine mediated the light-dependent modulation of the sodium channel activity by activating dopamine receptors in dim light with agonist applications.
A nonspecific dopamine receptor agonist, 2-amino-6,7-dihydroxy-l,2,3,4-tetrahydro-naphthalene (ADTN; 20–50 μm), mimicked the effects of bright conditions. In dim conditions, ADTN markedly reduced the sodium currents, compared with control conditions (Fig. 4A), decreasing both the peak amplitude (Fig. 4D) and the charge transfer (Fig. 4E). The specific D1 antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH23390; 50 μm) reversed the suppressive effects of ADTN (Fig. 4B,D,E), and sodium currents were now similar to those in control conditions, suggesting that they were mediated by D1 receptors. In bright-light conditions, dopamine levels are postulated to be high, which should attenuate sodium currents. If dopamine levels are high, then the D1 receptor antagonist SCH23390 should block the suppressive effect of bright light. When SCH23390 was applied before light adaptation, the sodium currents were no longer attenuated in bright light (Fig. 4C–E), suggesting that dopamine release in bright conditions diminishes sodium channel activity in bipolar cells.
Figure 4.

Dopamine D1 receptors mediated the effects of light adaptation on voltage-gated sodium currents. A, A dopamine agonist, ADTN (20 μm), reduced the sodium currents in dim conditions (n = 5) compared with control current in dim light (dotted line, same as Fig. 3A, top). B, A D1 receptor blocker, SCH23390 (SCH; 50 μm), blocked the ADTN effect (n = 3). The dotted line is control current in dim light from Figure 3A (top). C, Bright illumination did not reduce the sodium current when D1 receptors were blocked (50 μm SCH23390; n = 3). The dotted line is the control current in bright conditions from Figure 3A (bottom). D, E, Effects of agonists and antagonists on peak amplitude (D) and charge transfer (E). *Significant differences from control (p < 0.05); #significant differences from ADTN (dim; p < 0.05).
We then determined whether dopamine modulated the light-evoked synaptic responses in bipolar cells by recording L-EPSPs in dim- and bright-light-adapted conditions, in the presence of D1 receptor agonists or antagonists. In dim conditions, the D1 agonist 1-phenyl-2,3,4,5-tetrahydro-1H-3-benzodiazepine-7,8-diol hydrochloride (SKF38393; 20 μm) mimicked the effects of bright illumination, reducing the L-EPSPs and occluding the suppressive effects of TTX (Fig. 5A,B). In contrast, pretreatment with D1 receptor antagonist SCH23390 (50 μm) blocked the effects of bright illumination, and TTX now reduced the L-EPSPs in bright conditions (Fig. 5C,D). In dim conditions, D1 agonist application did not reduce L-EPSPs in sustained ON bipolar cells that lack sodium currents (97 ± 1% of control amplitude; n = 2). These findings suggest that the D1 agonist did not reduce L-EPSPs in ON transient bipolar cells by acting upstream of the sodium channels, either on cone photoreceptors or mGluR6 receptors. Together, our data suggest that dopamine, released in bright light, attenuates the sodium channel-dependent amplification of L-EPSPs.
Figure 5.

D1 dopamine receptor agonist and antagonist mimic light and darkness, respectively. A, In dim conditions, a D1 agonist, SKF38393 (20 μm), reduced the peak amplitude of L-EPSPs, mimicking bright conditions. The D1 agonist occluded the TTX (0.5 μm) effect on peak amplitude. B, Graph summarizes effects of D1 agonist and TTX on L-EPSP peak amplitude (n = 6). C, Cells were pretreated with D1 receptor antagonist [50 μm SCH23390 (SCH)], and then the preparation was exposed to bright light for 10 min. TTX reduced the L-EPSP, indicating that the D1 receptor mediated the suppressive effect of background illumination on sodium currents in bipolar cells. D, Summary of TTX effects on L-EPSPs in bright conditions, pretreated with D1 antagonist (n = 4). *p < 0.01, different from control level.
Bipolar cell sodium channels boost ganglion cell light responses only in dim-light conditions
Distinct subtypes of bipolar cells provide excitatory drive to different classes of ganglion cells. Bipolar cells that possess voltage-gated sodium currents make contacts with ON–OFF ganglion cells with dendrites that ramified in the mid-IPL (Ichinose et al., 2005). To determine how bipolar cell sodium channels affected retinal information processing in different illumination conditions, we recorded light-evoked EPSCs (L-EPSCs) at light onset from ON–OFF ganglion cells that were adapted to different light levels (Fig. 6A–C, black traces). L-EPSCs were isolated by voltage clamping the cell at −60 mV (ECl) to null inhibitory synaptic inputs. Lidocaine N-ethyl bromide (QX-314; 10 mm) was included in the pipette to block sodium channels in the recorded ganglion cell. L-EPSCs recorded from ganglion cells in dark-adapted retinas (>12 h in darkness, scotopic) were mediated by rods, as suggested by their slow times to peak (905 ± 106 ms) and low thresholds (5.1 × 10−11, relative attenuation) (Schnapf and Copenhagen, 1982). In contrast, L-EPSCs recorded in retinas exposed to dim background illumination showed shorter times to peak (147 ± 7.9 ms; p < 0.01 vs scotopic) and higher thresholds (1.3 × 10−6; relative attenuation), consistent with cone-mediated signaling (Schnapf and Copenhagen, 1982). Brighter background illumination further accelerated the L-EPSC kinetics (time to peak, 120 ± 6.0 ms; p < 0.01 vs scotopic and dim; threshold intensity, 9.4 × 10−3, relative attenuation).
In scotopic conditions, TTX did not reduce rod-mediated L-EPSCs (Fig. 6A), consistent with the notion that sodium channels are only found in cone-dominant transient ON bipolar cells (Ichinose et al., 2005). Instead, TTX enhanced the L-EPSCs, most likely attributable to the blockade of GABA-mediated inhibition in the IPL (Cook and McReynolds, 1998; Bloomfield and Xin, 2000), because TTX did not affect rod-mediated L-EPSCs when GABAA and GABAC receptors were blocked by bicuculline (200 μm), I4AA (20 μm), and picrotoxin (200 μm; p = 0.2; n = 8) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast, cone-mediated L-EPSCs recorded in dim conditions were reduced by TTX (Fig. 6B). The TTX-mediated reduction of the L-EPSCs was attributed to its direct actions at ON bipolar cells because ganglion cell sodium channels were blocked by including QX-314 (10 mm) in the recording pipette and because we previously showed that this reduction was not mediated by amacrine cell transmission (Ichinose et al., 2005). In bright conditions, TTX did not affect L-EPSCs (Fig. 6C), as expected from bipolar cell L-EPSP results (Fig. 2C). These results suggest that bipolar cell sodium channel activity increases ganglion cell L-EPSCs exclusively in cone-mediated, dim-light conditions.
Bipolar cell sodium channels enhance ganglion cell sensitivity to mean intensity and contrast
Light adaptation occurs by adjusting retinal network sensitivity to mean light intensity and contrast (the range of intensities around the mean luminance) in different ambient light conditions (Shapley and Enroth-Cugell, 1984; Smirnakis et al., 1997; Dunn and Rieke, 2006). The retinal network increases its sensitivity in low-light conditions to detect small visual inputs and decreases its sensitivity in bright-light conditions to prevent saturation. Sodium channels in bipolar cells may contribute to network light adaptation by increasing light sensitivity in dim conditions but not in bright conditions.
The ganglion cell sensitivity to mean light intensity was calculated for each cell (see Material and Methods) to determine whether it was affected by bipolar cell sodium channels. In dim-light-adapted conditions, TTX shifted the intensity–response curve for ganglion cell L-EPSCs to brighter intensities and reduced the maximum response (Fig. 6D, Dim). In bright-light-adapted conditions, TTX did not affect the intensity–response curve (Fig. 6D, Bright), whereas in scotopic conditions TTX shifted the intensity–response curve to dimmer intensities (Fig. 6D, Scotopic), attributable to blocking IPL inhibition, as noted above. The ratio of the sensitivity in the absence and the presence of TTX in each light condition is plotted in Figure 6E. In dim conditions, bipolar cell sodium channels strikingly enhanced the sensitivity, whereas in bright conditions, as well as scotopic conditions, the sensitivity was not affected, demonstrating that the sodium channels only boosted the sensitivity to dim cone signals.
The ganglion cell sensitivity to contrast, or the range of intensities around the mean luminance (Demb, 2002), was calculated in two ways (see Material and Methods): response/log intensity ratio from the intensity–response curve (Fig. 6F) or the slope of the contrast–response curve (Fig. 6G,H). We found that the contrast sensitivity was increased by bipolar cell sodium channels in dim-light-adapted conditions (Fig. 6F,G) but not in bright-light-adapted or scotopic conditions (Fig. 6F,H), suggesting that sodium channels also enhanced contrast sensitivity in dim, cone conditions.
Discussion
Light adaptation occurs in the retina to optimize its performance over a wide range of ambient light conditions (Dunn and Rieke, 2006). The pupil, photoreceptors, and retinal network all contribute to light adaptation. The retina adapts by adjusting its sensitivity to mean light intensity and contrast. Adaptation to mean light intensity occurs in both photoreceptors and the retinal network, whereas contrast adaptation only occurs in the retinal network (Green et al., 1975; Naka et al., 1979; Green and Powers, 1982; Shapley and Enroth-Cugell, 1984; Smirnakis et al., 1997; Pugh et al., 1999; Fain et al., 2001; Page-McCaw et al., 2004; Dunn and Rieke, 2006). Bipolar cells have been implicated as sites of network adaptation to both mean light intensity and contrast (Green et al., 1975; Naka et al., 1979; Shapley and Enroth-Cugell, 1984; Smirnakis et al., 1997; Rieke, 2001; Baccus and Meister, 2002; Page-McCaw et al., 2004); however, the cellular mechanisms that mediate these forms of adaptation are poorly understood. Our findings suggest that sodium channels, found in a subset of bipolar cells, regulate network adaptation by increasing retinal sensitivity to both mean intensity and contrast in dim-light conditions but not in bright conditions when they were inactivated by light-evoked dopamine release. This allows bipolar cell sodium channels to dynamically control retinal signal gain.
Dopamine and bright light contribute to network adaptation by reducing sodium currents in bipolar cells
Dopamine is “a messenger of light adaptation” that is released from amacrine cells by illumination and is likely a key factor in network adaptation (Witkovsky, 2004). Dopamine reduces rod-mediating signaling and enhances cone-mediated signaling in dim-light-adapted conditions (Witkovsky, 2004). Recent molecular studies suggest that dopamine reduces light sensitivity in bright-light conditions. Retinal network adaptation is perturbed in mutant zebrafish that cannot synthesize dopamine, and, unlike control fish, bright conditions do not reduce light sensitivity (Page-McCaw et al., 2004).
Our findings demonstrate that light-evoked dopamine release modulates bipolar cell sodium channel activity. First, we found that application of the dopamine agonist ADTN mimicked the effect of light adaptation in dim conditions. ADTN and bright conditions both reduced sodium currents and L-EPSPs in transient ON bipolar cells. Second, a D1 receptor antagonist reduced the effects of light adaptation (Figs. 4, 5). When D1 antagonists were applied before light adaptation, bipolar cell L-EPSPs and sodium currents were similar to those in dark-adapted conditions. These findings suggest that bright illumination caused dopamine release from amacrine cells that modulated bipolar cell sodium channels. L-EPSPs in sustained ON bipolar cells, which do not have sodium currents, were not affected by dopamine agonists, suggesting that dopamine reduced L-EPSPs in transient ON bipolar cells by acting on sodium channels and not on cones or mGluR6 receptors, which mediate responses in both bipolar cell classes.
In agreement with our results, Myhr et al. (1994) show that dopamine release in salamander retina is increased when illumination that predominantly affects cones is increased from dim to bright levels, consistent with biochemical experiments demonstrating that dopamine levels are two to four times higher in light, compared with dark levels (Godley and Wurtman, 1988; Boatright et al., 1989; Puppala et al., 2004). Our preliminary results show that the suppressive effects of bright light on bipolar sodium currents developed over several minutes, similar to other forms of dopamine-dependent light adaptation (Myhr et al., 1994). Figure 7 summarizes how bipolar cell sodium channels contribute ganglion cell light sensitivity at different background light levels. The TTX-sensitive changes in ganglion cell sensitivity are greatest when dopamine levels are thought to be low (dim mesopic) and lowest when dopamine levels are thought to be high (photopic). Ganglion cell light sensitivity during cone signaling conditions decreases as a function of background light intensity (Fig. 7). These findings, along with our observations that dopamine agonists mimic bright-light conditions, suggest that increases in dopamine release with increasing light levels are responsible for decreasing ganglion cell sensitivity, modulated by bipolar cell sodium channels.
Figure 7.

The enhancement of ganglion cell (GC) light sensitivity is decreased by increasing adaptation levels. The enhancement of ganglion cell sensitivity to mean illumination attributed to bipolar cell (BC) sodium channels was plotted as a function of background light (same dataset as in Fig. 6E). In scotopic conditions, light sensitivity in ganglion cells was not affected by bipolar cell sodium channels. In mesopic, dim-light-adapted conditions, light sensitivity was dramatically enhanced. The enhancement was decreased by increased ambient light levels and was not observed in photopic conditions. The decrease in light sensitivity was attributed to decreased bipolar cell sodium channel contributions and may be correlated with dopamine release by light adaptation (see Discussion).
Dopamine acted directly on bipolar cell sodium channels
Dopamine receptors are found on other types of retinal neurons in addition to bipolar cells, but the effects that we observed were not attributable to the activation of dopamine receptors on other cell types. This is because we eliminated the polysynaptic effects of dopamine on L-EPSPs by pharmacologically isolating the recorded bipolar cell from the network. Our results also suggest that dopamine affected bipolar cell L-EPSPs by acting directly on sodium channels, because calcium and potassium channels, which can be affected by dopamine (Pfeiffer-Linn and Lasater, 1993; Liu and Lasater, 1994; Fan and Yazulla, 1999), were pharmacologically blocked during the sodium current recordings.
It is possible that other modulatory substances in addition to dopamine were released and modulated sodium channels, because we adapted to bright light or perfused dopamine before applying a mixture of receptor antagonists to block network effects. Acetylcholine (ACh) release in the retina might be affected by dopamine or bright light (Yeh et al., 1984; Hensler and Dubocovich, 1986; Hensler et al., 1987). ACh could reduce sodium currents by activating muscarinic receptors, as reported for hippocampal neurons (Cantrell et al., 1996). Muscarinic receptors are present on a subset of bipolar cells in chick and primate retinas (Fischer et al., 1998; Yamada et al., 2003). However, our preliminary experiments showed that the muscarinic receptor antagonist atropine did not alter the effects of bright illumination on sodium currents, ruling out this possibility. Nitric oxide (NO) may also be released by light and/or dopamine application in the retina (Eldred and Blute, 2005; Sekaran et al., 2005) and could have modulated the bipolar cell sodium currents. However, in other parts of the CNS, NO increases voltage-gated sodium currents (Hammarstrom and Gage, 1999), which is opposite to our results. This suggests that bipolar cell sodium channels were not affected by nitric oxide.
The function of voltage-gated sodium channels can be modulated by dopamine
Voltage-gated sodium channels that are located on the dendrites and soma boost synaptic responses in the CNS. Sodium channels nonlinearly increase the amplitude of EPSPs in cerebral cortex pyramidal neurons and hippocampal cells (Schwindt and Crill, 1995; Stuart and Sakmann, 1995; Lipowsky et al., 1996; Gonzalez-Burgos and Barrionuevo, 2001). We previously reported that sodium channels in a subset of retinal bipolar cells boost light-evoked synaptic responses (Ichinose et al., 2005). Voltage-gated sodium channels are regulated by G-protein-coupled receptor (GPCR)-mediated phosphorylation (for review, see Cantrell and Catterall, 2001). Dopamine (Surmeier et al., 1992; Ma et al., 1994; Cantrell et al., 1997; Cantrell et al., 1999) and serotonin (Carr et al., 2003) modulate the generation of sodium spikes. However, it was not known whether EPSP amplification by sodium channels is affected by GPCR activation. Our results show that D1 dopamine GPCR activation in bright-light conditions reduced the activity of sodium channels in bipolar cells, attenuating the amplification of L-EPSPs.
Dopamine modulates voltage-gated sodium channels through diverse mechanisms. In retinal ganglion cells, dopamine accelerates the rate of entry of sodium channels into an inactivated state and slows their recovery from the inactivated state (Hayashida and Ishida, 2004). In cortical neurons, sodium currents are reduced by a reduction in the peak open probability and also by a negative shift in the voltage dependence of steady-state inactivation (Maurice et al., 2001). In hippocampal neurons, dopamine reduces sodium currents but did not affect channel inactivation (Cantrell et al., 1997). We found that dopamine reduced bipolar cell sodium currents by shifting the steady-state inactivation and activation of sodium channels, which reduced the availability of the channel at the resting membrane potential and also reduced the peak amplitude when activated (Fig. 3).
Our findings demonstrate that sodium channels in bipolar cells dynamically control retinal sensitivity to mean light and to contrast over a wide range of ambient light conditions. In dim-light-adapted conditions, the sodium channels boost retinal sensitivity, whereas in bright-light-adapted conditions, our results strongly suggest that dopamine suppresses the excitatory effect of the sodium channels to avoid saturation. We propose that bipolar cell sodium channels contribute to a novel mechanism of network light adaptation.
Footnotes
This work was supported by National Institutes of Health Grants EY08922 (to P.D.L.) and EY02687 (to the Department of Ophthalmology, Washington University), Research to Prevent Blindness, and The M. Bauer Foundation. We thank Drs. Erika Eggers, Paul Witkovsky, Andrew Ishida, Laura Frishman, Christopher Lingle, Russell Van Gelder, and Botir Sagdullaev for constructive comments on this manuscript and Dr. Mae Gordon for her comments on statistical analysis.
References
- Baccus SA, Meister M. Fast and slow contrast adaptation in retinal circuitry. Neuron. 2002;36:909–919. doi: 10.1016/s0896-6273(02)01050-4. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA, Xin D. Surround inhibition of mammalian AII amacrine cells is generated in the proximal retina. J Physiol (Lond) 2000;523:771–783. doi: 10.1111/j.1469-7793.2000.t01-1-00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright JH, Hoel MJ, Iuvone PM. Stimulation of endogenous dopamine release and metabolism in amphibian retina by light- and K+-evoked depolarization. Brain Res. 1989;482:164–168. doi: 10.1016/0006-8993(89)90555-6. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Ma JY, Scheuer T, Catterall WA. Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron. 1996;16:1019–1026. doi: 10.1016/s0896-6273(00)80125-7. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci. 1999;19 doi: 10.1523/JNEUROSCI.19-17-j0003.1999. RC21(1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA, Surmeier DJ. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- Cook PB, McReynolds JS. Lateral inhibition in the inner retina is important for spatial tuning of ganglion cells. Nat Neurosci. 1998;1:714–719. doi: 10.1038/3714. [DOI] [PubMed] [Google Scholar]
- Cook PB, Lukasiewicz PD, McReynolds JS. Action potentials are required for the lateral transmission of glycinergic transient inhibition in the amphibian retina. J Neurosci. 1998;18:2301–2308. doi: 10.1523/JNEUROSCI.18-06-02301.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demb JB. Multiple mechanisms for contrast adaptation in the retina. Neuron. 2002;36:781–783. doi: 10.1016/s0896-6273(02)01100-5. [DOI] [PubMed] [Google Scholar]
- Dunn FA, Rieke F. The impact of photoreceptor noise on retinal gain controls. Curr Opin Neurobiol. 2006;16:363–370. doi: 10.1016/j.conb.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Eldred WD, Blute TA. Imaging of nitric oxide in the retina. Vision Res. 2005;45:3469–3486. doi: 10.1016/j.visres.2005.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey PK, Burkhardt DA. Effects of light adaptation on contrast processing in bipolar cells in the retina. Vis Neurosci. 2001;18:581–597. doi: 10.1017/s0952523801184087. [DOI] [PubMed] [Google Scholar]
- Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- Fan SF, Yazulla S. Modulation of voltage-dependent K+ currents (IK(V)) in retinal bipolar cells by ascorbate is mediated by dopamine D1 receptors. Vis Neurosci. 1999;16:923–931. doi: 10.1017/s095252389916512x. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, McKinnon LA, Nathanson NM, Stell WK. Identification and localization of muscarinic acetylcholine receptors in the ocular tissues of the chick. J Comp Neurol. 1998;392:273–284. [PubMed] [Google Scholar]
- Godley BF, Wurtman RJ. Release of endogenous dopamine from the superfused rabbit retina in vitro: effect of light stimulation. Brain Res. 1988;452:393–395. doi: 10.1016/0006-8993(88)90046-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Barrionuevo G. Voltage-gated sodium channels shape subthreshold EPSPs in layer 5 pyramidal neurons from rat prefrontal cortex. J Neurophysiol. 2001;86:1671–1684. doi: 10.1152/jn.2001.86.4.1671. [DOI] [PubMed] [Google Scholar]
- Green DG, Powers MK. Mechanisms of light adaptation in rat retina. Vision Res. 1982;22:209–216. doi: 10.1016/0042-6989(82)90120-1. [DOI] [PubMed] [Google Scholar]
- Green DG, Dowling JE, Siegel IM, Ripps H. Retinal mechanisms of visual adaptation in the skate. J Gen Physiol. 1975;65:483–502. doi: 10.1085/jgp.65.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom AK, Gage PW. Nitric oxide increases persistent sodium current in rat hippocampal neurons. J Physiol. 1999;520:451–461. doi: 10.1111/j.1469-7793.1999.t01-1-00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida Y, Ishida AT. Dopamine receptor activation can reduce voltage-gated Na+ current by modulating both entry into and recovery from inactivation. J Neurophysiol. 2004;92:3134–3141. doi: 10.1152/jn.00526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensler JG, Dubocovich ML. D1-dopamine receptor activation mediates [3H]acetylcholine release from rabbit retina. Brain Res. 1986;398:407–412. doi: 10.1016/0006-8993(86)91506-4. [DOI] [PubMed] [Google Scholar]
- Hensler JG, Cotterell DJ, Dubocovich ML. Pharmacological and biochemical characterization of the D-1 dopamine receptor mediating acetylcholine release in rabbit retina. J Pharmacol Exp Ther. 1987;243:857–867. [PubMed] [Google Scholar]
- Ichinose T, Shields CR, Lukasiewicz PD. Sodium channels in transient retinal bipolar cells enhance visual responses in ganglion cells. J Neurosci. 2005;25:1856–1865. doi: 10.1523/JNEUROSCI.5208-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C. Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol. 1996;76:2181–2191. doi: 10.1152/jn.1996.76.4.2181. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lasater EM. Calcium currents in turtle retinal ganglion cells. II. Dopamine modulation via a cyclic AMP-dependent mechanism. J Neurophysiol. 1994;71:743–752. doi: 10.1152/jn.1994.71.2.743. [DOI] [PubMed] [Google Scholar]
- Ma JY, Li M, Catterall WA, Scheuer T. Modulation of brain Na+ channels by a G-protein-coupled pathway. Proc Natl Acad Sci USA. 1994;91:12351–12355. doi: 10.1073/pnas.91.25.12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora-Ferrer C, Yazulla S, Studholme KM, Haak-Frendscho M. Dopamine D1-receptor immunolocalization in goldfish retina. J Comp Neurol. 1999;411:705–714. doi: 10.1002/(sici)1096-9861(19990906)411:4<705::aid-cne14>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Myhr KL, Dong CJ, McReynolds JS. Cones contribute to light-evoked, dopamine-mediated uncoupling of horizontal cells in the mudpuppy retina. J Neurophysiol. 1994;72:56–62. doi: 10.1152/jn.1994.72.1.56. [DOI] [PubMed] [Google Scholar]
- Naka KI, Chan RY, Yasui S. Adaptation in catfish retina. J Neurophysiol. 1979;42:441–454. doi: 10.1152/jn.1979.42.2.441. [DOI] [PubMed] [Google Scholar]
- Nguyen-Legros J, Simon A, Caille I, Bloch B. Immunocytochemical localization of dopamine D1 receptors in the retina of mammals. Vis Neurosci. 1997;14:545–551. doi: 10.1017/s0952523800012207. [DOI] [PubMed] [Google Scholar]
- Normann RA, Werblin FS. Control of retinal sensitivity. I. Light and dark adaptation of vertebrate rods and cones. J Gen Physiol. 1974;63:37–61. doi: 10.1085/jgp.63.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw PS, Chung SC, Muto A, Roeser T, Staub W, Finger-Baier KC, Korenbrot JI, Baier H. Retinal network adaptation to bright light requires tyrosinase. Nat Neurosci. 2004;7:1329–1336. doi: 10.1038/nn1344. [DOI] [PubMed] [Google Scholar]
- Pfeiffer-Linn C, Lasater EM. Dopamine modulates in a differential fashion T- and L-type calcium currents in bass retinal horizontal cells. J Gen Physiol. 1993;102:277–294. doi: 10.1085/jgp.102.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh EN, Jr, Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol. 1999;9:410–418. doi: 10.1016/S0959-4388(99)80062-2. [DOI] [PubMed] [Google Scholar]
- Puppala D, Maaswinkel H, Mason B, Legan SJ, Li L. An in vivo microdialysis study of light/dark-modulation of vitreal dopamine release in zebrafish. J Neurocytol. 2004;33:193–201. doi: 10.1023/b:neur.0000030694.88653.d6. [DOI] [PubMed] [Google Scholar]
- Rieke F. Temporal contrast adaptation in salamander bipolar cells. J Neurosci. 2001;21:9445–9454. doi: 10.1523/JNEUROSCI.21-23-09445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnapf JL, Copenhagen DR. Differences in the kinetics of rod and cone synaptic transmission. Nature. 1982;296:862–864. doi: 10.1038/296862a0. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- Sekaran S, Cunningham J, Neal MJ, Hartell NA, Djamgoz MB. Nitric oxide release is induced by dopamine during illumination of the carp retina: serial neurochemical control of light adaptation. Eur J Neurosci. 2005;21:2199–2208. doi: 10.1111/j.1460-9568.2005.04051.x. [DOI] [PubMed] [Google Scholar]
- Shapley RM, Enroth-Cugell C. Visual adaptation and retinal gain controls. Prog Retin Res. 1984;3:263–346. [Google Scholar]
- Smirnakis SM, Berry MJ, Warland DK, Bialek W, Meister M. Adaptation of retinal processing to image contrast and spatial scale. Nature. 1997;386:69–73. doi: 10.1038/386069a0. [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci USA. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Wassle H. Immunohistochemical localization of dopamine D1 receptors in rat retina. Eur J Neurosci. 1996;8:2286–2297. doi: 10.1111/j.1460-9568.1996.tb01192.x. [DOI] [PubMed] [Google Scholar]
- Werblin FS. Control of retinal sensitivity. II. Lateral interactions at the outer plexi form layer. J Gen Physiol. 1974;63:62–87. doi: 10.1085/jgp.63.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40. doi: 10.1023/b:doop.0000019487.88486.0a. [DOI] [PubMed] [Google Scholar]
- Wu SM, Gao F, Maple BR. Functional architecture of synapses in the inner retina: segregation of visual signals by stratification of bipolar cell axon terminals. J Neurosci. 2000;20:4462–4470. doi: 10.1523/JNEUROSCI.20-12-04462.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada ES, Dmitrieva N, Keyser KT, Lindstrom JM, Hersh LB, Marshak DW. Synaptic connections of starburst amacrine cells and localization of acetylcholine receptors in primate retinas. J Comp Neurol. 2003;461:76–90. doi: 10.1002/cne.10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HH, Battelle BA, Puro DG. Dopamine regulates synaptic transmission mediated by cholinergic neurons of the rat retina. Neuroscience. 1984;13:901–909. doi: 10.1016/0306-4522(84)90104-0. [DOI] [PubMed] [Google Scholar]