

Abstract
The microRNA-200 (miR-200) family is part of a gene expression signature that predicts poor prognosis in lung cancer patients. In a mouse model of K-ras/p53-mutant lung adenocarcinoma, miR-200 levels are suppressed in metastasis-prone tumor cells, and forced miR-200 expression inhibits tumor growth and metastasis, but the miR-200 target genes that drive lung tumorigenesis have not been fully elucidated. Here, we scanned the genome for putative miR-200 binding sites and found them in the 3′-untranslated region (3′-UTR) of 35 genes that are amplified in human cancer. Mining of a database of resected human lung adenocarcinomas revealed that the levels of one of these genes, Flt1/VEGFR1, correlate inversely with duration of survival. Forced miR-200 expression suppressed Flt1 levels in metastasis-prone lung adenocarcinoma cells derived from K-ras/p53-mutant mice, and negatively regulated the Flt1 3′-UTR in reporter assays. Cancer-associated fibroblasts (CAFs) isolated from murine lung adenocarcinomas secreted abundant VEGF and enhanced tumor cell invasion in coculture studies. CAF-induced tumor cell invasion was abrogated by VEGF neutralization or Flt1 knockdown in tumor cells. Flt1 knockdown decreased the growth and metastasis of tumor cells in syngeneic mice. We conclude that miR-200 suppresses lung tumorigenesis by targeting Flt1.
Introduction
Metastasis is the primary cause of death from lung cancer. The lack of curative treatment options for patients with metastatic disease emphasizes the need for a better understanding of the biological processes that drive metastasis. Toward that goal, mouse models have been generated that develop lung adenocarcinomas with defined metastatic potential. Mice that express K-rasG12D conditionally, inducibly, or somatically develop lung adenocarcinomas that become locally advanced but do not metastasize, whereas mice that express the same K-ras mutation and a Tp53 mutation (p53R172H) that is found in patients with Li–Fraumeni syndrome and sporadic lung cancers develop widely metastatic lung adenocarcinoma (1–5). Studies on these metastasis-prone mice have revealed that their lung tumors recapitulate transcriptional features of tumors from poor-prognosis lung adenocarcinoma patients (6).
Accompanying metastasis in K-ras/p53-mutant mice is a suppression of the microRNA-200 (miR-200) family, which includes 5 family members (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) clustered in 2 genomic loci (200b-200a-429 and 200c-141) that target members of the Zeb family of transcriptional repressors that drive epithelial-to-mesenchymal transition (EMT; ref. 7). Forced expression of the miR-200b-200a-429 cluster abrogates the tumorigenic and metastatic capacities of these tumor cells and induces broad transcriptional changes (>1,000 genes increased or decreased), conferring transcriptional features of metastasis-incompetent tumor cells (7). Demonstrating the relevance of these findings to human lung cancer, low miR-200b levels are part of a microRNA signature in early-stage lung cancer specimens that predicts disease recurrence in patients (8). The effects of miR-200 on tumor cell invasion are clearly contextual, as reports on other tumor types have shown that miR-200 family members increase with neoplastic transformation and promote tumor cell invasion (9, 10). In addition to regulating EMT and invasion, the miR-200 family governs other tumor cell biologic processes including the acquisition of stem cell features, apoptosis, proliferation, and chemoresistance (11–16). Collectively, these findings indicate that miR-200 is a central regulator of tumorigenesis and demonstrate a critical need to identify miR-200 target genes involved in these processes.
The metastatic capacity of tumor cells is governed in part by extracellular signals emanating from infiltrating stromal cells (fibroblasts, inflammatory cells, endothelial cells, and others). These cells secrete a broad spectrum of ligands (growth factors, chemokines, and cytokines) that bind to cognate receptors on tumor cells and drive tumor cell proliferation and invasion (17). These ligands also direct intratumoral inflammation and angiogenesis owing to cognate receptors on inflammatory cells and endothelial cells (17). Thus, intratumoral ligands establish a complex network of cell–cell interactions within the tumor microenvironment. Although microRNAs regulate the meta-static capacity of tumor cells by targeting genes involved in a variety of processes (18), microRNAs that target receptors for prometastatic ligands have not been reported. Here we scanned the genome for putative miR-200 binding sites and found that Flt1/VEGFR1 is a candidate miR-200 target gene, leading us to posit that miR-200 suppresses lung adenocarcinoma metastasis by targeting Flt1 in tumor cells. Findings from these studies suggest that miR-200 suppresses metastasis by targeting Flt1 and support a growing body of evidence that, in addition to stimulating angiogenesis, VEGF promotes tumorigenesis through direct effects on tumor cells.
Material and Methods
Animal husbandry
Before their initiation, all mouse experiments were submitted to and approved by the Institutional Animal Care and Use Committee at the University of Texas M.D. Anderson Cancer Center. Mice received standards of care and were euthanized according to the standards set forth by the IACUC. 129/SV syngeneic mice were injected with tumor cells (1 × 106) subcutaneously in the right flank using RPMI containing 10% fetal bovine serum as vehicle. Mice were monitored daily for 6 weeks, at which time necropsies were performed to isolate and weigh the primary tumors and count lung metastases.
Establishment of murine lung adenocarcinoma cell lines
The methods used to establish lung adenocarcinoma cell lines in culture from murine tumors have been described previously (7). Cell lines were named according to the mouse number and site of derivation (e.g., 393P, denotes primary lung tumor; 344SQ, subcutaneous metastasis). These cells have alveolar type II cell properties and variable propensities to undergo EMT and metastasize following injection into syngeneic mice (5, 7).
Isolation of primary lung fibroblasts
Murine lung tissues isolated at necropsy were immediately perfused with 2% fetal bovine serum in Hank’s buffered salt solution (FBS-HBSS) and dispersed into single cell suspension by immersion in 3 μg/mL of Collagenase and DispaseII (Roche) on a gentleMACS Dissociator (Miltenyi Biotec) using the lung tissue dissociation programs (Lung_01 and Lung_02). Dispersed cells were centrifuged, washed with FBS-HBSS, and subjected to red blood cell lysis by adding RBC Buffer (BioLegend). The remaining cells were centrifuged, washed, filtered (70 and 40 μm), counted (Countess, Invitrogen), and mixed twice with antibody-conjugated magnetic beads (Dynal-Magnetic beads; Invitrogen) on a rotator, each time for 45 minutes at 4°C, to first deplete leukocytes (anti-CD45 and anti-CD68), endothelial cells (anti-CD31), and epithelial cells (anti-EPCAM) and then to isolate fibroblasts (anti-Thy-1) from the supernatants. Fibroblasts were eluted off the anti-Thy-1-conjugated beads by incubation in FBS-HBSS, 0.5% BSA, and 2 mmol/L EDTA, centrifuged, washed, and cultured in RPMI 1640 containing 10% FBS and 100 μg/100 U penicillin–streptomycin (GIBCO).
3′-UTR reporter assays
Flt1 3′-UTR (2.0 kb) was isolated from a mouse BAC clone (BACPAC Resource Center at Children’s Hospital Oakland Research Institute, RP23-375F2) by PCR and ligated into pCI-neo-hRL vector. ZEB1 3′-UTR (1.9 kb) subcloned into the same vector was used as a control. These reporters were cotransfected with synthetic miR-200 precursors (5 nmol/L, Ambion) into 344SQ cells (1 × 105 cells per well). After 48 hours, luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. To construct miR200-binding site mutants, a PCR-based site-directed mutagenesis strategy (19) was carried out using the following primers: (mut1) 5′-CCAGCCCCTGACAGGACTATACATCTATGAG-3′ and (mut2) 5′-GGTTTTATCTCAAGGACTAATATATA-GACAA-3′ (mutation sites are underlined).
Antibodies and recombinant peptides
For preparation of antibody-conjugated magnetic beads, we purchased Magnetic Sheep anti-Rat Dynabeads (Invitrogen) and the following rat anti-mouse antibodies: IgG (Abcam), CD31 (BD Pharmigen and Abcam), CD326 (BD Pharmigen), F4/80 (AbD Serotec and Abcam), CD90 (BD Pharmigen and Abcam), and CD68 (Abcam). Western blot analysis and immunofluorescence studies were performed using anti-VEGFR1 (R&D Systems) and anti-VEGFR3 (Santa Cruz Biotechnology) antibodies. For VEGF treatment and neutralization experiments, cells were treated with anti-mouse VEGF neutralizing antibodies, control IgG, or recombinant murine VEGF (R&D Systems).
Invasion assays
As described previously (7), cells were seeded first in the lower chambers (105 cells) and next in the upper chambers (5 × 104) of 24-well Transwell invasion plates (BD Biosciences). As indicated, antibodies (1 μg/mL) or recombinant peptides (10 ng/mL) were added to the lower chambers. Cells in the upper chambers were allowed to invade for 14 to 16 hours, at which time conditioned media samples were collected and stored at −80°C and the inserts in the upper chambers were removed for analysis. Cells on the inserts were fixed with 90% ethanol, stained with 0.1% crystal violet blue, and washed with ddH2O. Noninvaded cells on the upper side of the inserts were wiped off with a cotton swab. Invaded cells were counted in 5 microscopic fields at 4× magnification, which were averaged.
Cytokine quantification in conditioned media samples
Triplicate samples from 3 independent experiments (9 samples per condition) were simultaneously subjected to Magnetic Luminex Bead Pro Bioplex Assays (Bio-Rad Laboratories). For each cytokine calibration curve, 8 standards were performed ranging from 2.0 to 32,000 pg/mL. Data acquisition and analysis were completed by using the Bio-Plex 200 system with workstation (Bioplex Manager Software Version 5.0). Mean values were calculated from replicates (FSD) and a Dunnett’s test was performed to find significant differences.
VEGFR1 shRNA transfections
HuSH shRNA plasmid panels (29-mer) expressing murine VEGFR1 shRNA (TG514364) or scrambled control shRNA (TR30013)in pGFP-V-RS vector were purchased(OriGENE) that contained the following VEGFR1-specific shRNAmir sequences: 5′-GAAACCACAGCAGGAAGACGGTCCT-ATCG-3′ and 5′-TGAAGCGGTTCACCTGGACTG-AGACCAAG-3′. 344SQ cells were transfected using Lipofectamine and Opti-MEM (Invitrogen). Transfection media and reagents were replaced 24 hours after transfection, and cells were selected in puromycin (15 μg/mL) for up to 14 days to generate transfectant pools from which single cells were isolated, expanded, and used for experimentation.
Colony formation assays
Cells (5 × 104 in 0.3% agar) were seeded onto a layer of 0.8% agar in 6-well plates, allowed to proliferate for 21 days, and stained with 0.5 mg/mL nitrotetrazolium blue(Sigma-Aldrich). Colonies were visualized by light microscopy(4×) and counted. Results were expressed as mean values from triplicate wells.
Real-time quantitative PCR assays
RNA was isolated from cells using Trizol Reagent (Invitrogen), and 2 μg of each RNA sample was reverse-transcribed. RT-PCR analysis was performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems) using the comparative threshold method with L32 ribosomal protein mRNA as an endogenous reference housekeeping gene. For each reaction, a standard curve was performed using serial dilutions of a mixture of cDNA samples. SYBR green I (Applied Biosystems) was used as the fluorophore. All experiments were performed in triplicate. The primer sequences used are listed in Supplementary Table 1.
Western blot analysis
Lysates from cell lines were harvested using RIPA buffer and were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride nitrocellulose membrane (Bio-Rad Laboratories). Membranes were immunoblotted overnight at 4°C with primary antibodies in TBST containing 5% nonfat dry milk. Antibody binding was detected with an enhanced chemiluminescence kit according to the manufacturer’s instructions (Amersham).
Results
Identification of candidate miR-200 target genes
We used 2 prediction tools (www.microRNA.org and “conserved targets” at www.targetscan.org) to scan the genome for putative miR-200 binding sites and identified 432 genes in common to both programs, of which 183 had high (>0.80) probability of conserved targeting (Targetscan PCT) scores (20). To focus on those theoretical gene targets that might confer a selective advantage to cancer cells, the 432 genes were merged with the list of genes in regions of amplification in human cancer (21), which yielded 35 genes in common to both lists (Supplementary Table 2). These 35 genes were enriched in the Gene Ontology classifications “protein kinase activity” (6 genes, 1-sided Fisher’s exact test, P = 2.2E-4), “protein amino acid phosphorylation” (6 genes, 4.2E-4), and “regulation of progression through cell cycle” (4 genes, 4.9E-4). One of the genes on that list is the receptor tyrosine kinase Flt1/VEGFR1, which was of interest based on the underlying hypothesis of this study that miR-200 suppresses lung adenocarcinoma metastasis by targeting receptors for prometastatic ligands. Supporting a potential role for Flt1 in this regard, mining of publicly available gene expression profiles from 2 cohorts of lung adenocarcinoma patients (22, 23) revealed that intratumoral Flt1 expression correlated inversely with duration of survival in one (n = 117, P = 0.007, log-rank test; P = 0.016, univariate Cox) and with relapse-free survival in the other (n = 193, P = 0.01, log-rank test; P = 0.08, univariate Cox; Fig. 1A).
Figure 1.

Flt1 is a miR-200 target gene. A, Kaplan–Meier analysis of 2 cohorts of lung denocarcinoma patients on the basis of VEGFR1 expression (top quartile vs. lower 3 quartiles) in resected primary tumor specimens. The number of patients analyzed (n; top right corner) and the results of statistical tests (P; bottom left corner) are indicated for each cohort. B, miR-200 targets sequences in the Flt1 gene 3′-UTR. Reporter constructs containing wild-type and mutant Flt1 3′UTR are illustrated diagrammatically. 344SQ cells were transiently cotransfected with pre-miRs (10 nmol/L) and reporter plasmids (500 ng) that express luciferase alone (control) or are linked to the full-length 3′-UTR of Zeb1 (Zeb1) or Flt1 that is wild-type (Flt1) or mutated at the 5′ (mut1) or 3′ (mut2) putative miR-200 binding site. Results were normalized on the basis of renilla luciferase and expressed as the mean values of triplicate wells. *, P < 0.05. C, miR-200 suppresses VEGFR1 expression. Western blot analysis (left) of 344SQ cells and murine lung endothelial cells (MEC) as a positive control. Quantitative RT-PCR analysis (bar graphs) of 344SQ cells stably transfected with miR-200 (200b) or empty (control) lentiviral vectors normalized on the basis of L32 ribosomal protein mRNA levels and expressed as mean values of triplicate cultures relative to control transfectants, which were set at 1.0. Western blot analysis (right) of VEGFR1 in the same transfectants, which was quantified densitometrically, normalized on the basis of actin, and expressed relative to control, which was set at 1.0.
Experimental validation of Flt1 as a miR-200 target
Flt1 has 2 putative miR-200b/c/429 binding sites in its 3′-UTR, one of which is located from nucleotides 33 to 39 and has a probability of conserved targeting (PCT) score of 0.84, which is similar to scores for the bona fide miR-200b/c/ 429 binding sites within the Zeb1 3′-UTR, and the other is located from nucleotides 847 to 853 and has a PCT score < 0.10 (diagrammatically illustrated in Fig. 1B). To determine whether Flt1 is a direct target of miR-200, a lung adenocarcinoma cell line derived from KrasLA1/+; p53R172HΔG/+ mice that expresses low endogenous miR-200 levels and is metastasis-prone (344SQ, ref. 7), was cotransfected with synthetic miR-200 precursors (200a, 200b, 200c, 141, or 429) and a reporter plasmid containing luciferase fused to 3′-UTR sequences from Flt1, Zeb1 (a positive control), or nothing (a negative control). Relative to the effect of scrambled control oligomers, miR-200b and 200c suppressed the activity of the Flt1 3′-UTR reporter by 50% to 60%, which was similar to the effect of these oligomers on the Zeb1 3′-UTR reporter, and had no effect on the control luciferase reporter (Fig. 1B). This suppressive effect was abrogated by site-directed mutagenesis of the 5′ putative miR-200 binding site but not the 3′ binding site (Fig. 1B). Furthermore, other miR-200 family members (200a and 429) had no effect on Flt1 3′-UTR reporter activity (data not shown), suggesting that Flt1 regulation is specific to miR-200b and 200c.
Regulation of endogenous Flt1 expression by miR-200 was examined by stably transfecting lentiviral vectors expressing the miR-200b/a/429 (miR-200b) cluster into 344SQ cells, which expressed VEGFR1 and VEGFR3 (Fig. 1C) but no detectable VEGFR2 (data not shown). Forced miR-200b expression decreased Flt1 mRNA and VEGFR1 protein levels by 73% and 38%, respectively (Fig. 1C), whereas the mRNA levels of VEGFR3 did not change (data not shown). Interestingly, VEGF-A levels in conditioned media samples from 344SQ_miR200b cells (258.36 pg/mL) were 3.8-fold higher than that of 344SQ_vector cells (66.87 pg/mL), raising the possibility that VEGFR1 suppression by miR-200 activated a feedback loop involving VEGF that maintains VEGFR1 activity.
miR-200 inhibits lung adenocarcinoma cell invasion by targeting Flt1
On the basis of the aforementioned findings, we posited that miR-200 abrogates lung adenocarcinoma cell invasion and metastasis by targeting VEGFR1 on tumor cells. To test this hypothesis, we cocultured lung adenocarcinoma cells with CAFs, which reportedly secrete VEGF (24). CAFs and (as a comparison) normal lung fibroblasts (LF) were isolated from the lungs of KrasLA1 mice and wild-type littermates, respectively. Inflammatory cells and endothelial cells were first negatively selected from dispersed lung tissues by incubation with magnetic beads linked to antibodies against CD45 and CD31, respectively. Fibroblasts were isolated from the residual tissues by incubation with magnetic beads linked to antibodies against Thy-1 (CD90), a glycosylated cell surface protein expressed on lung fibroblasts, inflammatory cells, and neural cells (25). Thy-1pos cells isolated from normal lungs expressed vimentin but not markers of endothelial cells (CD31), macrophages (CD68), or leukocytes (CD45; Fig. 2A and B), establishing the specificity of these conditions for the isolation of lung fibroblasts. Examination of CAFs and LFs in culture revealed clear differences. CAFs were smaller and more rounded, had fewer cellular projections (Fig. 2C), expressed α-SMA at higher levels (Fig. 2B), and had stress fibers arranged in parallel sheets, whereas LFs had stress fibers that were interwoven, flat, and branching (Fig. 2D). The secretomes of CAFs and LFs were compared by performing multiplexed antibody-based assays on conditioned media samples collected from these cells (n = 9 samples per condition). The concentrations of 22 (64%) of the 34 proteins analyzed were at least 2-fold higher in monocultured CAFs than they were in monocultured LFs or 344SQ cells, including monocyte chemotactic protein-1 (MCP-1/CCL2), the murine functional homologues of human CXCL1 (KC and MIP2), VEGF-A, interleukin-6, and others (Table 1). VEGF-A was one of the most abundant proteins detected in CAF conditioned media samples (409.68 pg/mL), whereas VEGF-D, a VEGFR3 ligand, was present at extremely low levels (3.82 pg/mL).
Figure 2.

Characterization of LFs and CAFs. A, quantitative RT-PCR analysis of LFs. Values normalized on the basis of L32 ribosomal protein mRNA levels. B, Detection of fibroblast markers in LFs and CAFs by immunocytochemistry imaged by fluorescent or light microscopic analysis (20× magnification) of identical fields. Bar indicates 200 μm. C, morphology of primary lung fibroblasts (light microscopic images at 20× magnification). Bar indicates 100 μm. D, distinct patterns of stress fibers in LFs and CAFs. Immunofluorescent staining and laser scanning confocal imaging of α-smooth muscle actin in 3D matrices produced by fibroblasts after extraction of cellular debris. Bar indicates 100 μm.
Table 1.
Protein concentrations in conditioned media from monocultures and cocultures
| Proteins | 344SQ | LF | CAF | LF/344SQ | CAF/344SQ |
|---|---|---|---|---|---|
| MCP-1 | 9.11 | 282.46 | 2,524.16 | 1,182.22a | 2,711.63a |
| MIP-1α | 21.29 | 21.67 | 59.85 | 33.2 | 177.97a |
| MIP-1β | 0.75 | 2.71 | 60.64 | 14.16a | 142.34a |
| RANTES | 0.5 | 1.22 | 19.65 | 9.40a | 29.24 |
| Eotaxin | 76.12 | 36.8 | 149.24 | 86.01 | 196.3 |
| KC | 486.38 | 52.69 | 1,191.99 | 809.42 | 2,370.50a |
| MIP-2 | 2.24 | 3.9 | 118.74 | 31.07a | 1,119.31a |
| MIG | 2.51 | 1.58 | 31.82 | 10.08a | 69.44a |
| bFGF | 1.94 | 2.07 | 12.57 | 5.28a | 19.08a |
| PDGF-β | 38.86 | 22.36 | 17.41 | 13.67 | 13.58 |
| VEGF-A | 61.94 | 8.3 | 409.68 | 85.75a | 712.91a |
| VEGF-D | 0.92 | 6.33 | 3.82 | 0.5 | 11.75 |
| IL-1α | 0.2 | 0.33 | 1.71 | 0.43 | 1.56 |
| IL-1β | 6.87 | 3.36 | 23.35 | 10.15a | 34.51a |
| IL-2 | 0.75 | 0.7 | 1.55 | 0.69 | 1.64 |
| IL-3 | 0.03 | 0.18 | 0.24 | 0.09 | 0.29 |
| IL-4 | 0.04 | 1.67 | 0.21 | 0.04 | 0.27 |
| IL-5 | 0.41 | 0.6 | 1.28 | 0.49 | 1.66 |
| IL-6 | 1.29 | 30.2 | 1,838.95 | 128.90a | 675 |
| IL-9 | 5.15 | 2.35 | 23.67 | 4.42 | 24.96 |
| IL-10 | 0.87 | 0.48 | 2.08 | 0.85 | 2.95a |
| IL-12(p40) | 0.43 | 0.32 | 1.65 | 0.54a | 2.21a |
| IL-12(p70) | 22.89 | 22.64 | 39.19 | 28.82a | 41.69a |
| IL-13 | 34.08 | 27.36 | 53.69 | 30.29 | 61.19 |
| IL-17 | 0.23 | 0.34 | 0.97 | 0.25 | 1.22 |
| IL-15 | 2.38 | 1.78 | 40.17 | 12.2a | 76.29a |
| IL-18 | 16.02 | 18.35 | 55.4 | 19.44 | 78.42* |
| TGF-β1 | 852.39 | 110.68 | 1,875.37 | 903.56a | 2,687.85a |
| G-CSF | 10.09 | 3.67 | 47.43 | 32.76a | 122.32a |
| GM-CSF | 16.37 | 3.93 | 12.22 | 16.21 | 27.56a |
| M-CSF | 13.29 | 32.54 | 106.95 | 59.80a | 132.21a |
| IFN-γ | 3.24 | 2 | 8.58 | 3.48 | 10.85 |
| TNF-α | 7.67 | 5.82 | 15.7 | 8.61 | 20.92a |
| LIF | 30.62 | 7.74 | 103.94 | 36.23 | 171.74a |
NOTE: Protein concentrations are pg/mL. Comparisons (coculture vs. either of 2 monocultures) were based on Dunnett’s test.
Protein concentrations that were significantly different (P < 0.05) in coculture relative to that of either of the monocultures.
Four different lung adenocarcinoma cells derived from KrasLA1/+; p53R172HΔG/+ mice were cocultured in Boyden chambers with LFs or CAFs. Tumor cells were more invasive in coculture with CAFs than they were in coculture with LFs (Fig. 3A). Several proteins increased sharply in the coculture conditioned media samples relative to their levels in monocultures, including TGFβ1, VEGF-A, monokine induced by interferon-γ (MIG/CXCL9), CXCR2 ligands (KC and MIP2), macrophage inflammatory protein (MIP-1α/CCL3), MIP-1β/CCL4, MCP-1, and leukemia inhibitory factor (LIF), which increased more prominently in the CAF cocultures than they did in the LF cocultures (Table 1). Examination of the mRNA levels of receptors for those ligands revealed that Flt1 was one of the most abundant receptors expressed in 344SQ cells (Supplementary Fig.); therefore, we examined whether VEGF-A was required for CAF-induced tumor cell invasion. The cocultures were treated with an anti-VEGF neutralizing antibody, which abrogated CAF-induced 344SQ cell invasion (Fig. 4A). Of note, VEGF neutralization (Fig. 4A) and forced miR-200 expression (Fig. 4B) suppressed 344SQ cell invasion to similar degrees, arguing in favor of VEGFR1 as a downstream mediator of miR-200–induced tumor cell invasion. Conversely, when added to the lower chamber instead of CAFs, recombinant VEGF enhanced the invasion of 4 of the 5 lung adenocarcinoma cell lines examined (Fig. 4C). Together, these findings demonstrated that VEGF-A was required for CAF-induced tumor cell invasion.
Figure 3.
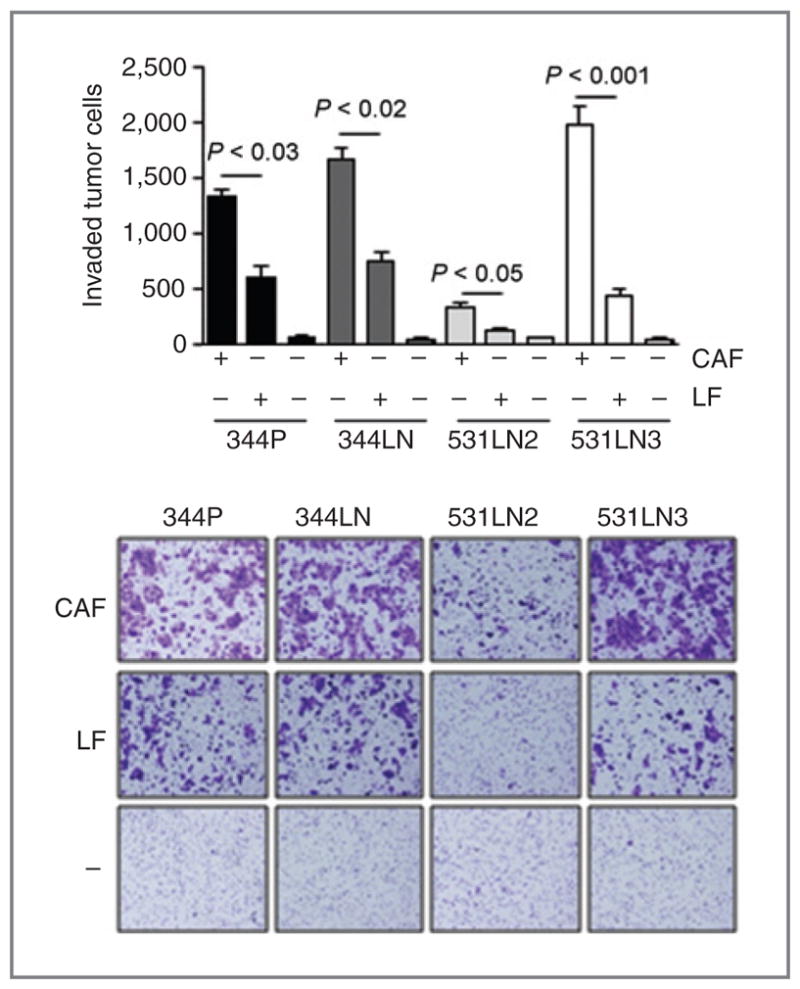
Fibroblast-induced tumor cell invasion. 344SQ cells were monocultured (−) or cocultured with LFs or CAFs. Invasive tumor cells were photographed (images) and counted in at least 5 separate microscopic fields per well, which were averaged, and the mean values per well ± SD were calculated from replicate wells (bar graphs).
Figure 4.

VEGF mediates CAF-induced lung adenocarcinoma cell invasion. A, 344SQ cells were cocultured with CAFs in the presence (+) or absence (−) of anti-VEGF-A neutralizing antibodies. Invasive tumor cells were photographed (images) and counted in at least 5 separate microscopic fields per well, which were averaged, and the mean values per well ± SD were calculated from replicate wells (bar graphs). B, miR-200 suppresses CAF-induced tumor cell invasion. Transwell invasion assays of 344SQ cells that stably express miR-200b (200b) or empty (control) lentiviral vectors in coculture with CAFs. Invaded cells were stained, photographed (image), and counted in at least 5 separate microscopic fields per well, which were averaged, and the mean values per well ± SD were calculated from replicate wells (bar graph). C, The indicated tumor cell lines derived from KrasLA1/+; p53R172HΔG/+ mice were monocultured in the upper chamber of a dual-chamber well, and recombinant VEGF-A was added (+) or not added (−) to the lower chamber. Invasive tumor cells were photographed (images) and counted in at least 5 separate microscopic fields per well, which were averaged, and the mean values per well ± SD were calculated from replicate wells (bar graphs).
Flt1 is required for lung adenocarcinoma cell invasion and metastasis
Following subcutaneous injection into syngeneic mice, 344SQ cells form tumors that prominently express Flt1 (Fig. 5A). To assess its role in these tumors, Flt1 was knocked down in 344SQ cells by introduction of short hairpin RNA (shRNA). Two 344SQ_Flt1 shRNA clones (#2–3 and #4–1) were selected for further study on the basis of prominent reductions in Flt1 mRNA and VEGFR1 protein levels (Fig. 5B). Relative to scrambled shRNA transfectants, the Flt1-depleted cells exhibited decreased proliferation in monolayer culture (Fig. 5C), decreased colony formation in soft agar (Fig. 5D), decreased invasion in coculture with CAFs (Fig. 5E), and reduced metastatic potential following subcutaneous injection into syngeneic mice (Fig. 5F).
Figure 5.

Flt1 regulates multiple biologic properties of lung adenocarcinoma cells. A, Flt1/VEGFR1 detected in 344SQ syngeneic tumor. Immunofluorescent staining of a 344SQ syngeneic tumor with anti-VEGFR1 antibody (green) and 4′,6-diamidino-2-phenylindole (DAPI, blue) to detect nuclei. Adjacent tissue section stained with hematoxylin and eosin. B, suppression of Flt1 expression by Flt1 shRNA. Quantification of Flt1 in 2 independent clones of 344SQ transfected with Flt1 shRNA (#2–3, #4–1) or scrambled shRNA (SCR) by quantitative RT-PCR of triplicate RNA samples (bar graph) and Western blot analysis of protein (gels); bands were quantified densitometrically. Flt1 RNA and protein levels were normalized on the basis of L32 ribosomal protein mRNA and actin levels, respectively, and expressed relative to controls, which were set at 1.0. C, Flt1 knockdown inhibits 344SQ proliferation. Transfectants were seeded in monolayer cultures, counted, and expressed as mean values (± SD) of quadruplicate samples. D, Flt1 knockdown inhibits 344SQ anchorage-independent growth. Transfectants were seeded in soft agar, and visible colonies were photographed (images) and counted (bar graph) at 21 days. Results expressed as mean values ± SD of triplicate samples. E, Flt1 knockdown inhibits CAF-induced tumor cell invasion. Transfectants were cocultured with CAF; invasive tumor cells were photographed (images) and counted in 5 separate microscopic fields per well, averaged, and mean values per well ± SD were calculated from replicate wells (bar graphs). F, Flt1 knockdown inhibits tumor metastasis. Syngeneic mice were injected subcutaneously with tumor cells and necropsied at 6 weeks to measure primary tumor size and count lung metastases (scatter plots). Mean values, long horizontal lines; standard deviations, short horizontal lines.
Discussion
Findings from mouse models of human cancer support a central role for miRs in tumor progression, and studies to identify miR target genes have identified novel oncogenes and tumor suppressor genes (26, 27), demonstrating that miRs are valuable tools to better understand cancer biology. Herein, we scanned the genome for theoretical miR-200 target genes to identify ones that might be required for tumor cells to respond to metastasis-promoting extracellular signals and made the discovery that Flt1 is a miR-200 target gene that is required for tumor cell invasion and metastasis.
The putative miR-200 target genes identified here are numerous and have diverse biological functions, which is consistent with the diverse transcriptional changes induced by forced miR-200 expression in tumor cells (7) and reflects the functional diversity of the miR-200 target genes validated thus far, which include, among others, Zeb1, Zeb2, Friend of GATA 2 (FOG2), Sox2, Klf4, and the polycomb repressor Bmi1 (28–30). Among the 35 theoretical target genes that overlapped with regions of gene amplification in human cancer, Flt1 had the highest high PCT score. We posited that miR-200 regulates tumor cell sensitivity to extracellular signals by targeting Flt1, and several salient findings reported here support this hypothesis. First, forced miR-200 expression suppressed Flt1 levels in a metastasis-prone lung adenocarcinoma cell line and negatively regulated the Flt1 3′-UTR in reporter assays. Second, CAF-induced tumor cell invasion was abrogated by Flt1 knockdown or VEGF-A neutralization. Third, Flt1 knockdown abrogated invasion and metastasis in a syngeneic tumor model. However, reconstitution of Flt1 expression in 344SQ_miR200b cells was not sufficient to rescue tumor growth and metastasis in mice (data not shown), indicating that additional miR-200 target genes must be reexpressed to reverse the phenotypic changes induced by forced miR-200 expression. Collectively, these findings suggest that miR-200 opposes metastasis by coordinately repressing multiple target genes, a scenario that has been described for miR-31, which coordinates repression of a cohort of metastasis-promoting genes (31).
Findings presented here show that Thy-1pos CAFs are a rich source of secreted factors that have diverse biological functions. The concentrations of CAF-derived factors were several-fold higher than they were in 344SQ cell cultures (Table 1), arguing that cells in the tumor stroma are a primary source of extracellular factors required for tumor cell invasion and metastasis. Many of these factors are immunomodulatory (KC, MIP2, TGFβ1, MIG, MIP-1α, MIP-1β, and MCP-1) and proangiogenic (KC, MIP2, and VEGF; ref. 32), suggesting that CAFs regulate multiple biological processes in this model. From these studies, it is unclear which stages of lung neoplasia are regulated by CAF because CAFs were isolated from KrasLA1 mice at 10 to 12 months of age when lung lesions are multifocal and have differing histologic severities (atypical alveolar hyperplasia, adenoma, and adenocarcinoma). To address this question, in situ studies are needed to localize CAFs in lung tumors and correlate their presence with histologic severity and associated inflammatory and endothelial cells as an indicator of the cell types that CAFs recruit. These studies will require markers that reliably distinguish CAFs from LFs, which is a challenge given that many candidates, such as α-smooth muscle actin, are expressed in other cell types such as pericytes (33).
Several findings presented here complement a growing body of evidence that, in addition to its role as an angiogenic factor, VEGF promotes tumorigenesis through direct effects on tumor cells. First, intratumoral Flt1 levels correlated inversely with duration of survival in lung adenocarcinoma patients. We can not exclude the possibility that contaminating stromal elements contributed to the high Flt1 expression, but the specimens chosen for analysis in these cohorts had more than 70% tumor cell purity (22, 23), which argues against this possibility. Second, studies on Flt1-deficient lung adenocarcinoma cells demonstrated that VEGFR1 was required for cell proliferation, invasion, and metastasis. Similarly, in colorectal and pancreatic cancer cells, VEGFR1 maintains cell survival through cross-talk with Wnt-1/Frizzled, and VEGFR1 is required for tumor cell migration and invasion (34–38). Studies performed to address the tumor cell–specific effects of VEGF have revealed that VEGFR1 activation induces tumor cell EMT and increases the phosphorylation of Src family members (34, 36). The lung adenocarcinoma cells in this study, however, did not exhibit morphologic or biochemical evidence of EMT following treatment with recombinant VEGF (data not shown), suggesting that VEGF enhances tumor cell invasion through distinct mechanisms that are tumor cell–type specific. In contrast to its active role in tumor cells, VEGFR1 functions as a VEGF sink in endothelial cells and opposes VEGFR2-induced endothelial cell division (39, 40).
These findings expand our understanding of the mechanisms by which the VEGFR1 axis is activated in tumor cells. In renal cell carcinomas, inactivating VHL mutations increase expression of hypoxia-inducible factor-1α, which increases VEGF-A secretion, enhancing tumor angiogenesis (41). On the basis of findings reported here, we propose that miR-200 suppression raises VEGFR1 levels, enhancing the sensitivity of tumor cells to VEGF in the tumor microenvironment. miR-200 is commonly repressed in human tumor types other than lung cancer (42–44), suggesting that the findings presented here are broadly applicable. Given the exquisite sensitivity of renal cell carcinomas to VEGF antagonists (41), it is tempting to speculate that the subset of lung adenocarcinomas with low miR-200 expression might be similarly sensitive to treatment with these agents.
Supplementary Material
Acknowledgments
We thank all the members of the Kurie lab for assistance and comments on the work.
Grant Support
NIH grants 5 T32 CA009666 (D.L. Gibbons); P50 CA70907 (Lung Cancer SPORE), R01 grants CA132608 and CA117965, David M. Sather Memorial Fund, and The Armour Family Lung Cancer Research Fund (J.M. Kurie); P30 CA125123 and the Dan L. Duncan Cancer Center at Baylor College of Medicine (C.J. Creighton). D.L. Gibbons was also supported by a Young Investigator Award from The ASCO Cancer Foundation and an International Association for the Study of Lung Cancer (IASLC) Fellow Grant. J.M. Kurie is the Elza A. and Ina Shackelford Freeman Endowed Professor in Lung Cancer.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 2.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 4.Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M, Mims B, El-Naggar AK, et al. High metastatic potential in mice inheriting a targeted p53 missense mutation. Proc Natl Acad Sci U S A. 2000;97:4174–9. doi: 10.1073/pnas.97.8.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 6.Gibbons DL, Lin W, Creighton CJ, Zheng S, Berel D, Yang Y, et al. Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PLoS One. 2009;4:e5401. doi: 10.1371/journal.pone.0005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–51. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patnaik SK, Kannisto E, Knudsen S, Yendamuri S. Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res. 2010;70:36–45. doi: 10.1158/0008-5472.CAN-09-3153. [DOI] [PubMed] [Google Scholar]
- 9.Elson-Schwab I, Lorentzen A, Marshall CJ. MicroRNA-200 family members differentially regulate morphological plasticity and mode of melanoma cell invasion. PLoS One. 2010;5:e13176. doi: 10.1371/journal.pone.0013176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761–72. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 13.Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–17. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Schickel R, Park SM, Murmann AE, Peter ME. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol Cell. 2010;38:908–15. doi: 10.1016/j.molcel.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leskela S, Leandro-Garcia LJ, Mendiola M, Barriuso J, Inglada-Perez L, Munoz I, et al. miR-200 family controls {beta}-tubulin III expression and is associated with paclitaxel-based treatment response and progression-free survival in ovarian cancer patients. Endocr Relat Cancer. doi: 10.1677/ERC-10-0148. [Epub 2010 Nov 4] [DOI] [PubMed] [Google Scholar]
- 17.Chiodoni C, Colombo MP, Sangaletti S. Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010;29:295–307. doi: 10.1007/s10555-010-9221-8. [DOI] [PubMed] [Google Scholar]
- 18.Valastyan S, Weinberg RA. MicroRNAs: Crucial multi-tasking components in the complex circuitry of tumor metastasis. Cell Cycle. 2009;8:3506–12. doi: 10.4161/cc.8.21.9802. [DOI] [PubMed] [Google Scholar]
- 19.Meza R, Nunez-Valdez ME, Sanchez J, Bravo A. Isolation of Cry1Ab protein mutants of Bacillus thuringiensis by a highly efficient PCR site-directed mutagenesis system. FEMS Microbiol Lett. 1996;145:333–9. doi: 10.1111/j.1574-6968.1996.tb08597.x. [DOI] [PubMed] [Google Scholar]
- 20.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chitale D, Gong Y, Taylor BS, Broderick S, Brennan C, Somwar R, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28:2773–83. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomida S, Takeuchi T, Shimada Y, Arima C, Matsuo K, Mitsudomi T, et al. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J Clin Oncol. 2009;27:2793–9. doi: 10.1200/JCO.2008.19.7053. [DOI] [PubMed] [Google Scholar]
- 24.Ito TK, Ishii G, Chiba H, Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- 25.Rege TA, Hagood JS. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim Biophys Acta. 2006;1763:991–9. doi: 10.1016/j.bbamcr.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L, Weinberg RA. MicroRNAs in malignant progression. Cell Cycle. 2008;7(5):570–2. doi: 10.4161/cc.7.5.5547. [DOI] [PubMed] [Google Scholar]
- 27.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 28.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 29.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyun S, Lee JH, Jin H, Nam J, Namkoong B, Lee G, et al. Conserved MicroRNA miR-8/miR-200 and its target USH/FOG2 control growth by regulating PI3K. Cell. 2009;139:1096–108. doi: 10.1016/j.cell.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–46. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol. 2010;85:593–8. doi: 10.1002/ajh.21745. [DOI] [PubMed] [Google Scholar]
- 34.Yang AD, Camp ER, Fan F, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- 35.Wey JS, Fan F, Gray MJ, Bauer TW, McCarty MF, Somcio R, et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer. 2005;104:427–38. doi: 10.1002/cncr.21145. [DOI] [PubMed] [Google Scholar]
- 36.Lesslie DP, Summy JM, Parikh NU, Fan F, Trevino JG, Sawyer TK, et al. Vascular endothelial growth factor receptor-1 mediates migration of human colorectal carcinoma cells by activation of Src family kinases. Br J Cancer. 2006;94:1710–7. doi: 10.1038/sj.bjc.6603143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallas NA, Fan F, Gray MJ, Van Buren G, 2nd, Lim SJ, Xia L, et al. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev. 2007;26:433–41. doi: 10.1007/s10555-007-9070-2. [DOI] [PubMed] [Google Scholar]
- 38.Zeitlin BD, Ellis LM, Nor JE. Inhibition of vascular endothelial growth factor receptor-1/Wnt/{beta}-catenin crosstalk leads to tumor cell death. Clin Cancer Res. 2009;15:7453–5. doi: 10.1158/1078-0432.CCR-09-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–30. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 40.Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004;164:1531–5. doi: 10.1016/S0002-9440(10)63711-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep. 2009;11:94–101. doi: 10.1007/s11912-009-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu X, Schwarz JK, Lewis JS, Jr, et al. A microRNA expression signature for cervical cancer prognosis. Cancer Res. 2010;70:1441–8. doi: 10.1158/0008-5472.CAN-09-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saydam O, Shen Y, Wurdinger T, Senol O, Boke E, James MF, et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol Cell Biol. 2009;29:5923–40. doi: 10.1128/MCB.00332-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, et al. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res. 2009;15:5060–72. doi: 10.1158/1078-0432.CCR-08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.