

Abstract
Microsomal prostaglandin E synthase-1 (mPGES-1) is the terminal synthase responsible for the synthesis of the pro-tumorigenic prostaglandin E2 (PGE2). mPGES-1 is overexpressed in a wide variety of cancers. Since its discovery in 1997 by Bengt Samuelsson and collaborators, the enzyme has been the object of over 200 peer-reviewed articles. Although today mPGES-1 is considered a validated and promising therapeutic target for anticancer drug discovery, challenges in inhibitor design and selectivity are such that up to this date there are only a few published records of small-molecule inhibitors targeting the enzyme and exhibiting some in vivo anticancer activity. This review summarizes the structures, and the in vitro and in vivo activities of these novel mPGES-1 inhibitors. Challenges that have been encountered are also discussed.
Prostaglandin E2 (PGE2), the pivotal prosta-glandin (PG) produced by most mammalian tissues, regulates multiple biological processes under both normal and pathological conditions. PGE2 is the chief mediator of inflammation and represents one of the most abundant prostanoid. The final step in the biosynthesis of PGE2 is catalyzed by prostaglandin E synthases (PGESs), a family of oxido-reductases, which has generated increasing interest as a therapeutic target in the treatment of inflammatory-related diseases. Although this family of enzymes plays an important role in inflammatory-related diseases, this review focuses on microsomal PGE synthase-1 (mPGES-1), the inducible PGES and its role in cancer specifically. Structural and biological properties of the enzyme are briefly summarized in the first part of this review since this protein has been the object of many detailed reviews [1–4]. In the second part of this review, compounds that have been described in the literature to inhibit mPGES-1 activity are presented and challenges regarding their selectivity and in vivo activity are also discussed.
Structure, function & regulation of mPGES-1
Structure of mPGES-1
Microsomal prostaglandin E synthase-1 is a member of the membrane-associated proteins involved in eicosanoid and glutathione metabolism (MAPEG) superfamily [5] and exhibits a significant sequence homology with micro-somal glutathione-S-transferase (GST)-1-like 1 (MGST-1), 5-lipoxygenase (LOX)-activating protein (FLAP) and leukotriene C4 synthase (LTC4S). All MAPEG proteins are small proteins of 14–18 kDa and have a similar 3D structure [6]. An electron crystallographic structure (3.5 Å) of mPGES-1 was published in 2008 (PDB: 3DWW) [7] and confirmed the trimeric structure of the protein as predicted by Xing et al._[8] and suggested by Hetu et al. [9]. Similarly to MGST-1, FLAP and LTC4S, the protein folds into four transmembrane helices (TM1–4) (Figure 1A). As MGST-1, mPGES-1 requires glutathione (GSH) as an essential cofactor for its activity [10]. Consequently, the protein was crystallized in the presence of GSH, which binds in the active site of the enzyme defined mostly by TM1 and TM4 for each of the subunits. GSH interacts in a ‘U-shape’ mainly with Arg126, Arg110 and Glu77 from TM4 and His72 from TM1 of another subunit [7,8,11,12]. It should be stressed that the mPGES-1 structure obtained by Jegerschöld et al. represents a closed conformation of the protein [7]. A model of the open conformation reveals that prostaglandin endoperoxide (PGH2) could fit into the cleft defined by TM1 and TM4, allowing the synthesis of PGE2 [7]. The homology model published by Xing et al. predicted a 3:3 binding stochiometry of mPGES-1 and its substrate [8]. A co-crystal of mPGES-1 with a small-molecule inhibitor would confirm these predictions and facilitate drug design for this interesting therapeutic target (see later discussion). Of note are also the structural similarities with other crystallized proteins (Figure 1B) such as the Huntingtin interacting protein 12 (PDB: 1R0D), the V-type sodium ATP syn-thase subunit K (PDB: 2BL2), or the protein tyrosine kinase 2 β (β3GM3) (Figure 1B & Table 1). Part of these structural similarities should be taken in consideration perhaps when selective inhibitor design is undertaken.
Figure 1. Microsomal prostaglandin E synthase-1 and structural homologies with other proteins.

(A) View from the top of the trimeric complex. The structure was downloaded from the PDB database (3DWW). GSH is shown in ball and sticks. (B) Structural similarities between mPGES-1 (3DWW, in orange), and MGST-1 (2H8A.A, in cyan), FLAP (2Q7M.F, in cyan), Huntingtin interacting protein 12 or HIP-12 (1R0D.A, in cyan) and the protein tyrosine kinase 2 β or PTK2 (3GM3.A, in cyan).
Table 1.
Sequences and structure similarities with microsomal prostaglandin E synthase-1 (PDB: 3DWW).
| Rank | Chain | Title† | p-value | RMSD‡ | Len1§ | Len2¶ | ID# (%) | COV1†† (%) | COV2‡‡ (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2H8A.A | Microsomal glutathione S-transferase 1 | 3.41E-13 | 1.70 | 142 | 121 | 17 | 82 | 97 |
| 2 | 2Q7M.F | Arachidonate 5-lipoxygenase-activating protein | 2.35E-8 | 3.13 | 142 | 149 | 5 | 89 | 85 |
| 3 | 1R0D.A | Huntingtin-interacting protein 12 | 1.17E-7 | 3.21 | 142 | 194 | 5 | 84 | 61 |
| 4 | 2BL2.J | V-type sodium ATP synthase subunit K | 1.35E-7 | 3.49 | 142 | 158 | 8 | 82 | 74 |
| 5 | 2OKU.B | Acyl-CoA dehydrogenase family protein | 2.85E-7 | 3.21 | 142 | 116 | 5 | 75 | 92 |
| 6 | 3GM3.A | Protein tyrosine kinase 2β | 3.06E-7 | 3.10 | 142 | 133 | 2 | 64 | 68 |
| 7 | 1XG2.B | Pectinesterase inhibitor | 7.32E-7 | 3.23 | 142 | 151 | 5 | 63 | 59 |
| 8 | 1X91.A | Invertase/pectin methylesterase inhibitor family protein | 7.78E-7 | 3.29 | 142 | 149 | 5 | 63 | 60 |
| 9 | 2QYW.A | Vesicle transport through interaction with t-SNAREs 1B homolog | 1.02E-6 | 2.69 | 142 | 96 | 6 | 46 | 69 |
| 10 | 1NFN.A | Apolipoprotein E3 | 1.05E-6 | 2.99 | 142 | 132 | 5 | 62 | 67 |
| 2827 | 1Z9H.A | Membrane-associated prostaglandin E synthase-2 polypeptide (L) | 0.0257 | 8.45 | 142 | 247 | -1 | 82 | 42 |
| 16724 | 1EJF.B | Human co-chaperone P23 (cPGES) | 0.946 | 3.65 | 142 | 110 | -1 | 26 | 34 |
Title for protein chain description.
RMSD value of the alignment.
The length of the chain 1.
The length of the chain 2.
The percentage sequence identity in the alignment.
The coverage, or percentage, of aligned residues in chain 1.
The coverage, or percentage, of aligned residues in chain 2.
Other PGE2 synthases
There are two other prostaglandin E2 synthases that have been identified. We present a short summary of their structure, activity and function below. Throughout the rest of the review, we specifically focus on mPGES-1.
Microsomal PGE synthase-2
The membrane-bound mPGES-2 has a broad substrate specificity, and bears similarity to glutaredoxin and thioredoxin [13]. mPGES-2 is expressed constitutively in a variety of human tissues and, unlike mPGES-1, it is not induced by pro-inflammatory signals. The crystal structure of mPGES-2 was also recently published as a cocrystal with indomethacin (PDB: 1Z9H), an anti-inflammatory drug, at 2.6-Å resolution [14]. mPGES-1 and mPGES-2 only share 43% of homology. GSH is not an essential cofactor for mPGES-2 activity [15], but mPGES-2 activity can be stimulated experimentally by various GSH-reducing agents such as dithiothreitol. mPGES-2 is expressed in several tissues in which mPGES1 expression is relatively low [16]. The enzyme seems to have no real preference for cyclooxygenase 1 (COX-1) or cyclooxygenase 2 (COX-2) but can serve as a terminal PGES for both [17]. mPGES-2 can be cleaved (N-terminal cleavage) to a soluble catalytically competent form [17]. mPGES-2-deficient mice showed no specific phenotype and no alteration in PGE2 levels in several tissues (including liver, kidney, heart and brain) or in lipopolysaccharide (LPS)-stimulated macrophages [18].
Cytosolic PGE synthase
Cytosolic PGES (cPGES) is thought to mediate constitutive PGE2 biosynthesis as it couples preferentially with COX-1 [15]. cPGES is also a GSH-requiring enzyme, constitutively expressed in a variety of cells as a 23-kDa protein and is identical to p23, a heat-shock protein 90 (Hsp90)-binding protein [15]. cPGES was crystallized at 2.49 Å only after cleaving 35 amino acids from the acidic C-terminal part of the protein (PDB: 1EJF) [19]. cPGES can be regulated by casein kinase 2 (CK-II)-dependent phosphorylation, which increases the cPGES activity [20]. Activation of CK-II by upstream signals (serum stimulation) triggers dual phos-phorylation of Ser113 and Ser118 on cPGES and promotes the recruitment of cPGES into the Hsp90 complex, which finally leads to the full activation of cPGES. LPS or interleukin-1 (IL-1) can stimulate the production of PGE2 via the selective activation of COX-1 and cPGES pathway [15,21]. Knockout of cPGES in mice has been shown to be peri-natally lethal [22].
Function of mPGES-1
Microsomal prostaglandin E synthase-1 is the terminal enzyme in the biosynthesis of PGE2 (Figure 2) . In the first step, membrane-bound and secretory phospholipase A2 (PLA2) isoforms convert phospholipids (PL) to arachidonic acid (AA). Next, the COXs convert AA into the unstable intermediate, PGH2. Finally, terminal PGESs isomerize PGH2 into PGE2. PGH2 is the precursor for several structurally related PGs, which are formed by the action of specialized prostaglandin synthases [23]. The PGs synthesized by this pathway include the afore-mentioned PGE2, as well as prostaglandin D2 (PGD2), prostaglandin F2α (PGF2α), prosta-glandin I2 (PGI2, also known as prostacyclin) and thromboxane A2 (TXA2). Consequently, it is thought that inhibition of COX-2 activity affects the synthesis of all prostanoids down-stream of PGH2, whereas selective targeting of mPGES-1 would only reduce PGE2 production. It should be noted that shunting towards other PG has been observed and that dual inhibitors for the 5-LOX and mPGES-1 are considered as a novel excellent avenue to inhibit the pathway. These inhibitors are discussed in the section later.
Figure 2. Prostaglandin E2 synthesis pathway.

The initial step of PGE2 synthesis is the stimulus-induced liberation of AA from the membrane phospholipids by PLA2 enzymes. AA is then sequentially metabolized into PGG2 and then to PGH2 by either COX-1 or COX-2. PGH2 is an unstable intermediate prostanoid, which is rapidly converted into various prostanoids by specific terminal prostaglandin synthases, of which prostaglandin E synthases (mPGES-1, mPGES-2 and cPGES) generate PGE2 from PGH2. The other synthases include PGIS that forms PGI2, PGDS for PGD2, PGFS for PGF2α and TXS for TXA2. PGI2 is non-enzymatically metabolized to the more stable 6-keto PGF1α. PGE2 mediates inflammation, stimulates cell growth and angiogenesis, and also inhibits apoptosis.
†Upregulated in cancers.
AA: Arachidonic acid; PGDS: Prostaglandin D synthase; PGE2: Prostaglandin E2; PGFS: Prostaglandin F synthase; PGIS: Prostaglandin I synthase; PLA2: Phospholipase A2; TXA2: Thromboxane A2; TXS: Thromboxane synthase.
Regulation of mPGES-1 expression & activity
Microsomal prostaglandin E synthase-1 expression is low in most normal tissues, although abundant and constitutive expression is detected in a limited number of organs, such as the lung, kidney and reproductive organs. The induction of COX-2 and mPGES-1 by pro-inflammatory factors and their cooperation in converting AA to PGE2 in vitro [10] suggests that both enzymes are important for PGE2 biosynthesis and that inhibition of either is sufficient to inhibit PGE2 production [24,25]. The kinetics of induction of mPGES-1 and COX-2 has been reported to be different [24,26,27] suggesting a differential regulation of the enzymes. mPGES-1 expression can be specifically induced by LPS, IL-1β and TNF-α in various cell types with or without induction of COX-2 [5,28,29]. The putative promoter of human mPGES-1 gene is GC-rich, lacks a TATA box and contains binding sites for C/EBP and AP-1, two tandem GC boxes, two progesterone receptor and three GRE elements [30]. Of these sites, the GC boxes are critical for the promoter activity where the transcription factor early growth response protein 1 (Egr-1) binds to the proximal GC box and triggers mPGES-1 transcription. Mice genetically deficient in mPGES-1 have shown that the enzyme is a key mediator of inflammation, pain, angiogenesis, fever, bone metabolism and tumorigenesis [25,31–33], thus making this protein an attractive target for the treatment of osteoarthritis, rheumatoid arthritis, acute or chronic pain and cancer, which is the focus of this review.
Role of mPGES-1 in diseases
Role in cancer
Experimental observations developed from cell culture studies, together with the well-recognized role of PGE2 during tumor promotion, have provided the rationale for several recent in vivo studies focused on the impact of mPGES-1 on tumorigenesis. Table 2 summarizes the cancers in which mPGES-1 expression has been shown to be increased compared with normal tissues. mPGES-1 is overexpressed in gastrointestinal (GI) cancers (including esophageal [34], gastric [35–38], colorectal [39,40], liver [41,42] and pancreatic cancers [43]), brain cancers (gliomas and medulloblastomas [44,45]), breast cancer [46], kidney cancer [47], thyroid cancer [48] and several cancers derived from the epithelium (including head and neck [49,50], penis [51], lungs [52–54], larynx [55], cervix [56], endometrium [57] and ovary [58]). In a recent review by Nakanishi et al., we summarized the role of mPGES-1 in colon carcinogenesis, where data appear to be somewhat contradictory in knockout mice [59]. Another recent review by Radmark and Samuelsson has also detailed the various results obtained in knockout models [3]. In brief, deletion of mPGES-1 led to the inhibition of colon tumorigenesis in APCΔ14/+ mice [2]. A significant reduction of the number of polyps/mouse in the colon as well as in the small intestine was observed. It was also shown that mPGES-1 knockout decreased adenomatous polyps in the intestine and colon of APCΔ14/+ mice [2]. Interestingly, there has also been another report showing that genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in the APCmin/+ mice [60]. However, the effects were small and limited to an increase in the number of intestinal but not colon tumors, without an increase in tumor size. Although these differences may well have been due to the choice of animal models and their background, the conclusions from these studies are that mPGES-1 plays a role in colorectal carcinogenesis. Other reports have shown in vitro the importance of mPGES-1 in other cancer types where using siRNA the authors demonstrate a reduction in colony formation, cell migration and a slower growth of xenograft tumors in nude mice [61,62].
Table 2.
Microsomal prostaglandin E synthase-1 overexpression in human cancers.
| Cancer type | Percentage | Detection methods | Ref. |
|---|---|---|---|
| Colorectal cancer | 83% (15/18)† | IHC‡+WB§ | [39] |
| 96% (81/84) | IHC | [40] | |
| Non-small-cell lung cancer | 79% (15/19) | IHC+WB | [52] |
| 66% (61/93) | IHC+WB+RT-PCR¶ | [54] | |
| 70% (55/79) | IHC+WB | [53] | |
| Gastric adenoma | 79% (23/29)# 82% (14/17)†† 100%‡‡ |
IHC | [38] |
| Gastric cancer (Helicobacter pylori related) | 44% (22/50)§§ 66% (55/84)¶¶ |
IHC | [35] |
| Gastric cancer | 33% (15/45) | IHC+WB | [36] |
| 47% (60/129) | IHC+WB | [37] | |
| Head and neck squamous cell carcinoma | 79% (11/14) | IHC+WB | [50] |
| 84% (21/25) | IHC+RT-PCR | [49] | |
| Squamous cell carcinoma of the penis | 100% (16/16) | IHC+WB | [51] |
| Squamous cell carcinoma of the larynx | 92% (23/24) | IHC | [55] |
| Squamous cell carcinoma of the uterine cervix | 46% (7/15) | IHC | [56] |
| Breast cancer | 79% (70/89) | IHC | [46] |
| Ovarian adenocarcinoma | Not specified | IHC+WB | [58] |
| Hepatocelluar carcinoma | Not specified 39% (7/18)## 35% (14/40)††† 0% (0/6)‡‡‡ |
IHC+WB+RT-PCR IHC |
[41] [42] |
| Renomedullary interstitial cell tumors | 86% (12/14) | IHC | [47] |
| Pancreatic cancer | 71% (5/7) | IHC+WB+RT-PCR | [43] |
| Barrett’s esophageal | 100% (123/123) | RT-PCR | [34] |
| Glioma | Not specified | IHC | [44] |
| Medulloblastoma | 100% (39/39) | IHC | [45] |
| Papillary thyroid carcinoma | 95% (19/20) | IHC | [48] |
| Endometrial adenocarcinoma | Not specified | IHC | [57] |
Adenoma plus cancer.
Immunohistochemistry.
Western blotting.
Reverse transcription PCR.
Overall.
In patients with synchronous carcinoma.
In patients with synchronous adenocarcinoma.
In patients responsive to eradication therapy.
In patients resistant to eradication therapy.
Well differentiated.
Moderately differentiated.
Poorly differentiated.
Role in pain
In 2003, the role of mPGES-1 in inflamma-tory and pain response was first studied [25]. The authors generated mPGES-1-deficient trans-genic mice and showed that reduced expression of mPGES-1 leads to decrease in writhing, an indicator of inflammatory pain. Other reports have further concluded that mPGES-1 is indeed involved in various types of inflamma-tion, including pain hyperalgesia, granulation associated with angiogenesis, and inflammatory arthritis accompanying bone destruction [33,63]. However, using the acetic acid stretching test with or without LPS stimulation, Kamei et al. demonstrated that mPGES-1 contributes more profoundly to LPS-primed inflammatory hyper-algesia than to basal acute pain perception [33].
Drug discovery with mPGES-1 as a molecular target
Given the known effects of PGs on cardiovascular function, there has been concern of the potential for cardiotoxicity with any inhibitor of PG biosynthesis as anticancer agents. For example, prostacyclin synthase is expressed in endothelial cells, its product, PGI2, is known for its cardio-protective properties that cause platelet de-aggregation and vessel dilation [23]. The recent history of cardiotoxicity associated with high doses of COX-2 inhibitors clearly illustrates the potential problem. For example, a study in patients taking selective COX-2 inhibitors showed that the increased risk of myocardial infarction (MI) and stroke [64,65], and increased mortality after MI [66] may be due to an imbalance of prothrombotic eicosanoids (increased TXA2) and antithrombotic eico-sanoids (decreased PGI2) [67]. Interestingly, the deletion of mPGES-1 did not have impact on blood pressure when the mice were crossed with low-density lipoprotein receptor (LDLR) knockout mice [63]. Moreover, Wu et al. [68] demonstrated absence or reduced levels of myocardial damage after coronary occlusion in mice lacking mPGES-1 compared to mice given COX-2 inhibitor (celecoxib) [69]. However, in contrast to work with COX-2 inhibitors mice with targeted deletion of the gene encoding mPGES-1 did not show any alteration the levels of TXA2 or PGI2 in the heart after MI [70]. Therefore, phar-macological inhibition of mPGES-1 may not be associated with the perturbations in TXA2 and PGI2 metabolism that increase the risk of arterial thrombosis in patients taking COX-2 inhibitors. Moreover, it was recently reported by Cheng et al. that mPGES-1 deletion, in contrast to deletion, disruption, or inhibition of COX-2, does not result in hypertension or a predisposition to thrombosis in normolipidemic mice [71]. These important findings suggest that selective mPGES-1 inhibitors should have very low, if any, cardiotoxic side effects typically associated with COX-2 inhibitors.
Identification & characterization of mPGES-1 inhibitors
There are several examples of compounds that were identified and developed to target mPGES-1. In this review, the compounds that have been described in the literature are classified into three different categories:
Endogenous lipid, fatty acids and PGH2 analogs (Table 3);
Known anti-inflammatory drugs and/or inhibitors of leukotrienes (LTs) biosynthesis (Table 4);
Natural compounds (Table 5).
Table 3.
Endogenous lipids, fatty acids and prostaglandin endoperoxide analogs.
| Number | Compound | Structure | IC50[μM]† | Ref. |
|---|---|---|---|---|
| 1 | LTC4 |
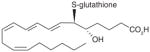
|
5 | [26] |
| 2 | 15-deoxy-Δ12,14-PGJ2 |
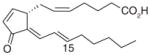
|
0.3 | [73] |
| 3 | U-51605 (PGH2 stable analog) |
|
<10 >100 |
[73] [26] |
| 4 | U-44069 (PGH2 stable analog) |
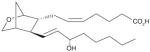
|
NI | [26] |
| 5 | U-46619 (PGH2 stable analog) |
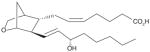
|
NI | [26] |
| 6 | Arachidonic acid |
|
0.3 | [73] |
| 7 | Docosahexaenoic acid |
|
0.3 | [73] |
| 8 | Eicosapentaenoic acid |
|
0.3 | [73] |
| 9 | Palmitic acid |
|
2 | [73] |
Determined by cell-free mPGES-1 activity assays.
NI: No significant inhibition; PGH2: Prostaglandin endoperoxide.
Table 4.
Known anti-inflammatory drugs and/or inhibitors of leukotrienes biosynthesis.
| Number | Compound | Structure | IC50 (μM)† | Ref. |
|---|---|---|---|---|
| 10 | Sulindac sulfide |
|
80 | [26] |
| 11 | NS-398 |
|
20 | [26] |
| 12 | Celecoxib |
|
22 | [74] |
| 13 | Dimethylcelecoxib |
|
16 | [74] |
| 14 | MK-886 |
|
1.6 | [76] |
| 15 | Licofelone (ML3000) |
|
6 | [80] |
Determined by cell-free mPGES-1 activity assays.
Table 5.
Natural compounds.
| Number | Compound | Structure | Cell-free assay
|
Effects in human blood cells or A549 cells
|
Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 against mPGES-1 | COX-1 | COX-2 | PGE2 | Other COX-1- derived prostanoids‡ | Other COX-2- derived prostanoids§ | ||||
| 16 | Curcumin |
|
0.3 | NI† | NI | Decrease in LPS-stimulated HWB‡(EC50= 15 μM) | Decrease in 12-HHT in unchallenged HWB (EC50= 19 μM) | Decrease in β6-keto in LPS-stimulated PGF1α HWB only at higher dose (30 μM) | [82] |
|
| |||||||||
| 17 | EGCG |
|
1.8 | IC50> 30 μM |
NI | Decrease in LPS-stimulated HWB (EC50> 30 μM) | ND | No effect on 6-keto and 12-HHT in PGF1α LPS-stimulated HWB (at 30 μM) | [83] |
|
| |||||||||
| 18 | Garcinol |
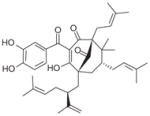
|
0.3 | IC50= 12 μM | NI | Decrease in IL-1β-stimulated A549 cells (EC50 ~10 μM) Decrease in LPS- stimulated HWB (EC50= 30 μM) |
Decrease in 12-HHT (EC50= 11 μM) and TXB2 (EC50 = 16 μM) in unchallenged human platelets No effect on 12-HHT in unchallenged HWB (up to 33 μM) |
No effect on 6-keto PGF1αin IL-1β-stimulated A549 cells (up to 33 μM) No effect on 6-keto PGF1αin LPS- stimulated HWB (up to 30 μM) | [84] |
|
| |||||||||
| 19 | Myrtu-commulone |
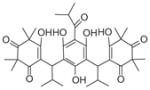
|
1 | IC50> 15 μM | NI | Decrease in IL-1β-stimulated A549 cells (EC50= 30 μM) Decrease in LPS-stimulated HWB |
No effect on 12-HHT in unchallenged HWB (up to 33 μM) | No effect on 6-oxo PGF1αin IL-1β-stimulated A549 cells (up to 33 μM) | [85] |
|
| |||||||||
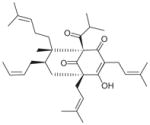
| 20 | Arzanol |
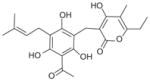
|
0.4 | IC50= 17.5 μM | NI | Decrease in LPS-stimulated human monocyte (EC50= 9 μM) Decrease in LPS-stimulated HWB (EC50 ~30 μM) |
Decrease in 12-HHT (EC50= 2.3 μM) and TBA2 (EC50= 2.9 μM) in unchallenged human platelets | Decrease in β6-keto PGF1αin IL-1β-stimulated A549 cells moderately (EC50≥ 30 μM) No effect on TXB2 and 6-keto PGF1αin LPS- stimulated HWB (at 30 μM) | [86] |
|
| |||||||||
| 21 | Boswellic acids | (AKBA)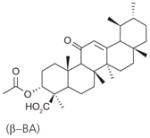
|
3 | ND | ND | Decrease in IL-1β-stimulated A549 cells (EC50 = 20–30 μM) Only β-BA decreases LPS- stimulated HWB (EC50= 10 μM) | β-BA: No effect on 12-HHT in unchallenged HWB (at 50 μM) | No effect on 6-keto PGF1αin IL-1β-stimulated A549 cells (at 30 μM) No effect on 6-keto PGF1α and TXB2in LPS- stimulated HWB (at 10 μM) | [87] |
| 22 | (β–BA)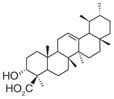
|
5 | ND | ND | |||||
| 23 | (KBA)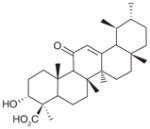
|
10 | ND | ND | |||||
|
| |||||||||
| 24 | Hyperforin |
|
1 | IC50= 12 μM | NI | Decrease in LPS- stimulated HWB (EC50 ~3 μM) Decrease in AA- stimulated HWB (EC50= 0.25 μM) | No effect on 12-HHT in unchallenged HWB (up to 33 μM) | No effect on 6-keto PGF1α and TXB2in LPS- stimulated HWB (up to 30 μM) No effect on 6-keto PGF1αin AA-stimulated HWB (up to 30 μM) | [88] |
NI: No significant inhibition up to 30 or 33 μM.
COX-1-derived prostanoids are measured in HWB or COX-1 expressing human platelets stimulated with Ca2+-inophore plus AA, but not challenged by LPS.
COX-2-derived prostanoids are measured in LPS-stimulated HWB or IL-1b-stimulated A549 cells (low COX-1 and induced COX-2).
AKBA: 3-O-acetyl-11-keto-β-boswellic acid; β-BA: β-boswellic acid; HWB: Human whole-blood; KBA: 11-keto-β-boswellic acid; ND: Not determined.
Compounds that were further improved based on their structure and cellular activities are also described in the next section (Tables 6 & 7).
Table 6.
Synthetic compounds.
| Scaffold (early leads) | Selected lead compound(s) | IC50 (μM)† | EC50 (μM)‡ | HWB | In vivo activity | Ref. |
|---|---|---|---|---|---|---|
| MK-886 (indole) |
25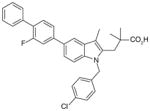
|
0.007 | 0.49 8.0¶ |
NI§ | ND | [76] |
26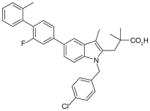
|
0.003 | 0.27 5.8¶ |
NI | ND | ||
|
| ||||||
| Phenanthrene imidazoles |
27 (MF63) 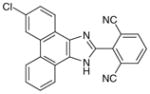
|
0.001 | 0.42¶ | 1.3 | Analgesic/antipyretic in guinea pigs and mPGES-1 KI mice (ED50= 100 mg/kg in guinea pig hyperalgesia model) | [92,94] |
28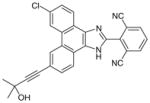
|
0.001 | 0.02¶ | 0.2 | Analgesic in guinea pigs (ED50= 30 mg/kg) | [95] | |
|
29 (MK-7285) 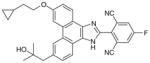
|
0.0009 | 0.01¶ | 0.14 | Analgesic in guinea pigs (ED50= 14 mg/kg) | [95] | |
|
| ||||||
| Biaryl imidazole |
30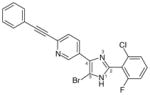
|
0.001 | 0.013 0.160¶ |
1.6 | ND | [97] |
|
| ||||||
31 (pirinixic acid)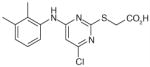
|
32 (YS121)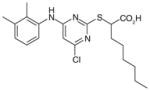
|
3.4 | 12 | 2 | Anti-inflammatory in carrageenan-induced rat pleurisy model | [99,101] |
33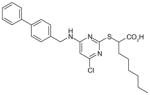
|
1.3 | 6 | ND | ND | [99] | |
34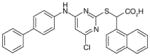
|
0.94 | ND | ND | ND | [100] | |
35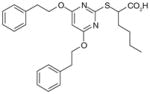
|
1.2 | ND | ND | ND | [102] | |
36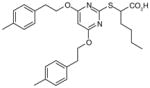
|
0.9 | ND | ND | ND | ||
|
| ||||||
| Trisubstituted benzene |
37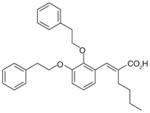
|
1.1 | ND | ND | ND | [102] |
|
| ||||||
| 2-mercaptohexanoic acids |
38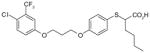
|
1.7 | ND | ND | ND | [103] |
39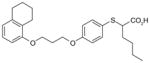
|
2.2 | ND | ND | ND | ||
40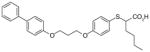
|
2.2 | ND | ND | ND | [103] | |
|
| ||||||
| Licofelone (arylpyrrolizines) |
41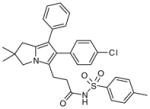
|
2.1 | ND | ND | ND | [104] |
|
| ||||||
| Benzo[g]indol-3-carboxylates |
42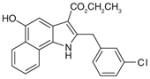
|
0.6 | 2 | ND | Anti-inflammatory in carrageenan- induced mouse paw edema and rat pleurisy models | [106] |
|
| ||||||
Oxicam 43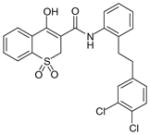
|
45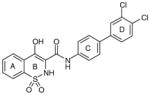
|
0.016 | 0.42 | 5 | ND | [107] |
| ↓ 44 (IC50 = 0.11 μM; EC50 = 0.46 μM) 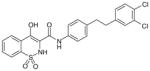
|
(PF-9184) | |||||
|
| ||||||
46 (trisubstituted ureas; 88% enzyme inhibition at 10 μM)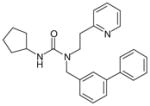
|
47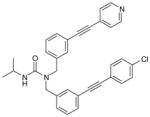
|
0.002 | 0.34¶ | 2.1 | ND | [110] |
|
| ||||||
| Carbazole benzamides |
48 (AF3442)
|
0.06 | 0.41 (LPS-stimulated monocytes) | 29 | ND | [111] |
IDetermined by cell-free assays measuring the conversion of prostaglandin E2 to prostaglandin E2 in vitro.
Determined by cell-based assays measuring the production of prostaglandin E2 by cells.
NI: No significant inhibition up to 30 or 33 μM.
In the presence of 50% FBS.
AA: Arachidonic acid; HWB: Human whole-blood; LPS: Lipopolysaccharide; ND: Not determined.
Table 7.
Other scaffolds identified by computational approaches.
| Number | Structure | IC50 (μM)† | Ref. |
|---|---|---|---|
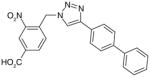
| 49 |
|
0.5 | [112] |
| 50 |
|
3.2 | [113] |
| 51 |
|
2.3 | [114] |
| 52 |
|
2.8 | [114] |
| 53 |
|
7.9 | [114] |
| 54 |
|
2.6 | [114] |
| 55 |
|
7.7 | [114] |
| 56 |
|
3.0 | [114] |
| 57 |
|
0.4 | [114] |
| 58 |
|
3.7 | [114] |
| 59 |
|
0.5 | [114] |
Determined by cell-free mPGES-1 activity assays.
Endogenous lipids, fatty acids & PGH2 analogs
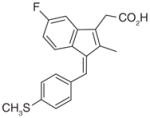
It has been reported that mPGES-1 is weakly inhibited (IC50 = 5 μM) by cysteinyl leukotriene C4 (LTC4, (1), Table 3) [26], which also inhibits the structurally related MGST-1 with higher potency (IC50 = 50 nM) [72]. With a GSH moiety, LTC4 has been shown to inhibit MGST-1 by competing with GSH [72]. Because of the structural homology between the members of MAPEG family of enzymes, inhibition of mPGES-1 activity by LTC4 may be due to a similar mechanism. Other lipid mediators such as PGs have also been tested for mPGES-1 inhibition. The anti-inflammatory 15-deoxy-Δ 12,14-PGJ2 (2) is found to be the most potent inhibitor of mPGES-1 (IC50 = 0.3 μM) compared with PGE2, PGF2α, TXB2 and PGJ2[73]. The fact that 15-deoxy-Δ12,14-PGJ2 is much more potent than its analogs PGJ2 or Δ12-PGJ2 (IC50 > 50 μM) suggests that the hydroxyl group at C15 position impairs mPGES-1 inhibition. Beside naturally occurring PGs, stable PGH2 analogs have also been tested as potential mPGES-1 inhibitors, among which U-51605 (3) inhibits mPGES-1 activity to some extent. However, the potency is inconsistent between the studies [26,73]. Unlike U-51605, two other stable PGH2 analogs U-44069 (4) and U-46619 (5) fail to inhibit mPGES-1 [26,73]. The activity of mPGES-1 is also inhibited by a number of fatty acids such as AA (6), docosahexaenoic acid (DHA, 7), eicosapentaenoic acid (EPA, 8) (IC50 = 0.3 μM for each), and palmitic acid (9) (IC50 = 2 μM) [73]. These results suggest that the anti-inflammatory properties of 15-deoxy-Δ12,14-PGJ2, DHA and EPA can be partly attributed to mPGES-1 inhibition.
Known anti-inflammatory drugs &/or inhibitors of LTs biosynthesis
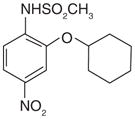
The only traditional nonsteroidal anti-inflammatory drug (NSAID) that exhibits inhibitory effect for mPGES-1 is sulindac. Its active metabolite sulindac sulfide (10; Table 4) has been shown to weakly inhibit mPGES-1 activity (IC50 = 80 μM) [26]. There are several examples of selective COX-2 inhibitors that also found to inhibit mPGES-1 activity. For instance, NS-398 (11) is a COX-2 inhibitor that also inhibits mPGES-1 with an IC50 value of 20 μM [26]. Similarly, some other coxibs such as celecoxib (12) (IC50 = 22 μM), lumiracoxib (IC50 = 33 μM), and valdecoxib (IC50 = 75 μM) also moderately inhibit mPGES-1 activity, whereas the other tested coxibs (etoricoxib and rofecoxib) fail to inhibit mPGES-1 activity even when used up to 200 μM [74]. Interestingly, the celecoxib derivative dimethylcelecoxib (DMC, 13) loses the COX-2 inhibitory effect, while obtaining slightly better potency for mPGES-1 inhibition (IC50 = 16 μM) as measured in a cell-free assay [74].
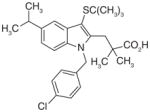
MK-886 (14), an LT suppressor acting through inhibition of FLAP (IC50 = 26 nM), is also found to inhibit mPGES-1 in vitro (IC50 = 1.6 μM) [75,76]. This result reinforces the similarity among the members of MAPEG (mPGES-1 versus FLAP, see Table 1 and Figure 1B). In intact cells, however, MK-886 has limited inhibitory effects on PGE2. At 100 μM, MK-886 only slightly reduces (~20%) LPS-induced PGE2 in human whole-blood, and does not show further inhibition with higher concentration [77]. In cytokine-stimulated gin-gival fibroblasts, MK-886 does not significantly reduce PGE2 synthesis at 2–4 μM, although the protein level of mPGES-1 is slightly reduced. When used at higher concentration (8 μM), it even increases PGE2 production in these gingival fibroblasts, with a concomitant upregulation of COX-2 protein [78]. In Caco-2 and HT-29 colon cancer cells, 10 μM of MK-886 significantly increases PGE2 production, which may be due to a shunt of AA metabolism to the PG pathway, since MK-886 is an inhibitor targeting the 5-LOX pathway [79]. Taken together, the lack of inhibitory effect of MK-886 on cellular PGE2 synthesis suggests that this compound is unlikely to serve as an mPGES-1 inhibitor in vivo to reduce PGE2 production. Nevertheless, MK-886 has been used as a basis for the development of more potent and selective mPGES-1 inhibitors (see later discussion).
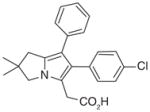
Another anti-inflammatory drug licofelone (ML3000, 15; Table 4), originally identified as a dual inhibitor blocking both COX and 5-LOX pathways, has also been shown to inhibit mPGES-1 activity with an IC50 value of 6 μM [80]. It dose-dependently reduces PGE2 production (EC50= 0.1 μM) in IL-1β-stimulated A549 cells, a system where COX-1 is undetectable [26], without affecting the generation of PGI2 (as detected by its stable metabolite 6-keto PGF1α using an ELISA assay) [80]. However, the in vivo effect of licofelone on PGE2 reduction is also contributed by COX-1 inhibition, because licofelone is a potent COX-1 inhibitor as tested in vitro (IC50 = 0.8 μM) and in intact human platelets (EC50 = 0.24 μM for 12-hydroxy-5,8,10-heptadecatrienoic acid (12-HHT) reduction [80]. Interestingly, it has been shown by flexible alignment that licofelone shares pharmacophore features with MK-886 [81]. In line with this observation, it acts primarily on FLAP rather than 5-LOX itself [81]. Licofelone is currently evaluated as a treatment for osteoarthritis, as it can suppress both PGE2 and LTs biosynthesis, which offers benefits over traditional NSAIDs and selective COX-2 inhibitors. In fact, licofelone derivatives have also been developed as selective mPGES-1 inhibitors by further structure–activity relationship (SAR) studies (see later discussion).
Natural compounds
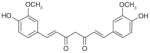
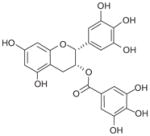
Recently, the Werz group based in Germany published a series of reports on natural anti-inflammatory compounds as novel mPGES-1 inhibitors, providing a novel mechanism by which these compounds act [82–88]. As summarized in Table 5, these include curcumin (16) from turmeric (IC50 = 0.3 μM) [82], epi-gallocatechin gallate (17) from green tea (IC50 = 1.8 μM) [83], garcinol (18) from the fruit rind of Guttiferae species (IC50 = 0.3 μM) [84], myrtucommulone (MC, 19 ) from myrtle (IC50 = 1 μM) [85], arzanol (20) from Helichrysum italicum (IC50 = 0.4 μM) [86], boswellic acids (21–23) from frankincense (IC50 = 3–10 μM) [87], and the acylphloroglucinol hyperforin (24) from St. John’s wort (IC50 = 1 μM) [88]. These natural compounds inhibit mPGES-1 without affecting COX-2 activity in vitro, although some of them inhibit COX-1 to some extent (Table 5). In LPS-stimulated human whole-blood or IL-1β-stimulated A549 cells, the compounds reduce PGE2 production after a short period of exposure (with EC50 values in the range of 10–30 μM), but do not significantly alter other prostanoids that are derived mainly from COX-2 under these experimental conditions. However, it is not clear whether mPGES-1 inhibition is a predominant mechanism of PGE2 reduction in vivo for some of these compounds, because the effects are usually multi-faceted with longer exposure (≥24 h). For example, curcumin has been shown to down-regulate the expression of COX-2. In fact, transcriptional regulation of COX-2 has been considered as a major mechanism of curcumin [89]. In addition, hyperforin significantly and dose-dependently inhibits the expression of both COX-2 and mPGES-1 proteins in IL-1 β-stimulated A549 cells after 24 h incubation [88]. Therefore, the suppression of PGE2 formation by these natural compounds may be due to the inhibition of COX-2 or mPGES-1 expression during prolonged exposure times. Interestingly, all of these compounds have also been shown to inhibit 5-LOX activity in vitro.
Further development of mPGES-1 inhibitors
MK-886 derivatives (indole carboxylic acids)
As previously noted, a series of molecules based on the indole FLAP inhibitor, MK-866, has been developed as selective mPGES-1 inhibitors [76]. The two most potent compounds are shown in Table 6 (25 & 26, IC50 = 7 and 3 nM, respectively). They show mPGES-1 selectivity compared with their ability to inhibit mPGES-2 or TXA2 syn-thase in vitro, and no apparent FLAP binding is observed as measured using a competitive ligand-binding assay [90]. However, there is a shift in potency when these compounds are tested in cell-based assays in the presence of fetal bovine serum (FBS). This may be caused by a high degree of plasma protein binding, which makes these compounds ineffective in reducing PGE2 in LPS-stimulated human whole-blood and preclude them from in vivo testing.
Phenanthrene imidazoles
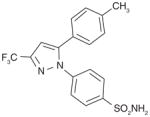
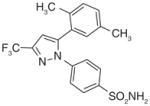
From an high-throughput screening (HTS) campaign using a mPGES-1 cell-free assay [91], the JAK inhibitor azaphenanthrenone was discovered at Merck Frosst as a hit (IC50 value of 0.14 μM). In further SAR studies and lead optimization, the phenanthrene imidazole MF63 (27) was then identified as a potent selective mPGES-1 inhibitor by removal of the pyridine moiety essential for JAK inhibition and by ortho-di-substitution of the imidazole-bonded 2-phenyl moiety [92]. MF63 potently inhibits the human mPGES-1 enzyme (IC50 value of 1.3 nM), with a high degree (>1000-fold) of selectivity over human mPGES-2 and thromboxane synthase (TXS). Unlike the indole carboxylic acids described earlier, this compound remains potent and selective in cell-based assays under high plasma protein conditions. In the presence of 50% FBS, MF63 inhibits PGE2 production with an EC50 value of 0.42 μM in A549 lung cancer cells. Further, it inhibits PGE2 production when tested in LPS-stimulated human whole-blood (EC50 = 1.3 μM) without concomitant inhibition of TXB2 (EC50 > 40 μM). However, MF-63 does not inhibit the rat ortholog of mPGES-1 in the cell-free assay (IC50 > 40 μM), and this discrepancy is also observed for mice [92]. According to a recently proposed model, the species selectivity may be due to the substitutions of amino acid residues Thr131, Leu135 and Ala138 (human amino acid sequence) with bulky aromatic residues in rat mPGES-1, which occlude the entrance to the active site and therefore prevent some inhibitors from binding [93]. Interestingly, rat and mouse mPGES-1 are the only rodent orthologs found to have this feature. Therefore, the anti-inflammatory property of MF63 has been tested in guinea pigs (the IC50 value against guinea pig mPGES-1 is 0.9 nM [92]) as well as knockin mice expressing human mPGES-1, instead of the well-established rat models for inflammatory pain. In these preclinical animal models, MF63 administered orally suppresses PGE2 synthesis and showed efficient analgesic and antipyretic effects, without causing GI toxicity and PGI2 reduction, which are typically associated with NSAIDs [94]. Furthermore, MF63 demonstrates desirable pharmacokinetic properties in guinea pigs, where the concentrations are 3.0, 4.1, and 3.2 μM in the plasma at 1, 2, and 6 h, respectively, and 20 μM in the brain at 6 h after dosing (30 mg/kg orally) [94]. However, it has a short half-life in rats and rhesus monkeys (1.5 and 1.3 h, respectively) when intravenously administered [95]. Thus, further SAR studies aiming for favorable pharmacokinetic profiles while improving the potency of mPGES-1 inhibition were conducted. As shown in Table 6, two other phenanthrene imidazoles (28 & 29) have been identified as selective mPGES-1 inhibitors. Both compounds are equipotent to MF63 against human mPGES-1 in vitro, yet superior to MF63 when tested in human whole-blood, A549 cells with 50% FBS, and in vivo [95]. Compound 28, which has a tertiary alcohol substituted alkyne at the 9’ position of the phenanthrene backbone, shows greatest whole cell and whole-blood activity as compared with other tested 9’-substituted chlo-rophenanthrene imidazoles. The compound 28 also demonstrates analgesic activity in the LPS-induced hyperalgesia guinea pig model (ED50 = 30 mg/kg) when administered orally. According to pharmacokinetic studies in rat, 28 has a slow absorption rate (Cmax at 6 h) and very long half-life (20 h) after single intravenous dosing at 5 mg/kg. Consistently, the compound is barely metabolized when incubated with rat or human hepatocytes for 2 h at 37°C (only 3% metabolism in both cases). In order to avoid an excessive long half-life and low metabolism in human, another phenanthrene imidazole (compound 29, also known as MK-7285 [96]) has been developed by additional SAR studies. Overall, MK-7285 shows excellent activity in cell-free and cell-based assays without concomitant reduction of other prostanoids. Importantly, it has an appropriate rat half-life (2.3 h), higher degree of metabolism in rat (32%) and human (19%) hepatocytes, faster absorption rate (Cmax at 1 h), and good bioavailability (68%). Finally, MK-7285 demonstrates greater in vivo efficacy than MF63 and compound 28 in the LPS-induced hyperalgesia guinea pig model (ED50 = 14 mg/kg) [95].
Biarylimidazoles
Biarylimidazole, a scaffold that resides in the structure of MF63, has been described in a series of SAR studies looking for more potent mPGES-1 inhibitors [97]. Four segments in the biarylimidazole backbone were explored: the 2-imidazole group, the imidazole core, the 4- imidazole group, and the 5-imidazole group. The SAR analysis led to the identification of 30, which is highly potent in the mPGES-1 enzymatic cell-free assay (IC50 = 1 nM) as well as in intact cell assay using A549 cells with EC50 values of 13 nM and 0.16 μM in the presence of 2% and 50% FBS, respectively. In addition, compound 30 inhibits LPS-induced PGE2 formation in human whole-blood with an EC50 value of 1.6 μM, which approaches the EC50 of rofecoxib (EC50 = 0.53 μM). Furthermore, 30 exhibits great bioavailability (127%) and an adequate half-life (4.8 h) in rats, indicating a good pharmacological profile [97]. However, the in vivo efficacy of this compound remains to be determined or has not yet been reported.
Pirinixic acid derivatives
A class of dual mPGES-1/5-LOX inhibitors derived from the structure of pirinixic acid (31) has been described recently by the Werz group. Although pirinixic acid itself does not show inhibitory effects on 5-LOX or mPGES-1, bulky lipophilic in a α-substitution (n-hexyl, n-octyl, or naphthyl, regardless of the absolute configuration) of the carboxylic acid group results in a reduction of 5-LOX products in peripheral blood mononuclear leukocytes (PMNL) [98], as well as mPGES-1 inhibition as measured in a cell-free assay [99]. As exemplified in Table 6, the α-(n-hexyl)-substituted compound 32 (also named as YS121) inhibits mPGES-1 (IC50 = 3.4 μM) and 5-LOX (IC50 = 6.5 μM) in the corresponding cell-free assays. Overall, esterification of the carboxylic acid group is detrimental to mPGES-1 inhibition but favorable to 5-LOX inhibition. However, bulky lipophilic substituent (e.g., biphenyl-4-methane amine moiety) at C6 of the pyrimidine ring circumvents the preference of esterification for 5-LOX inhibition, while also improving the efficacy of mPGES-1 inhibition. This led to the discovery of 33, which is the most potent mPGES-1 inhibitor within this series with an IC50 value of 1.3 and 2.0 μM in cell-free assays for mPGES-1 and 5-LOX, respectively [98]. Although lipophilic acids could potentially inhibit COX-1/2 due to AA mimicry, these two compounds and most of the other pirinixic acid derivatives exhibit only moderate inhibitory effects (IC50 > 10 μM) on COX-1 or COX-2 in vitro. Moreover, in IL-1β-stimulated A549 cells, YS121 and 33 dose- dependently reduce PGE2 production with EC50 = 12 and 6 μM, respectively [99]. Recently, further SAR studies based on α-naphthyl substituted pirinixic acid were conducted, yielding the identification of compounds with higher potency for both mPGES-1 and 5-LOX by replacing the 2,3-dimethylphenyl group with larger aromatic substituents. For example, compound 34 is a dual mPGES-1/5-LOX inhibitor with an IC50 value of 0.94 μM (mPGES-1) and 2.3 μM (5-LOX) [100]. However, the efficacy of PGE2 reduction in cells was not determined.
Compound YS121 (32; Table 6) has been further characterized and has been tested in an animal model of inflammation [101]. In human whole-blood, YS121 reduces PGE2 formation in a dose-dependent manner (EC50 = 2 μM) without affecting the levels of other prostanoids (up to 30 μM). In a carrageenan-induced rat pleurisy model, YS121 (1.5 mg/kg intraperitoneal, 30 min before carrageenan) was shown to remarkably inhibit exudate formation and leukocyte infiltration 4 h after carrageenan injection to the pleural cavity. In addition, the pleural levels of PGE2 and LTB4 were significantly reduced (36% and 48% inhibition, respectively). However, YS121 also suppressed the generation of 6-keto PGF1α (45% reduction) in the exudates, which might be due to its effect on COX-2 expression as observed in IL-1β-stimulated A549 cells (treated with 10 μM of YS212). The underlying mechanism could be peroxisome proliferator-activated receptors (PPAR)-α/γ agonism shown to downregulate COX-2 expression, because YS121 and other α-alkyl-substituted pirinixic acid derivatives are found to be dual agonists of PPAR-α and -γ. Furthermore, YS121 is barely active in murine RAW264.7 cells, indicating the species selectivity also seen in other mPGES-1 inhibitors. Therefore, the contribution of mPGES-1 inhibition to the in vivo efficacy is not clear.
Based on the structure of a compound within the α-substituted pirinixic acid series (35) (IC50 = 1.2 μM), another comprehensive SAR study has very recently been conducted with the aim of identifying novel and more simplified scaffolds of dual mPGES-1/5-LOX inhibitors [102]. Modifications of the two phenethoxy substituents at the pyrimidine core of 35 reveal that elongation of ethylene spacers or introduction of para-methyl substituents (36) improves the potency of mPGES-1 inhibition, whereas replacement of phenyl group by various aliphatic moieties reduces the potency. Among these novel derivatives of 35, 36 shows great and well-balanced activity against mPGES-1 (IC50 = 0.9 μM) and 5-LOX (IC50 = 3.1 μM in cell-free assay, and EC50 = 0.5 μM in PMNL). Notably, good activities are retained when the pyrimidine core including the thioether of 36 is replaced by a benzyl or benzylidene moiety, leading to the identification of a novel and simpler structural scaffold (trisubstituted benzene, e.g., 37). In this class of compounds, 37 is the most potent lead with an optimized pattern of phenethoxy substitutions on the central benzene ring. It inhibits mPGES-1 in vitro with an IC50 value of 1.1 μM. On the other hand, the 5-LOX inhibition of 37 is much more pronounced in the cell-based assay (EC50 = 0.8 μM in A549 cells) than in the cell-free assay (remaining 5-LOX activity at 10 μM is 91.3±10.5%), implying a non-direct interference of 5-LOX activity. Importantly, none of the identified lead compounds inhibits more than 50% of COX-1 or COX-2 activity at 10 μM [102].
2-mercaptohexanoic acids
Like the α-substituted pirinixic acid derivatives, 2-mercaptohexanoic acid derivatives were originally identified as activators of PPAR-α and -γ and later shown to be dual inhibitors of mPGES-1 and 5-LOX [103]. Compounds 38–40 are the most potent representatives of this series with mPGES-1 IC50 values of 1.7, 2.2 and 2.2 μM, respectively. They also efficiently inhibit 5-LOX both in cell-free and cell-based systems with IC50 and EC50 values in the low micromolar range. From the structural point of view, the 2-mercaptohexanoic acids with elongated lipophilic aryloxy moieties might target other enzymes within the AA metabolic cascade by mimicking the structure of AA (6). However, none of these compounds significantly inhibit isolated COX-1 or COX-2. In addition, at least for 40, the activity of isolated cytosolic PLA2 is not affected at 10 μM. Finally, 40 does not suppress the production of 12(S)-hydroperoxy-5,8-cis-10-trans-14-cis-eicosatetraenoic acid (12(S)-HpETE)) and 15(S)-HpETE by 12-LOX and 15-LOX in human PMNL up to 10 μM.
Licofelone derivatives (arylpyrrolizines)
Structure–activity relationship studies have been performed by using licofelone as a lead structure, with the aim of discovering more selective and potent dual mPGES-1/5-LOX inhibitors. As a result, introduction of a substituted sulfonamide group at the free acid function is found to improve the potency toward mPGES-1 in vitro, while retain the sub-micromolar 5-LOX inhibitory potency determined by cellular LTB4 formation in PMNL. One example of this series is compound 41 with an IC50 value of 2.1 μM in the cell-free mPGES-1 assay, which is 3.2-fold more potent than the lead compound licofelone. Of note, is that both COX-1 and COX-2 activities are less inhibited by 41 than by licofelone, indicating a better specificity. To improve chemical stability, the acetic acid moiety at the pyrrolizin C-5 position is extended by one CH2 unit to generate propionic analogs, which are shown to be more stable than the corresponding acetic derivatives. In addition, elongation of the alkyl chain length seems to diminish COX inhibition effects to some extent. Although these compounds are predicted to be active in cell-based assays on the basis of previous data for licofelone, such information is not available [104]. The compounds were not yet reported to have in vivo activity.
Benzo[g]indol-3-carboxylates
In efforts to develop dual mPGES-1/5-LOX inhibitors, indole-3-carboxylates were screened for their ability to inhibit mPGES-1, as they have previously been shown to potently suppress LTs production by inhibiting 5-LOX [105]. Structural optimization led to the identification of benzo[g] indol-3-carboxylates as mPGES-1 inhibitors exemplified by 42 (Table 6). Compound 42 exhibits an IC50 value of 0.6 μM in the cell-free mPGES-1 assay, and inhibits PGE2 formation in intact A549 cells with EC50 = 2 μM. In spite of showing marked inhibitory effects on isolated COX-1/2, 42 does not cause a significant reduction of COX-2-derived 6-keto PGF1α in A549 cells and only moderately (~43%) inhibits COX-1-derived 12-HHT in human platelets at higher concentration (≥10 μM). To assess the anti-inflammatory efficacy and the effect on PGE2 formation in vivo, 42 has been tested in carrageenan-induced paw edema in mice and carrageenan-induced pleurisy in rats. In the mouse paw edema model, intraperitoneal administration of 42 (4 mg/kg, 30 min prior to carrageen) reduced paw swelling by 61% compared with the vehicle control. In addition, 42 (4 mg/kg, intraperitoneal) suppressed inflammatory responses associated with the rat pleurisy model. Moreover, 42 reduced pleural levels of both PGE2 and LTB4, but only slightly and non-significantly lowered the 6-keto PGF1α level, which was efficiently suppressed by indomethacin. These results suggest that the in vivo anti-inflammatory effects could be attributed to the reduction of both PGE2 and LTB4 levels by dually inhibiting mPGES-1 and 5-LOX [106].
Oxicams
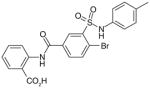
By high-throughput screening of compounds against human recombinant mPGES-1, Pfizer identified a series of benzo-thiopyran S-dioxides (e.g., 43; Table 6), which showed moderate mPGES-1 inhibition while exhibited selectivity for mPGES-1 over COX-2. Subsequently, the benzo-thiopyran group was replaced by dioxo-benzo-thiazinone (oxicam type, 44) and further SAR was explored, resulting in the identification of 45, which was later named as PF-9184 [107,108]. Briefly, the length and/or the nature of the linker between the C ring and D ring is not an important determinant of mPGES-1 inhibition activity, whereas the nature and positions of the substituents on the D ring value are critical for potency. PF-9184 with 3,4-dichloro substitution on the D ring inhibits human mPGES-1 in vitro with an IC50 of 0.016 μM. In addition, it suppresses PGE2 production in IL-1β =-stimulated fetal fibroblast cells (EC50 0.42 μM) and in LPS-stimulated human whole-blood (EC50 = 5 μM). Because oxicams (originally developed as NSAIDs) are COX inhibitors [109], it is necessary to evaluate the inhibitory effect of these oxicam analogs on COX inhibition. In fact, PF-9184 shows > 238-fold selectivity for mPGES-1 over COX-2, which is determined by the ratio of EC50 for reducing PGE2 and PGF2α (a stable PGH2 metabolite) in the IL-1 β-stimulated fetal fibroblast cell assay. This is also confirmed by cell-free COX inhibition assays (hCOX-1 IC50 = 118 μM, hCOX-2 IC50 = 263 μM) [107]. When further characterized in inflammation-relevant cell systems, PF-9184 reduces PGE2 while sparing other prostanoids within a short time frame (<1 h). However, longer exposures (> 16 h) result in unexpected inhibition of 6-keto PGF1α synthesis in IL-1β stimulated synovial fibroblasts derived from patients with rheumatoid arthritis, though only at higher doses. This is presumably due to the feedback regulation of mPGES-1 or COX-2 expression. Like other mPGES-1 inhibitors, PF-9184 shows low potency against rat mPGES-1 and fails to inhibit mPGES-1 in the rat air pouch inflammatory model where mPGES-1 and COX-2 are induced by carrageenan injection [108]. Finally, a major problem of PF-9184 is the poor aqueous solubility (<3 μM in water). Unfortunately, any effort to improve the solubility by manipulating the chemical structures led to a reduction in potency [107].
Trisubstituted ureas
From another high-throughput screening campaign against recombinant human mPGES-1 enzyme, a trisubstituted urea (46) was recently identified as a moderate mPGES-1 inhibitor by Merck Frosst. Subsequently, Subsequently, potency optimization was conducted by exploring SAR on all three substituents based on the urea core, resulting in the discovery of potent and selective mPGES-1 inhibitors [110]. As exemplified by 47, the bis-tolane inhibitors show great potency as measured in cell-free enzymatic assay and selectivity as evaluated by TXB2 inhibition in human whole-blood using an ELISA-based assay. More specifically, the upper tolane has a terminal pyridyl substituent and the lower tolane contains an electron-withdrawing group on the terminal phenyl group, as shown by 47. Compound 47 potently inhibits recombinant human mPGES-1 enzyme (IC50= 2 nM), and reduces PGE2 production in A549 cells in the presence of 50% FBS (EC50 = 0.34 μM). Importantly, it suppresses PGE2 in human whole-blood assay (EC50 = 2.1 μM) without reducing TXB2 (EC50 > 40 μM). Further pharmacokinetic studies need to be carried and in vivo efficacy has to be yet evaluated in other animal models.
Carbazole benzamides
Among a series of carbazole benzamides synthesized and characterized by Bruno et al., 48 (AF3442; Table 6) was identified as a potent inhibitor against recombinant mPGES-1 with an IC50 value of 0.06 μM [111]. In addition, AF3442 dose-dependently inhibits PGE2 production (EC50 = 0.41) in LPS-stimulated human monocytes where COX-2 and mPGES-1 expression are both induced. On the other hand, other prostanoids (TXB2, PGF2α and 6-keto-PGF1α) remain unaffected by AF3442. However, AF3442 at higher concentration (100 μM) significantly suppresses TXB2 and slightly reduce PGF2α and 6-keto-PGF1α , indicating a potential COX inhibition effect associated with higher doses. In fact, at 100 μM AF3442 reduces COX-2 expression by approximately 30% in these LPS-stimulated human monocytes. In human whole-blood assays, AF3442 inhibits the generation of PGE2 (EC50 = 29 μM) without affecting other prostanoids when tested up to 100 μM. Notably, AF3442 has no effect on thrombin-stimulated prostanoids (PGE2, TXB2, PGF2α 6-keto-PGF1) that are produced and mainly by COX-1 and constitutive terminal synthases in platelets. Together, AF3442 seems to be a selective mPGES-1 inhibitor.
Other scaffolds identified by computational approaches
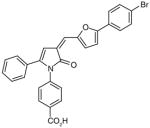
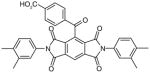
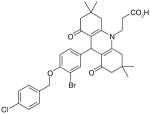
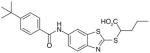
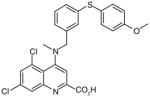
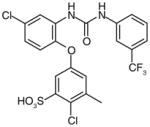
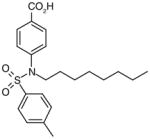
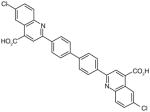
There are a number of novel scaffolds identified by virtual screening, although none of them has been tested in intact cells or animals yet (Table 7). For example, a multistep, ligand-based virtual screening of a library containing 360,169 compounds has led to the identification of 49 with an IC50 value of 0.5 μM in mPGES-1 cell-free assay [112]. Importantly, 49 (up to 30 μM) does not inhibit the activity of either COX isoforms. As another example, 50 (IC50 = 3.2 μM) was identified by molecular docking studies on 26 triazole-based compounds. This small set of compounds was designed by taking into account the binding requirements to the active site of MGST-1, which shares significant homology with mPGES-1 [113]. As revealed by molecular modeling, the carboxy group of 50 forms a hydrogen bond with the highly conserved Arg131 in MGST-1 (corresponding to Arg110 in mPGES-1), which is critical for PGH2 binding. Interestingly, this in silico screen also resulted in the identification of compounds targeting 5-LOX and FLAP. Finally, virtual screening of the National Cancer Institute and the Specs database was performed using a ligand-based pharmacophore model built upon the structure information of six acidic indole inhibitors of mPGES-1 [114]. Among 34 identified compounds, 29 of them with full solubility were selected for biological testing. As a result, nine compounds with diverse scaffolds (51–59, Table 7) were identified as mPGES-1 inhibitors (IC50 = 0.4 to 7.9 μM). Besides, most of them also inhibit 5-LOX in the cell-free system as well as in intact cells. Based on molecular docking studies, the most active compound (57) interacts with Arg126 and Glu77 of mPGES-1.
Future perspective
In this review, following a presentation of mPGES-1, we presented an overview of the compounds that have been described in the literature to inhibit the target. Figure 3 summarizes the number of peer-reviewed articles per year that have been published since the discovery of mPGES-1 as well as the number of patents that were issued per year. An exponential increase in the number of papers on mPGES-1 can be noticed since its discovery and clearly the pathway and the enzyme have generated a great interest in the field of research. Papers that describe small molecules that inhibit the activity of the enzyme have increased dramatically over the past 5 years. This observation can further be followed in the number of patents issued over these past 5 years as well. However, and interestingly, compounds that are subsequently found to inhibit mPGES-1 in cell-free assays and/or in vitro cellular assays, have been reported to exhibit in vivo anti-inflamma-tory activity only in rare cases in various animal models. One may wonder as to the explanation of such observation. There are several facts that could explain this. First, selectivity could be one of them. As stated in the first part of the review, the protein belongs to the MAPEG family of proteins and, thus, compounds that will inhibit the target will likely hit the other members of the family. The fact that one subunit of mPGES-1 also resembles other proteins such as the Huntingtin interacting protein 12 (PDB: 1R0D), the V-type sodium ATP synthase subunit K (PDB: 2BL2) or the protein tyrosine kinase 2 β (b3GM3) is also concerning. Only MK-886 (25) has been demonstrated to exhibit some anticancer properties in vivo, mostly due to its FLAP inhibitory properties [115]. However, increasing evidences suggest that dual inhibitors such as 5-LOX/mPGES-1 inhibitors would work well but clinical trials will further validate this novel concept. Second, amino acid sequence disparities between human, mouse and rat may have impaired research. Finally, from a modeling as well as a drug-design point of view, the trimeric target posses a very hydrophobic active site and has been proposed to exist in a open and closed conformation. The two facts increase the complications encountered during the development and/or discovery of novel selective inhibitors for mPGES-1. In conclusion, it is clear that mPGES-1 represents an attractive therapeutic target for cancer as well as other disease in which inflammation plays a role. How soon will a selective mPGES-1 be identified and tested in clinical trials will depend on the co-crystallization of a lead compound within the active or inactive protein and the selectivity that can be achieve within the MAPEG family of enzymes. These two challenges may eventually be bypassed should the biology show an increase efficacy and utility towards non-selective compounds regardless of their mechanism of action.
Figure 3. Number of publications and patents on microsomal prostaglandin E synthase-1.

Black bars are the number of papers containing microsomal prostaglandin E synthase-1 (mPGES-1) in their title (reviews included). Gray bars are the number of patents published on mPGES-1 (including usage patents).
Executive summary.
-
In summary, after its discovery in the late 1990s and at the point of writing:
Microsomal prostaglandin E synthase-1 has been validated as a novel and attractive therapeutic target for cancer drug discovery. For example, knockout mice have shown to develop less susceptible to colon carcinogenesis.
Several compounds have been identified as inhibitor of the enzyme. In vitro assays have been developed over the years and a wide variety of structures were shown to bind and inhibit the target.
Selectivity remains to be achieved.
Anticancer properties have yet to be shown in in vivo animal models for selective mPGES-1 inhibitors.
Acknowledgments
Financial & competing interests disclosure
This work was supported by grant CA138702 (to Emmanuelle J Meuillet) by the National Cancer Institute. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. No writing assistance was utilized in the production of this manuscript.
Key Terms
- mPGES-1
Terminal inducible synthase responsible for the production of PGE2. It is overexpressed in a variety of cancers
- COX-2
Rate-limiting key enzyme in the production of PGE2 from arachidonic acid. The enzyme produces PGH2, the substrate of mPGES-1 to produce the pro-tumorigenic PGE2
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Hara S, Kamei D, Sasaki Y, Tanemoto A, Nakatani Y, Murakami M. Prostaglandin E synthases: understanding their pathophysiological roles through mouse genetic models. Biochimie. 2010;92(6):651–659. doi: 10.1016/j.biochi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 2▪▪.Nakanishi M, Montrose DC, Clark P, et al. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. First evidence of a role for mPGES-1 knockout in colorectal carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 3.Radmark O, Samuelsson B. Microsomal prostaglandin E synthase-1 and 5-lipoxygenase: potential drug targets in cancer. J Intern Med. 2010;268(1):5–14. doi: 10.1111/j.1365-2796.2010.02246.x. [DOI] [PubMed] [Google Scholar]
- 4.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59(3):207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 5.Van Rees BP, Sivula A, Thoren S, et al. Expression of microsomal prostaglandin E synthase-1 in intestinal type gastric adenocarcinoma and in gastric cancer cell lines. Int J Cancer. 2003;107(4):551–556. doi: 10.1002/ijc.11422. [DOI] [PubMed] [Google Scholar]
- 6▪▪.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220. First report published on the identification and characterization of microsomal glutathione-dependent prostaglandin E2 synthase. First suggestion for microsomal prostaglandin E synthase-1 as a novel target for drug development in the area of inflammation and cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jegerschold C, Pawelzik SC, Purhonen P, et al. Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc Natl Acad Sci USA. 2008;105(32):11110–11115. doi: 10.1073/pnas.0802894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing L, Kurumbail RG, Frazier RB, et al. Homo-timeric structural model of human microsomal prostaglandin E synthase-1 and characterization of its substrate/inhibitor binding interactions. J Comput Aided Mol Des. 2009;23(1):13–24. doi: 10.1007/s10822-008-9233-4. [DOI] [PubMed] [Google Scholar]
- 9.Hetu PO, Ouellet M, Falgueyret JP, et al. Photo-crosslinking of proteins in intact cells reveals a dimeric structure of cyclooxygenase-2 and an inhibitor-sensitive oligomeric structure of microsomal prostaglandin E2 synthase-1. Arch Biochem Biophys. 2008;477(1):155–162. doi: 10.1016/j.abb.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 10.Murakami M, Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275(42):32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 11.Hammarberg T, Hamberg M, Wetterholm A, Hansson H, Samuelsson B, Haeggstrom JZ. Mutation of a critical arginine in microsomal prostaglandin E synthase-1 shifts the isomerase activity to a reductase activity that converts prostaglandin H2 into prostaglandin F2α. J Biol Chem. 2009;284(1):301–305. doi: 10.1074/jbc.M808365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamza A, Tong M, Abdulhameed MD, et al. Understanding microscopic binding of human microsomal prostaglandin E synthase-1 (mPGES-1) trimer with substrate and cofactor GSH: insights from PGH2 computational alanine scanning and site-directed mutagenesis. J Phys Chem B. 2010;114(16):5605–5616. doi: 10.1021/jp100668y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe K, Kurihara K, Tokunaga Y, Hayaishi O. Two types of microsomal prostaglandin E synthase: glutathione-dependent and -independent prostaglandin E synthases. Biochem Biophys Res Comm. 1997;235(1):148–152. doi: 10.1006/bbrc.1997.6708. [DOI] [PubMed] [Google Scholar]
- 14.Yamada T, Komoto J, Watanabe K, Ohmiya Y, Takusagawa F. Crystal structure and possible catalytic mechanism of microsomal prostaglandin E synthase type 2 (mPGES-2) J Mol Biol. 2005;348(5):1163–1176. doi: 10.1016/j.jmb.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275(42):32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 16.Murakami M, Kudo I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog Lipid Res. 2004;43(1):3–35. doi: 10.1016/s0163-7827(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 17.Murakami M, Nakashima K, Kamei D, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278(39):37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 18.Jania LA, Chandrasekharan S, Backlund MG, et al. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 2009;88(3–4):73–81. doi: 10.1016/j.prostaglandins.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver AJ, Sullivan WP, Felts SJ, Owen BA, Toft DO. Crystal structure and activity of human p23, a heat shock protein 90 co-chaperone. J Biol Chem. 2000;275(30):23045–23052. doi: 10.1074/jbc.M003410200. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi T, Nakatani Y, Tanioka T, et al. Regulation of cytosolic prostaglandin E synthase by phosphorylation. Biochem J. 2004;381(Pt 1):59–69. doi: 10.1042/BJ20040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanikawa N, Ohmiya Y, Ohkubo H, et al. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Comm. 2002;291(4):884–889. doi: 10.1006/bbrc.2002.6531. [DOI] [PubMed] [Google Scholar]
- 22.Nakatani Y, Hokonohara Y, Kakuta S, Sudo K, Iwakura Y, Kudo I. Knockout mice lacking cPGES/p23, a constitutively expressed PGE2 synthetic enzyme, are peri-natally lethal. Biochem Biophys Res Comm. 2007;362(2):387–392. doi: 10.1016/j.bbrc.2007.07.180. [DOI] [PubMed] [Google Scholar]
- 23.Cha YI, Solnica-Krezel L, Dubois RN. Fishing for prostanoids: deciphering the developmental functions of cyclooxygenase-derived prostaglandins. Dev Biol. 2006;289(2):263–272. doi: 10.1016/j.ydbio.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 24.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168(11):5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 25▪▪.Trebino CE, Stock JL, Gibbons CP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100(15):9044–9049. doi: 10.1073/pnas.1332766100. Demonstration for a role of mPGES-1 in both acute and chronic PGE2-dependent experimental models of inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thoren S, Jakobsson PJ. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur J Biochem. 2000;267(21):6428–6434. doi: 10.1046/j.1432-1327.2000.01735.x. [DOI] [PubMed] [Google Scholar]
- 27.Stichtenoth DO, Thoren S, Bian H, Peters-Golden M, Jakobsson PJ, Crofford LJ. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol. 2001;167(1):469–474. doi: 10.4049/jimmunol.167.1.469. [DOI] [PubMed] [Google Scholar]
- 28.Mustafa M, Wondimu B, Yucel-Lindberg T, Kats-Hallstrom AT, Jonsson AS, Modeer T. Triclosan reduces microsomal prostaglandin E synthase-1 expression in human gingival fibroblasts. J Clin Periodontol. 2005;32(1):6–11. doi: 10.1111/j.1600-051X.2004.00622.x. [DOI] [PubMed] [Google Scholar]
- 29.Yucel-Lindberg T, Hallstrom T, Kats A, Mustafa M, Modeer T. Induction of microsomal prostaglandin E synthase-1 in human gingival fibroblasts. Inflammation. 2004;28(2):89–95. doi: 10.1023/b:ifla.0000033024.13748.c1. [DOI] [PubMed] [Google Scholar]
- 30.Naraba H, Yokoyama C, Tago N, et al. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J Biol Chem. 2002;277(32):28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- 31▪▪.Kamei D, Murakami M, Nakatani Y, Ishikawa Y, Ishii T, Kudo I. Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J Biol Chem. 2003;278(21):19396–19405. doi: 10.1074/jbc.M213290200. Co-expression of COX-2 and mPGES-1 in HEK293 cells leads to tumorigenicity of these cells into mice. First demonstration of a role for mPGES-1 in tumorigenesis. [DOI] [PubMed] [Google Scholar]
- 32.Saha S, Engstrom L, Mackerlova L, Jakobsson PJ, Blomqvist A. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1100–R1107. doi: 10.1152/ajpregu.00872.2004. [DOI] [PubMed] [Google Scholar]
- 33.Kamei D, Yamakawa K, Takegoshi Y, et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279(32):33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 34.Von Rahden BH, Stein HJ, Hartl SA, et al. Expression of prostaglandin E synthase in Barrett’s cancer. Dis Esophagus. 2008;21(4):304–308. doi: 10.1111/j.1442-2050.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- 35.Nardone G, Rocco A, Vaira D, et al. Expression of COX-2, mPGE-synthase1, MDR-1 (P-gp), and Bcl-xL: a molecular pathway of H pylori-related gastric carcinogenesis. J Pathol. 2004;202(3):305–312. doi: 10.1002/path.1512. [DOI] [PubMed] [Google Scholar]
- 36.Jang TJ. Expression of proteins related to prostaglandin E2 biosynthesis is increased in human gastric cancer and during gastric carcinogenesis. Virchows Arch. 2004;445(6):564–571. doi: 10.1007/s00428-004-1104-3. [DOI] [PubMed] [Google Scholar]
- 37.Gudis K, Tatsuguchi A, Wada K, et al. Clinical significance of prostaglandin E synthase expression in gastric cancer tissue. Hum Pathol. 2007;38(12):1826–1835. doi: 10.1016/j.humpath.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 38.Rocco A, Caruso R, Toracchio S, et al. Gastric adenomas: relationship between clinicopathological findings, Helicobacter pylori infection, APC mutations and COX-2 expression. Ann Oncol. 2006;17(Suppl 7):103–108. doi: 10.1093/annonc/mdl961. [DOI] [PubMed] [Google Scholar]
- 39.Yoshimatsu K, Golijanin D, Paty PB, et al. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001;7(12):3971–3976. [PubMed] [Google Scholar]
- 40.Lim SC, Cho H, Lee TB, et al. Impacts of cytosolic phospholipase A2, 15-prostaglandin dehydrogenase, and cyclooxygenase-2 expressions on tumor progression in colorectal cancer. Yonsei Med J. 2010;51(5):692–699. doi: 10.3349/ymj.2010.51.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takii Y, Abiru S, Fujioka H, et al. Expression of microsomal prostaglandin E synthase-1 in human hepatocelluar carcinoma. Liver Int. 2007;27(7):989–996. doi: 10.1111/j.1478-3231.2007.01530.x. [DOI] [PubMed] [Google Scholar]
- 42.Nonaka K, Fujioka H, Takii Y, et al. mPGES-1 expression in non-cancerous liver tissue impacts on postoperative recurrence of HCC. World J Gastroenterol. 2010;16(38):4846–4853. doi: 10.3748/wjg.v16.i38.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasan S, Satake M, Dawson DW, et al. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37(2):121–127. doi: 10.1097/MPA.0b013e31816618ba. [DOI] [PubMed] [Google Scholar]
- 44.Mattila S, Tuominen H, Koivukangas J, Stenback F. The terminal prostaglandin synthases mPGES-1, mPGES-2, and cPGES are all overexpressed in human gliomas. Neuropathology. 2009;29(2):156–165. doi: 10.1111/j.1440-1789.2008.00963.x. [DOI] [PubMed] [Google Scholar]
- 45.Baryawno N, Sveinbjornsson B, Eksborg S, et al. Tumor-growth-promoting cyclooxygenase-2 prostaglandin E2 pathway provides medulloblastoma therapeutic targets. Neuro Oncol. 2008;10(5):661–674. doi: 10.1215/15228517-2008-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mehrotra S, Morimiya A, Agarwal B, Konger R, Badve S. Microsomal prostaglandin E2 synthase-1 in breast cancer: a potential target for therapy. J Pathol. 2006;208(3):356–363. doi: 10.1002/path.1907. [DOI] [PubMed] [Google Scholar]
- 47.Gatalica Z, Lilleberg SL, Koul MS, et al. COX-2 gene polymorphisms and protein expression in renomedullary interstitial cell tumors. Hum Pathol. 2008;39(10):1495–1504. doi: 10.1016/j.humpath.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Omi Y, Shibata N, Okamoto T, Obara T, Kobayashi M. Immunohistochemical demonstration of membrane-bound prostaglandin E2 synthase-1 in papillary thyroid carcinoma. Acta Histochem Cytochem. 2009;42(4):105–109. doi: 10.1267/ahc.09014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawata R, Hyo S, Araki M, Takenaka H. Expression of cyclooxygenase-2 and microsomal prostagalandin E synthase-1 in head and neck squamous cell carcinoma. Auris Nasus Larynx. 2010;37(4):482–487. doi: 10.1016/j.anl.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Cohen EG, Almahmeed T, Du B, et al. Microsomal prostaglandin E synthase-1 is overexpressed in head and neck squamous cell carcinoma. Clin Cancer Res. 2003;9(9):3425–3430. [PubMed] [Google Scholar]
- 51.Golijanin D, Tan JY, Kazior A, et al. Cyclooxygenase-2 and microsomal prostaglandin E synthase-1 are overexpressed in squamous cell carcinoma of the penis. Clin Cancer Res. 2004;10(3):1024–1031. doi: 10.1158/1078-0432.ccr-1032-3. [DOI] [PubMed] [Google Scholar]
- 52.Yoshimatsu K, Altorki NK, Golijanin D, et al. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001;7(9):2669–2674. [PubMed] [Google Scholar]
- 53.Wu YC, Su LJ, Wang HW, et al. Co-overexpression of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 adversely affects the postoperative survival in non-small cell lung cancer. J Thorac Oncol. 2010;5(8):1167–1174. doi: 10.1097/JTO.0b013e3181e2f4f5. [DOI] [PubMed] [Google Scholar]
- 54.Wang HW, Hsueh CT, Lin CF, et al. Clinical implications of microsomal prostaglandin e synthase-1 overexpression in human non-small-cell lung cancer. Ann Surg Oncol. 2006;13(9):1224–1234. doi: 10.1245/s10434-006-9001-4. [DOI] [PubMed] [Google Scholar]
- 55.Kawata R, Hyo S, Maeda T, Urade Y, Takenaka H. Simultaneous expression of cyclooxygenase-2 and microsomal prostaglandin E synthase in squamous cell carcinoma of the larynx. Acta Otolaryngol. 2006;126(6):627–632. doi: 10.1080/00016480500452541. [DOI] [PubMed] [Google Scholar]
- 56.Herfs M, Herman L, Hubert P, et al. High expression of PGE2 enzymatic pathways in cervical (pre)neoplastic lesions and functional consequences for antigen-presenting cells. Cancer Immunol Immunother. 2009;58(4):603–614. doi: 10.1007/s00262-008-0584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jabbour HN, Milne SA, Williams AR, Anderson RA, Boddy SC. Expression of COX-2 and PGE synthase and synthesis of PGE(2) in endometrial adenocarcinoma: a possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. Br J Cancer. 2001;85(7):1023–1031. doi: 10.1054/bjoc.2001.2033. [DOI] [PubMed] [Google Scholar]
- 58.Rask K, Zhu Y, Wang W, Hedin L, Sundfeldt K. Ovarian epithelial cancer: a role for PGE2-synthesis and signalling in malignant transformation and progression. Mol Cancer. 2006;5:62. doi: 10.1186/1476-4598-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59▪.Nakanishi M, Gokhale V, Meuillet EJ, Rosenberg DW. mPGES-1 as a target for cancer suppression: a comprehensive invited review “phospholipase A2 and lipid mediators”. Biochimie. 2010;92(6):660–664. doi: 10.1016/j.biochi.2010.02.006. Concise review of the role of mPGES-1 in colorectal carcinogenesis and the novel inhibitors developed for this target. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60▪.Elander N, Ungerback J, Olsson H, Uematsu S, Akira S, Soderkvist P. Genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in APC(Min/+) mice. Biochem Biophys Res Comm. 2008 doi: 10.1016/j.bbrc.2008.05.026. Controvertial report about the effects of mPGES-1 downregulation in intestinal tumorigenesis (in contradiction to [2]) [DOI] [PubMed] [Google Scholar]
- 61.Hanaka H, Pawelzik SC, Johnsen JI, et al. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc Natl Acad Sci USA. 2009;106(44):18757–18762. doi: 10.1073/pnas.0910218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62▪.Kamei D, Murakami M, Sasaki Y, et al. Microsomal prostaglandin E synthase-1 in both cancer cells and hosts contributes to tumour growth, invasion and metastasis. Biochem J. 2010;425(2):361–371. doi: 10.1042/BJ20090045. This report provides evidence that mPGES-1 in both cancer cells and host contributes to tumorigenesis in vitro and in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63▪.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, Fitzgerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci USA. 2006;103(39):14507–14512. doi: 10.1073/pnas.0606586103. Deletion of the mPGES-1 gene in low-density lipoprotein receptor knockout mice retards atherogenesis without affecting blood pressure. The report also confirms the Cheng work by [71] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343(21):1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 65.Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352(11):1081–1091. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- 66.Gislason GH, Jacobsen S, Rasmussen JN, et al. Risk of death or reinfarction associated with the use of selective cyclooxygenase-2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation. 2006;113(25):2906–2913. doi: 10.1161/CIRCULATIONAHA.106.616219. [DOI] [PubMed] [Google Scholar]
- 67.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286(8):954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 68.Wu D, Mennerich D, Arndt K, et al. Comparison of microsomal prostaglandin E synthase-1 deletion and COX-2 inhibition in acute cardiac ischemia in mice. Prostaglandins Other Lipid Mediat. 2009;90(1–2):21–25. doi: 10.1016/j.prostaglandins.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 69.Wu D, Mennerich D, Arndt K, et al. The effects of microsomal prostaglandin E synthase-1 deletion in acute cardiac ischemia in mice. Prostaglandins Leukot Essent Fatty Acids. 2009;81(1):31–33. doi: 10.1016/j.plefa.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 70.Degousee N, Fazel S, Angoulvant D, et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008;117(13):1701–1710. doi: 10.1161/CIRCULATIONAHA.107.749739. [DOI] [PubMed] [Google Scholar]
- 71▪.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116(5):1391–1399. doi: 10.1172/JCI27540. Reports that mPGES-1 deletion, in contrast to deletion, disruption, or inhibition of or cyclooxygenase 2 (COX-2), does not result in hypertension or a predisposition to thrombosis in normo-lipidemic mice. These important findings suggested that selective mPGES-1 inhibitors would have very low if any cardiotoxic side effects typically associated with COX-2 inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bannenberg G, Dahlen SE, Luijerink M, Lundqvist G, Morgenstern R. Leukotriene C4 is a tight-binding inhibitor of microsomal glutathione transferase-1. Effects of leukotriene pathway modifiers. J Biol Chem. 1999;274(4):1994–1999. doi: 10.1074/jbc.274.4.1994. [DOI] [PubMed] [Google Scholar]
- 73.Quraishi O, Mancini JA, Riendeau D. Inhibition of inducible prostaglandin E(2) synthase by 15-deoxy-Δ(12,14)-prostaglandin J(2) and polyunsaturated fatty acids. Biochem Pharmacol. 2002;63(6):1183–1189. doi: 10.1016/s0006-2952(02)00844-4. [DOI] [PubMed] [Google Scholar]
- 74.Wobst I, Schiffmann S, Birod K, et al. Dimethylcelecoxib inhibits prostaglandin E2 production. Biochem Pharmacol. 2008;76(1):62–69. doi: 10.1016/j.bcp.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 75.Mancini JA, Blood K, Guay J, et al. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276(6):4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- 76.Riendeau D, Aspiotis R, Ethier D, et al. Inhibitors of the inducible microsomal prostaglandin E2 synthase (mPGES-1) derived from MK-886. Bioorg Med Chem Lett. 2005;15(14):3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 77.Sciulli MG, Seta F, Tacconelli S, et al. Effects of acetaminophen on constitutive and inducible prostanoid biosynthesis in human blood cells. Br J Pharmacol. 2003;138(4):634–641. doi: 10.1038/sj.bjp.0705078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bage T, Modeer T, Kawakami T, Quezada HC, Yucel-Lindberg T. Regulation of prostaglandin E synthases: effects of siRNA-mediated inhibition of microsomal prostaglandin E synthase-1. Biochim Biophys Acta. 2007;1773(10):1589–1598. doi: 10.1016/j.bbamcr.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 79.Cianchi F, Cortesini C, Magnelli L, et al. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther. 2006;5(11):2716–2726. doi: 10.1158/1535-7163.MCT-06-0318. [DOI] [PubMed] [Google Scholar]
- 80.Koeberle A, Siemoneit U, Buhring U, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326(3):975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- 81.Fischer L, Hornig M, Pergola C, et al. The molecular mechanism of the inhibition by licofelone of the biosynthesis of 5-lipoxygenase products. Br J Pharmacol. 2007;152(4):471–480. doi: 10.1038/sj.bjp.0707416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koeberle A, Northoff H, Werz O. Curcumin blocks prostaglandin E2 biosynthesis through direct inhibition of the microsomal prostaglandin E2 synthase-1. Mol Cancer Ther. 2009;8(8):2348–2355. doi: 10.1158/1535-7163.MCT-09-0290. [DOI] [PubMed] [Google Scholar]
- 83.Koeberle A, Bauer J, Verhoff M, Hoffmann M, Northoff H, Werz O. Green tea epigallocatechin-3-gallate inhibits microsomal prostaglandin E(2) synthase-1. Biochem Biophys Res Comm. 2009;388(2):350–354. doi: 10.1016/j.bbrc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 84.Koeberle A, Northoff H, Werz O. Identification of 5-lipoxygenase and microsomal prostaglandin E2 synthase-1 as functional targets of the anti-inflammatory and anti-carcinogenic garcinol. Biochem Pharmacol. 2009;77(9):1513–1521. doi: 10.1016/j.bcp.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 85.Koeberle A, Pollastro F, Northoff H, Werz O. Myrtucommulone, a natural acylphloroglucinol, inhibits microsomal prostaglandin E(2) synthase-1. Br J Pharmacol. 2009;156(6):952–961. doi: 10.1111/j.1476-5381.2009.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bauer J, Koeberle A, Dehm F, et al. Arzanol, a prenylated heterodimeric phloroglucinyl pyrone, inhibits eicosanoid biosynthesis and exhibits anti-inflammatory efficacy in vivo. Biochem Pharmacol. 2011;81(2):259–268. doi: 10.1016/j.bcp.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 87.Siemoneit U, Koeberle A, Rossi A, et al. Inhibition of microsomal prostaglandin E2 synthase-1 as a molecular basis for the anti-inflammatory actions of boswellic acids from frankincense. Br J Pharmacol. 2011;162(1):147–162. doi: 10.1111/j.1476-5381.2010.01020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koeberle A, Rossi A, Bauer J, et al. Hyperforin, an anti-inflammatory constituent from St. John’s wort, inhibits microsomal prostaglandin E(2) synthase-1 and suppresses prostaglandin E(2) formation in vivo. Front Pharmacol. 2011;2:7. doi: 10.3389/fphar.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rao CV. Regulation of COX and LOX by curcumin. Adv Exp Med Biol. 2007;595:213–226. doi: 10.1007/978-0-387-46401-5_9. [DOI] [PubMed] [Google Scholar]
- 90.Charleson S, Prasit P, Leger S, et al. Characterization of a 5-lipoxygenase-activating protein binding assay: correlation of affinity for 5-lipoxygenase-activating protein with leukotriene synthesis inhibition. Mol Pharmacol. 1992;41(5):873–879. [PubMed] [Google Scholar]
- 91.Masse F, Guiral S, Fortin LJ, et al. An automated multistep high-throughput screening assay for the identification of lead inhibitors of the inducible enzyme mPGES-1. J Biomol Screen. 2005;10(6):599–605. doi: 10.1177/1087057105276083. [DOI] [PubMed] [Google Scholar]
- 92.Cote B, Boulet L, Brideau C, et al. Substituted phenanthrene imidazoles as potent, selectiveIdentification of a novel, potent and selective inhibitor of mPGES-1 activity, and orally active mPGES-1 inhibitors. Bioorg Med Chem Lett. 2007;17(24):6816–6820. doi: 10.1016/j.bmcl.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 93.Pawelzik SC, Uda NR, Spahiu L, et al. Identification of key residues determining species differences in inhibitor binding of microsomal prostaglandin E synthase-1. J Biol Chem. 2010;285(38):29254–29261. doi: 10.1074/jbc.M110.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu D, Rowland SE, Clark P, et al. MF63 [2-(6-chloro-1H-phenanthro[9,10-d] imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326(3):754–763. doi: 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]
- 95.Giroux A, Boulet L, Brideau C, et al. Discovery of disubstituted phenanthrene imidazoles as potent, selective and orally active mPGES-1 inhibitors. Bioorg Med Chem Lett. 2009;19(20):5837–5841. doi: 10.1016/j.bmcl.2009.08.085. [DOI] [PubMed] [Google Scholar]
- 96.Gosselin F, Lau S, Nadeau C, Trinh T, O’shea PD, Davies IW. A practical synthesis of m-prostaglandin E synthase-1 inhibitor MK-7285. J Org Chem. 2009;74(20):7790–7797. doi: 10.1021/jo901798d. [DOI] [PubMed] [Google Scholar]
- 97.Wu TY, Juteau H, Ducharme Y, et al. Biarylimidazoles as inhibitors of microsomal prostaglandin E2 synthase-1. Bioorg Med Chem Lett. 2010;20(23):6978–6982. doi: 10.1016/j.bmcl.2010.09.129. [DOI] [PubMed] [Google Scholar]
- 98.Werz O, Greiner C, Koeberle A, et al. Novel and potent inhibitors of 5-lipoxygenase product synthesis based on the structure of pirinixic acid. J Med Chem. 2008;51(17):5449–5453. doi: 10.1021/jm800588x. [DOI] [PubMed] [Google Scholar]
- 99.Koeberle A, Zettl H, Greiner C, Wurglics M, Schubert-Zsilavecz M, Werz O. Pirinixic acid derivatives as novel dual inhibitors of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Med Chem. 2008;51(24):8068–8076. doi: 10.1021/jm801085s. [DOI] [PubMed] [Google Scholar]
- 100.Hieke M, Greiner C, Thieme TM, Schubert-Zsilavecz M, Werz O, Zettl H. A novel class of dual mPGES-1/5-LO inhibitors based on the α-naphthyl pirinixic acid scaffold. Bioorg Med Chem Lett. 2011;21(5):1329–1333. doi: 10.1016/j.bmcl.2011.01.049. [DOI] [PubMed] [Google Scholar]
- 101.Koeberle A, Rossi A, Zettl H, et al. The molecular pharmacology and in vivo activity of 2-(4-chloro-6-(2,3-dimethylphenylamino) pyrimidin-2-ylthio)octanoic acid (YS121), a dual inhibitor of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Pharmacol Exp Ther. 2010;332(3):840–848. doi: 10.1124/jpet.109.160663. [DOI] [PubMed] [Google Scholar]
- 102.Hieke M, Greiner C, Dittrich M, et al. Discovery and biological evaluation of a novel class of dual microsomal prostaglandin E(2) synthase-1/5-lipoxygenase inhibitors based on 2-[(4,6-diphenethoxypyrimidin-2-yl)thio] hexanoic acid. J Med Chem. 2011 doi: 10.1021/jm200092b. [DOI] [PubMed] [Google Scholar]
- 103.Greiner C, Zettl H, Koeberle A, et al. Identification of 2-mercaptohexanoic acids as dual inhibitors of 5-lipoxygenase and microsomal prostaglandin E(2) synthase-1. Bioorg Med Chem. 2011;19(11):3394–3401. doi: 10.1016/j.bmc.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 104.Liedtke AJ, Keck PR, Lehmann F, Koeberle A, Werz O, Laufer SA. Arylpyrrolizines as inhibitors of microsomal prostaglandin E2 synthase-1 (mPGES-1) or as dual inhibitors of mPGES-1 and 5-lipoxygenase (5-LOX) J Med Chem. 2009;52(15):4968–4972. doi: 10.1021/jm900481c. [DOI] [PubMed] [Google Scholar]
- 105.Karg EM, Luderer S, Pergola C, et al. Structural optimization and biological evaluation of 2-substituted 5-hydroxyindole-3-carboxylates as potent inhibitors of human 5-lipoxygenase. J Med Chem. 2009;52(11):3474–3483. doi: 10.1021/jm900212y. [DOI] [PubMed] [Google Scholar]
- 106.Koeberle A, Haberl EM, Rossi A, et al. Discovery of benzo[g]indol-3-carboxylates as potent inhibitors of microsomal prostaglandin E(2) synthase-1. Bioorg Med Chem. 2009;17(23):7924–7932. doi: 10.1016/j.bmc.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 107.Wang J, Limburg D, Carter J, Mbalaviele G, Gierse J, Vazquez M. Selective inducible microsomal prostaglandin E(2) synthase-1 (mPGES-1) inhibitors derived from an oxicam template. Bioorg Med Chem Lett. 2010;20(5):1604–1609. doi: 10.1016/j.bmcl.2010.01.060. [DOI] [PubMed] [Google Scholar]
- 108▪.Mbalaviele G, Pauley AM, Shaffer AF, et al. Distinction of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition from cyclooxygenase-2 inhibition in cells using a novel, selective mPGES-1 inhibitor. Biochem Pharmacol. 2010;79(10):1445–1454. doi: 10.1016/j.bcp.2010.01.003. This study discusses technical assays to be performed in order to obtain mPGES-1 inhibitors profiles, disctinct from COX-2 inhibitor profiles. [DOI] [PubMed] [Google Scholar]
- 109.Olkkola KT, Brunetto AV, Mattila MJ. Pharmacokinetics of oxicam nonsteroidal anti-inflammatory agents. Clin Pharmacokinet. 1994;26(2):107–120. doi: 10.2165/00003088-199426020-00004. [DOI] [PubMed] [Google Scholar]
- 110.Chiasson JF, Boulet L, Brideau C, et al. Trisubstituted ureas as potent and selective mPGES-1 inhibitors. Bioorg Med Chem Lett. 2011;21(5):1488–1492. doi: 10.1016/j.bmcl.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 111.Bruno A, Di Francesco L, Coletta I, et al. Effects of AF3442 [N-(9-ethyl-9H-carbazol-3-yl)-2-(trifluoromethyl)benzamide], a novel inhibitor of human microsomal prostaglandin E synthase-1, on prostanoid biosynthesis in human monocytes in vitro. Biochem Pharmacol. 2010;79(7):974–981. doi: 10.1016/j.bcp.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 112.Rorsch F, Wobst I, Zettl H, et al. Nonacidic inhibitors of human microsomal prostaglandin synthase 1 (mPGES 1) identified by a multistep virtual screening protocol. J Med Chem. 2010;53(2):911–915. doi: 10.1021/jm9012505. [DOI] [PubMed] [Google Scholar]
- 113.De Simone R, Chini MG, Bruno I, et al. Structure-based discovery of inhibitors of microsomal prostaglandin E2 synthase-1, 5-lipoxygenase and 5-lipoxygenase-activating protein: promising hits for the development of new anti-inflammatory agents. J Med Chem. 2011;54(6):1565–1575. doi: 10.1021/jm101238d. [DOI] [PubMed] [Google Scholar]
- 114▪▪.Waltenberger B, Wiechmann K, Bauer J, et al. Pharmacophore modeling and virtual screening for novel acidic inhibitors of microsomal prostaglandin E(2) synthase-1 (mPGES-1) J Med Chem. 2011;54(9):3163–3174. doi: 10.1021/jm101309g. Most recent and compelling study that describes the identification of novel mPGES-1 inhibitor scaffolds from pharmacophore modeling and in silico studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schuller HM, Zhang L, Weddle DL, Castonguay A, Walker K, Miller MS. The cyclooxygenase inhibitor ibuprofen and the FLAP inhibitor MK886 inhibit pancreatic carcinogenesis induced in hamsters by transplacental exposure to ethanol and the tobacco carcinogen NNK. J Cancer Res Clin Oncol. 2002;128(10):525–532. doi: 10.1007/s00432-002-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]