

Abstract
Single nucleotide polymorphisms (SNPs) discovered by genome-wide association studies (GWASs) account for only a small fraction of the genetic variation of complex traits in human populations. Where is the remaining heritability? We estimated the proportion of variance for human height explained by 294,831 SNPs genotyped on 3,925 unrelated individuals using a linear model analysis, and validated the estimation method by simulations based upon the observed genotype data. We show that 45% of variance can be explained by considering all SNPs simultaneously. Thus, most of the heritability is not missing but has not previously been detected because the individual effects are too small to pass stringent significance tests. We provide evidence that the remaining heritability is due to incomplete linkage disequilibrium (LD) between causal variants and genotyped SNPs, exacerbated by causal variants having lower minor allele frequency (MAF) than the SNPs explored to date.
Genome-wide association studies in human populations have discovered hundreds of SNPs significantly associated with complex traits1,2, yet for any one trait they typically account for only a small fraction of the genetic variation. Where is the missing heritability, the so called dark matter of the genome3,4? Suggested explanations include the existence of gene-by-gene or gene-by-environment interactions5, the common disease-rare variant hypothesis6 and the possibility that inherited epigenetic factors cause resemblance between relatives7,8. However, the variance explained by the validated SNPs is usually much less than the narrow-sense heritability, the proportion of phenotypic variance due to additive genetic variance. Non-additive genetic effects do not contribute to the narrow-sense heritability, so explanations based on non-additive effects are not relevant to the problem of missing heritability (Supplementary Note). There are two explanations for the failure of validated SNP associations to explain the estimated heritability: either the causal variants each explain such a small amount of variation that their effects fail to reach stringent significance thresholds and/or the causal variants are not in complete linkage disequilibrium (LD) with the SNPs that have been genotyped. Lack of complete LD might, for instance, occur if causal variants have lower minor allele frequency (MAF) than genotyped SNPs. Here we test these two hypotheses and estimate the contribution of each to the heritability of height in humans as a model complex trait.
Height in humans is a classical quantitative trait, easy to measure and studied for well over a century as a model for investigating the genetic basis of complex traits9,10. The heritability of height has been estimated to be ~0.8 (refs. 9,11–13). Rare mutations that cause extreme short or tall stature have been found14,15, but these do not explain much of the variation in the general population. Recent GWASs on tens of thousands of individuals have detected ~50 variants that are associated with height in the population, but these in total account for only ~5% of phenotypic variance16–19.
Data from a GWAS that are collected to detect statistical associations between SNPs and complex traits are usually analysed by testing each SNP individually for an association with the trait. To account for the large number of significance tests carried out, a very stringent P value is used. This reduces the occurrence of false positives, but may cause many real associations to be missed, especially if individual SNPs have a small effect on the trait. An alternative approach designed to overcome this problem is to fit all the SNPs simultaneously20. The effects of the SNPs are treated statistically as random, and the variance explained by all the SNPs together is estimated. This approach, which we use here, does not attempt to test the significance of individual SNPs but provides an unbiased estimate of the variance explained by the SNPs in total.
RESULTS
Estimating genetic variance explained by genome-wide SNPs
From a number of GWASs, we selected 4,259 individuals who were not knowingly related to one another and confirmed this with SNP data. We then estimated their pairwise genetic relationships using all autosomal markers (MAF ≥ 0.01), and retained 3,925 individuals (3,248 adults and 677 16-year-olds) whose pairwise relationship was estimated at less than 0.025 (maximum relatedness approximately corresponding to cousins two to three times removed: Supplementary Fig. 1). We fitted a linear model to the height data and used restricted maximum likelihood (REML)21 to estimate the variance explained by the SNPs. (In Online Methods, we show how this can be conveniently implemented with a mathematically equivalent model that uses the SNPs to calculate the genomic relationship between pairs of subjects). Using this approach, we estimated the proportion of phenotypic variance explained by the SNPs as 0.45 (s.e. = 0.08, Table 1), a nearly tenfold increase relative to the 5% explained by published and validated individual SNPs.
Table 1.
Estimation of phenotypic variance explained from genetic relationships among unrelated individuals by restricted maximum likelihood method.
| # SNPs | L(H0)a | L(H1)b | LRTc | (s.e.) | (s.e.) | (s.e.) | h2 (s.e.)d | ||
|---|---|---|---|---|---|---|---|---|---|
| 295K SNPs | Raw | 294,831 | -1950.89 | -1936.12 | 29.53 | 0.445 (0.084) | 0.546 (0.082) | 0.991 (0.023) | 0.449 (0.083) |
| Adj.e | 294,831 | -1950.89 | -1936.12 | 29.53 | 0.532 (0.101) | 0.458 (0.098) | 0.991 (0.023) | 0.537 (0.100) | |
| 295K/516K SNPsf | Raw | 294,831/516,345 | -1950.89 | -1935.94 | 29.89 | 0.449 (0.085) | 0.536 (0.083) | 0.986 (0.022) | 0.456 (0.085) |
| Adj. | 294,831/516,345 | -1950.89 | -1935.87 | 30.04 | 0.536 (0.101) | 0.449 (0.099) | 0.985 (0.022) | 0.544 (0.101) | |
log-likelihood under the null hypothesis that ;
log-likelihood under the alternative hypothesis that ;
log-likelihood ratio test statistic, LRT = 2[L(H1) − L(H0)];
Estimate of variance explained by all SNPs with its standard error given in the parentheses;
Raw estimate of genetic relationship adjusted for prediction error by equation [9] (assuming c = 0);
The genetic relationships are estimated from 1,318 individuals with 516,345 SNPs and the other 2,607 individuals with 294,831 SNPs. See Online Methods for the notations.
Correcting for incomplete LD between SNPs and causal variants
Our estimate of 45% is still less than the 80% of phenotypic variance due to additive genetic effects (that is, the estimated heritability). One reason why the SNPs do not explain the full estimated heritability is that the SNPs on the arrays are not in complete LD with causal variants. The ability of the SNPs to explain the phenotypic variation caused by causal variants depends on the LD between all the causal variants and all the SNPs. Lack of complete LD is manifested as a difference between the genomic relationship between each pair of subjects j and k at the causal variants (Gjk) and the relationship between the same individuals calculated from the SNPs (Ajk). As causal variants are unknown, we cannot estimate their LD with observed SNPs directly. However, we can mimic it by considering the LD of the genotyped SNPs with one another. It is likely that the causal variants and the SNPs have different properties, so LD among SNPs is only a guide to LD between causal variants and SNPs. One way in which the causal variants may differ from the SNPs is in MAF. To investigate how the difference between Gjk and Ajk depends on the number of SNPs used and the MAF of the causal variants, we randomly sampled five sets of SNPs (50K, 100K, …, 250K, where K = 1,000) in the adult dataset and ten sets of SNPs in the adolescent dataset (50K, 100K, .., 500K). For each SNP set, we randomly split the SNPs into two groups, the first representing SNPs and the second representing causal variants, and estimated genetic relationships using all of the SNPs in the first group (Ajk) and using SNPs with MAF ≤ θ in the second group (proxy for Gjk), where θ = 0.1, 0.2, 0.3, 0.4 or 0.5. We calibrated the prediction error by calculating the regression of Gjk on Ajk. We established empirically that the regression coefficient (Fig. 1), where N is the number of SNPs used to calculate Ajk and the term in c depends on the MAF of the causal variants (Online Methods). If the causal loci that have the same spectrum of allele frequency as the genotyped SNPs (θ = 0.5), then c = 0, and 1/N can be interpreted as the sampling error for estimating the relationship over the whole genome from N random SNPs. The parameter c is > 0 if θ < 0.5 because the relationship at causal variants with low MAF is typically less than the average relationship over the whole genome.
Figure 1.
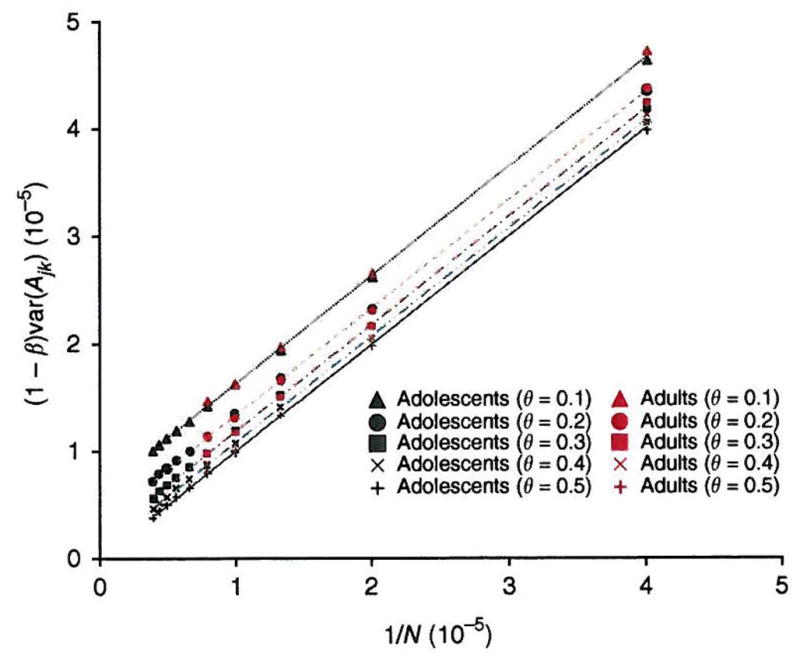
Prediction error of genetic relationship. The genetic relationship at unobserved causal loci is predicted, with error, from the relationship estimated from genotyped SNPs. The prediction error is calibrated by comparing the relationship at causal loci (mimicked by a set of random SNPs with MAF ≤ θ) to that estimated from another set of random SNPs. Values plotted on y-axis are (1−β) var(Ajk) (see Online Methods for the notations) calculated from different numbers of random SNPs (N) in both adult and adolescent datasets. The slope of each line is equal to 1.0, with R2 = 1.0. The intercept (c) is constant for a certain MAF threshold θ, and c = 6.2 × 10−6 (p-value = 2 × 10−14), 3.4 × 10−6 (p-value = 9 × 10−12), 1.8 × 10−6 (p-value = 4 × 10−10), 7.8 × 10−7 (p-value = 2 × 10−7) and 9.2 × 10−9 (p-value = 0.87, not significant) for θ = 0.1, 0.2, 0.3, 0.4 and 0.5, respectively.
Therefore, given the number of SNPs used, we can correct the estimate of the variance explained by the SNPs for incomplete LD with causal variants, if causal variants have the same allelic frequency spectrum as genotyped SNPs. Using the same linear model as above, but corrected for this incomplete LD (c = 0), we estimated the proportion of variance explained by causal variants to be 0.54 (s.e. = 0.10, Table 1). This estimate assumes that the LD between SNPs and causal variants is as strong as that between the genotyped SNPs. However, if the causal polymorphisms tend to have lower MAF than the SNPs that have been assayed, as expected from neutral and selection theories of quantitative genetic variation6,22, we expect the LD between SNPs and causal variants to be reduced. When we used SNPs with a MAF < 0.1 as proxies for causal variants we found c = 6.2 × 10−6. Using this value of c to correct for incomplete LD, we estimated the proportion of variance in height explained by causal variants to be 0.84 (s.e. = 0.16; Supplementary Table 1). Although the standard error is high, this result is consistent with causal variants being, on average, at lower frequency than the SNPs used on commercial arrays and therefore in less LD with these SNPs than the LD of the SNPs with other SNPs. This does not prove that the causal variants have MAF < 0.1, but it shows that if this were the case, they could explain the estimated heritability of height (~0.8).
Variance explained does not depend on number of SNPs
If our procedure for correcting for incomplete LD between SNPs and causal variants is correct, the variance explained by the causal variants should not depend on the number of SNPs used. To show that this is so, we randomly sampled 10%, 20%, …, and 100% of all the ~295K SNPs and estimated the variance explained by causal variants for each group of SNPs using both raw and adjusted estimates of relationships (assuming c = 0; Fig. 2). For the raw estimates of relationships, the proportion of variance explained increases with the number of SNPs because prediction error is reduced through inclusion of more SNPs. When the relationship estimates are adjusted for prediction error, the proportions of variance explained are independent from the number of SNPs and agree with an estimate of ~0.54 but have larger s.e. when fewer SNPs are used.
Figure 2.
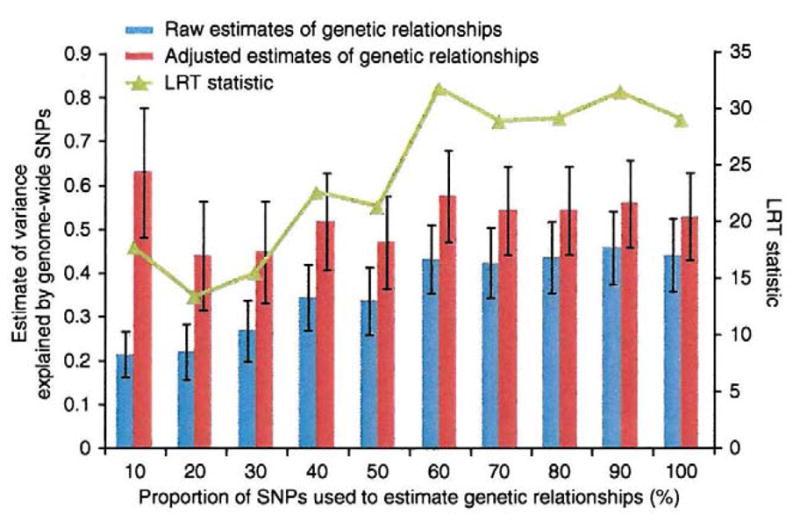
Estimates of variance explained by genome-wide SNPs from adjusted estimates of genetic relationships are unbiased. Results are shown as estimates of variance explained by different proportions of SNPs randomly selected from all the SNPs in the combined set. For each group of SNPs, the variance explained by genome-wide SNPs is estimated using both raw estimates of genetic relationships and adjusted estimates of genetic relationships correcting for prediction error (assuming c = 0). Error bars denote s.e. of the estimate of variance explained by genome-wide SNPs. The log-likelihood ratio test (LRT) statistic is calculated as twice the difference in log-likelihood between the full (h2 ≠ 0) and reduced (h2 = 0) models.
In addition, 1,318 of the 3,925 individuals were genotyped with ~516K SNPs, so we estimated relationships among these individuals (641 adults and 677 16-year-olds) with 516,345 SNPs and estimated the remaining pairwise relationships with 294,831 SNPs. We adjusted the two parts of the relationship matrix according to the number of SNPs used (assuming c = 0). The resulting estimate of proportion of variance explained by causal variants is no different from that using all the individuals with ~295K SNPs (Table 1).
Simulation studies
We used simulation studies to validate the method of estimating the variance explained by causal variants using genome-wide SNPs. We simulated a quantitative trait on the basis of the observed genotype data of 3,925 individuals and 294,831 SNPs in two ways: (i) randomly sampling causal variants from all the SNPs, and (ii) randomly sampling causal variants from the SNPs with MAF ≤ 0.1 (Supplementary Note). Table 2 shows that in case (i), if we included the causal variants in estimating the genetic relationships, we obtained an unbiased estimate of the proportion of phenotypic variance explained by the causal variants (in this case this is the heritability of the trait, because in a simulation we know that these causal variants explain all the genetic variance). When we excluded the causal variants, we underestimated heritability, as the relationship derived from SNPs overestimated the variation of the relationship at causal loci owing to imperfect LD. However, the heritability estimate recovered when we adjusted relationship estimates using equation [9] (Online Methods; c = 0). In case (ii), even if we included the causal variants in the analysis, we still underestimated heritability, because the causal variants have lower frequency than the SNPs, on average, and have less LD with the SNPs than the SNPs have with other SNPs. Similarly, when we adjusted the relationship estimates with equation [9] (c = 6.2 × 10−6), we obtained unbiased estimates of h2. These results are consistent with the inference we draw from the empirical data. The results show that the estimate of variance caused by causal variants is unbiased regardless of the number of SNPs used, provided the method proposed here is employed.
Table 2.
Heritability estimates averaged over 30 simulations based upon the observed genotype data.
| # Causal variants | h2 a | Est. h2 (s.e.m.)b | Est. h2 (s.e.m.)c | Est. h2 (s.e.m.)d | |
|---|---|---|---|---|---|
| MAF ≤ 0.5e | 2000 | 0.8 | 0.817 (0.014) | 0.678 (0.014) | 0.812 (0.014) |
| 2000 | 0.5 | 0.513 (0.015) | 0.428 (0.015) | 0.512 (0.015) | |
| 3000 | 0.8 | 0.831 (0.015) | 0.693 (0.016) | 0.831 (0.016) | |
| 3000 | 0.5 | 0.510 (0.016) | 0.424 (0.017) | 0.507 (0.017) | |
| MAF ≤ 0.1 | 2000 | 0.8 | 0.591 (0.015) | 0.433 (0.014) | 0.804 (0.026) |
| 2000 | 0.5 | 0.367 (0.016) | 0.271 (0.016) | 0.504 (0.030) | |
| 3000 | 0.8 | 0.620 (0.016) | 0.462 (0.016) | 0.856 (0.029) | |
| 3000 | 0.5 | 0.384 (0.020) | 0.287 (0.019) | 0.533 (0.036) | |
True heritability parameter.
Estimated h2 based on genetic relationship estimated from all of the SNPs (~295K), including the causal variants.
Estimated h2 based on relationship estimated from the SNPs, excluding the causal variants.
Estimated h2 based on relationship estimated from the SNPs, excluding the causal variants, and adjusted for prediction error with equation [9].
Minor allele frequencies of the causal variants. s.e.m. (standard error of the mean) was estimated over 30 simulations.
DISCUSSION
Highly significant and well-replicated SNPs identified to date explain only ~5% of the phenotypic variance for height19. Our results show that common SNPs in total explain another ~40% of phenotypic variance. Hence, 88% (40/45) of the variation due to SNPs has been undetected in published GWASs because the effects of the SNPs are too small to be statistically significant. Our results also suggest that the discrepancy between 80% heritability and 45% accounted for by all SNPs is due to incomplete LD between causal variants and the SNPs, possibly because the causal variants have a lower MAF on average than the SNPs typed on the array. We cannot tell from these results whether or not some of this discrepancy is due to causal variants with very low frequency – for example, MAF < 0.001 (ref. 4). However, the results show that the total genetic variance could be explained by causal variants similar to the SNPs, with MAF < 0.1. If causal variants affecting height had no effect on fitness, they would show a complete range of MAF but with a higher proportion at low MAF than the SNPs on commercial arrays. If variants affecting height are subject to selection for either allele, there will still be a spectrum of MAF, but with an even greater proportion at low MAF. Thus, we do not conclude that all causal variants have MAF <0.1, but that the spectrum of MAF at causal variants is more concentrated at low values than it is for the SNPs used as markers.
The power to detect individual SNPs as significantly associated with a trait such as height depends on the variance associated with the SNP. This, in turn, depends on the LD between the SNP and the causal variant, the effect of the causal variant and its frequency. Causal variants with small effects or rare alleles with large effects (including rare Mendelian variants) will explain little variance and so will tend not to be significant even if they are in high LD with an assayed SNP. However, the cumulative effect of these SNPs will be included as part of the 45% of phenotypic variance explained by the SNPs in our analysis. Despite the use of ~295K SNPs, many causal variants, especially if they have low MAF, will not be in perfect LD with the assayed SNPs. This reduces the power of a conventional GWAS to detect them and reduces the variance estimated for the SNPs collectively in our study. The results imply that most causal variants explain such a small proportion of the variance that many causal variants affecting height must exist. The results of published GWASs are consistent with this finding, as high test statistics are distributed over much of the genome16.
Could our results be biased because of ascertainment in the data, data analysis or interpretation? We carefully adjusted phenotypes for systematic differences and applied thorough quality control to the SNP data (Online Methods). We show by principal component analysis (PCA) of African, Asian and European populations that all of our samples are of European ancestry (Supplementary Fig. 2a,b). We demonstrate further by PCA of European populations only that our samples show close relationship to the UK population and do not show an apparent cline across Europe (Supplementary Fig. 2c,d). We performed REML analysis by fitting the first two, four and ten eigenvectors from the European-only PCA as covariates. The results show little to no systematic difference in the estimates of the variance explained by fitting up to ten eigenvectors (Supplementary Table 1). Furthermore, we performed single-SNP association analysis between 1,286 ancestry-informative markers (AIMs) and height, and did not detect a significant inflation of the test statistic for these AIMs (Supplementary Fig. 3; P = 0.219). All these results suggest that our estimate of variance explained by all SNPs is unlikely to be biased by population stratification. A subtle form of stratification in GWASs might occur because subjects are distantly related. We excluded any subjects with a relationship to another subject > 0.025. If distant pedigree relationships were an important cause of the estimated relationships, then all chromosomes of a pair of subjects should reflect this relationship. We found no correlation between relatedness estimated from different chromosomes (Supplementary Table 2). Thus, the relationships we estimate from SNPs are driven by LD among the SNPs. It is the same LD that causes a SNP that is not a causal variant to show an association with a trait such as height. In other words, our estimate of the variance explained by the SNPs is based on the same phenomenon as the SNP associations reported from GWASs (LD between SNPs and causal variants). However, we accumulate the variance explained by all SNPs and so are not limited by the need for individual SNPs to pass stringent significance tests.
We also verified that the estimates of variance explained by the SNPs are not driven by a few outlier individuals that are similar in height and in SNP genotypes (Fig. 3). We regressed the squared difference in height between each pair of individuals on the estimate of their relatedness. The intercept and slope are estimates of twice the phenotypic variance and minus twice the additive genetic variance explained by the SNPs, respectively23, so the estimate of variance explained by the SNPs from this regression analysis is ~0.51. The signal on the slope of the regression line comes from many points and is not due to a few outliers. Note that our maximum likelihood estimate is more accurate than this regression analysis; we show the latter only to illustrate the robustness of the estimate. In addition, we performed REML analysis using subsets of individuals by randomly splitting the whole sample into two and four groups and by sampling 1,000, 2,000 and 3,000 individuals with replacement for four replicates (Supplementary Fig. 4). The average estimates of variance explained by all SNPs are not affected by sample size, but, as expected, the sampling error increases as sample size decreases.
Figure 3.
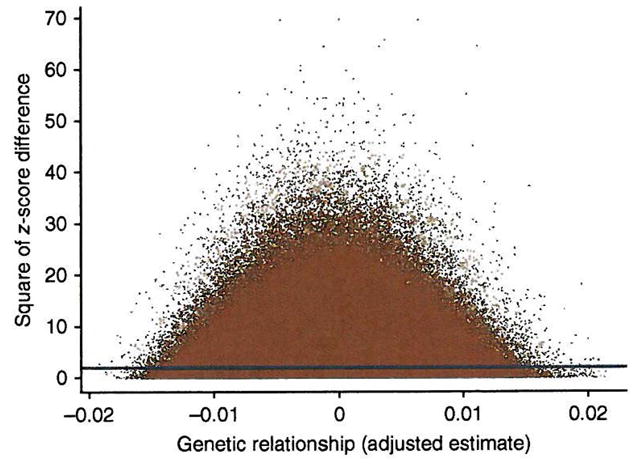
All pairwise comparisons contribute to the estimate of genetic variance. Shown are the squared z-score differences between individuals ( ) plotted against the adjusted estimates of genetic relationship ( ). The blue line is the linear regression line of on . The intercept and regression coefficient are estimates of twice the phenotypic variance and minus twice the genetic variances23, respectively. The intercept is 1.98 (s.e. = 0.001) and the regression coefficient is −1.01 (s.e = 0.27), consistent with estimates of the phenotypic and additive genetic variance of 0.990 and 0.505, respectively, and a proportion of variance explained by all SNPs of 0.51.
Heritability is the proportion of phenotypic variation due to additive genetic factors24; we therefore fitted a model in which SNPs have additive effects. Non-additive genetic variation and variation due to gene-environment interactions may exist, but they are not part of the missing heritability because they do not contribute to the heritability. Epigenetic mutations may cause resemblance between relatives and contribute to heritability if stably inherited, but in that case they would be equivalent to DNA sequence variants, would show LD with the assayed SNPs and would not contribute to missing heritability25.
The method we have presented could be misinterpreted as a method for estimating the heritability of height. Actually, we estimate the variance in height explained by the SNPs. We show that these SNPs do explain over half the estimated heritability of height and that the missing proportion can be explained by incomplete LD between the SNPs and causal variants.
If other complex traits in humans, including common diseases, have genetic architecture similar to that of height, then our results imply that larger GWASs will be needed to find individual SNPs that are significantly associated with these traits, because the variance typically explained by each SNP is so small. Even then, some of the genetic variance of the trait will be undetected because the genotyped SNPs are not in perfect LD with the causal variants. Deep resequencing studies are likely to uncover more polymorphisms, including causal variants that will be represented on future genotyping arrays. Our data provide strong evidence that the variation contributed by many of these causal variants is likely to be small and that very large sample sizes will be required to show that their individual effects are statistically significant. A similar conclusion was drawn recently for schizophrenia26. In some cases the small variance will be due to a large effect for a rare allele, but this will still require a large sample size to reach significance. Genome-wide approaches like those used in our study can advance our understanding of the nature of complex-trait variation and can be exploited for selection programs in agriculture27 and individual risk prediction in humans28.
ONLINE METHODS
Statistical framework
In a GWAS of a quantitative trait, we test for associations between individual SNPs and the trait by the following simple regression model,
| [1] |
where yj is the phenotypic value of the j-th individual; μ is the mean term; ai is the allele substitution effect of SNP i; xij is an indicator variable that takes value of 0, 1 or 2 if the genotype of the j-th individual at SNP i is bb, Bb or BB (alleles are arbitrarily called B or b), respectively; and ej is the residual effect, ., with σe2 being the residual variance.
Supposing that we could genotype subjects at the causal variants, we can include them all in the model
| [2] |
where gi is the total genetic effect of an individual j; m is the number of causal loci; ui is the scaled additive effect of the i-th causal variant; zij takes value of or if the genotype of the j-th individual at locus i is qq, Qq or QQ, respectively, with fi the frequency of Q allele at locus i (alleles are arbitrarily called Q or q)20,29; E(zij)= 0 and var(zij)= 1. In matrix notation, y=μ1+ g+ e and g= Zu. We treat u as random effects and assume , with σu2 being the variance of causal effects; then , where is variance of total additive genetic effects, and the variance-covariance matrix of y (the vector of observations) can be expressed as
| [3] |
where G is the genetic relationship matrix between pairs of individuals at causal loci. This equation shows the equivalence between the classical definition of heritability ( ) with σP2 being the phenotypic variance, and the proportion of phenotypic variance explained by the causal variants altogether.
In practice, we know little about the number and positions of the causal variants, so we are unable to obtain the G matrix directly. However, we can calculate the relationship from a genome-wide sample of SNPs (A) using the same formula as for G. That is.
| [4] |
where N is the number of SNPs and with pi the allele frequency at SNP i. This formula for A ignores the sampling error associated with each SNP. We can improve the estimate of A by calculating a weighted average across SNPs. For a SNP i, when j ≠ k (individuals j and k), ; in other words, it is the same for all SNPs regardless of allele frequency. When j = k, ; in other words, it is dependent on the allele frequency of the SNP. We therefore use the following equation to calculate Aij,
| [5] |
which provides an unbiased estimate of inbreeding coefficient (F) with mean of 1 + F, and has sampling variance of 1 when F = 0.
To obtain a genome-wide relationship, we combine Aijk for all of the SNPs using a common-sense weighting scheme,
| [6] |
Estimates of relationships are always relative to an arbitrary base population in which the average relationship is zero. We use the individuals in the sample as the base so that the average relationship between all pairs of individuals is zero and the average relationship of an individual with him/herself is 1.
Unbiased estimate of the relationship at the causal variants and the genetic variance
If we knew the genotypes at the causal variants, we could fit model [3] and estimate the genetic variance . Instead we will use a modified version (A*) of the relationship matrix based on the SNPs A. Although we will use REML to estimate , the requirements of A* to obtain an unbiased estimate of are more easily understood for the method illustrated in Fig. 3. In this method for each pair of subjects is regressed on Gjk. The slope of this regression is . If we replace Gjk by an estimate such that then , and the regression of on is still , so the estimate of remains the same −b/2. To obtain an unbiased estimate of Gjk with the required property, we use linear regression of Gjk on Ajk. We cannot calculate G, so instead we use one set of SNPs to mimic causal variants using the following steps:
Randomly sample 2N SNPs from all the SNPs across the genome and randomly split them into two groups (N SNPs in each group).
Calculate Ajk using all the SNPs in the first group.
Calculate Gjk using SNPs with MAF ≤ θ in the second group (mimicking the relationships at causal variants).
- Regress Gjk on Ajk for j ≤ k (use Gjk − 1 and Ajk − 1 when j = k). The regression coefficient is
[7] Repeat the procedure using different numbers of SNPs.
If the relationship at causal loci is predicted without error by the observed SNPs, β should equal one. When we applied this approach in our data, we found that for any of the MAF threshold θ, var(Ajk) is proportional to N whereas cov(Gjk, Ajk) is constant, irrespective of N (Supplementary Fig. 5). Consequently, we established an empirical linear relationship between β and the number of SNPs,
| [8] |
where c is constant for a certain MAF threshold θ —for example c = 6.2 × 10−6 when θ = 0.1 and c = 0 when θ = 0.5 (Fig. 1). The regression coefficient β is less than 1.0 because of two effects. First, the term in 1/N is due to the sampling error in estimating A from only N SNPs. This corresponds to the sampling error for Aijk at a single SNP calculated above as 1. If c = 0 and N = infinite, β = 1. In this case Ajk is the genomic relationship averaged over all positions in the genome. As the causal variants are a sample of such positions, Ajk is an unbiased estimated of Gjk. Second, the term in c occurs because the causal variants are not a random sample of all SNPs but a sample with low MAF. This causes the causal variants to have lower LD with the SNPs than random SNPs do with one another. Thus, even if Ajk was calculated from an infinite number of SNPs, it would still tend to overestimate the variance in relationships at the causal variants and consequently underestimate the genetic variance. We therefore adjust Ajk as
| [9] |
with the property of unbiasedness in the sense that .
Samples and genotyping
Height measurements, self-reported or clinically measured, from 35,189 Australian adults and 2,036 Australian adolescents (around 16-years-old) were collected by the Queensland Institute of Medical Research. Of these individuals, 8,884 adults and 1,668 adolescents have been genotyped using Illumina SNP chips in several genome-wide association studies. All the samples were collected with informed consent and appropriate ethical approval. The adult samples were genotyped by HumanCNV370-Quad v3.0 BeadChips (~351K SNPs) or Human610-Quad v1.0 BeadChips (~582K SNPs), and the adolescent samples were all genotyped by Human610-Quad v1.0 BeadChips.
We included only the genotyped individuals of European descent, as verified by ancestry analysis using genome-wide SNP data.30,31 We selected a set of 3,535 “unrelated” adults (1,421 males and 2,114 females; from 18 to 91 years old, with mean of 45) and 724 “unrelated” 16-year-old adolescents (354 males and 370 females), for a combined dataset of 4,259 “unrelated” individuals according to the pedigree information.
Quality control
We excluded SNPs in each individual dataset that had a mean GenCall Score < 0.7, missingness > 5%, a minor allele frequency (MAF) < 0.01 or a Hardy-Weinberg equilibrium (HWE) test P-value < 10−6, using PLINK32. A total of 304,013 SNPs in the adult dataset and 529,379 SNPs in the adolescent dataset passed this process, but only those in the autosomes were included in the analysis (295,400 SNPs for the adult dataset, 516,345 SNPs for the adolescent dataset and an intersect of 294,831 SNPs for the combined dataset).
We estimated the genetic relationships among all of the 4,259 individuals in the combined dataset by equation [6]. The estimated relationships (off-diagonal elements of the relationship matrix) ranged from −0.024 to 0.585, suggesting that some close relatives still remained. The mean of genetic relationships of “unrelated” individuals should be close to zero, so the lower-bound of the range can be roughly regarded as the maximum deviation of an estimate from the mean. We estimated the two-tailed 95% confidence interval of relationships (adjusted for multiple tests by Bonferroni correction) to be from about −0.027 to 0.027. Therefore, to avoid having any close relatives in the data, we chose a cut-off value of 0.025 and selectively excluded one of any pair of individuals with an estimated relationship > 0.025 to maximize the remaining sample size. We excluded 287 individuals from the adult dataset and 47 individuals from the adolescent dataset. A total of 3,248 “unrelated” adults and 677 “unrelated” adolescents, with a combined dataset of 3,925 “unrelated” individuals, was retained for analysis.
The phenotypes were corrected for age and sex, and standardized to z-scores in each adult and adolescent dataset separately. We used a two-tailed 90% Winsorisation33 to adjust the z-scores of four individuals in the adult dataset with absolute values greater than 4.17, the (100 – 5/3248)th percentile of the standard normal distribution based on Bonferroni correction, and combined the z-scores in both adult and adolescent datasets for the combined dataset of height (Supplementary Fig. 1e).
Supplementary Material
Acknowledgments
We are grateful to the twins and their families for their generous participation in these studies. We would like to thank staff at the Queensland Institute of Medical Research: Dixie Statham, Ann Eldridge and Marlene Grace for sample collection, Megan Campbell, Lisa Bowdler, Steven Crooks and staff of the Molecular Epidemiology Laboratory for sample processing and preparation, Belinda Cornes for height data preparation, David Smyth and Harry Beeby for IT support, and Allan McRae and Hong Lee for discussions. We thank Naomi Wray for helpful comments on the manuscript. We acknowledge funding from the Australian National Health and Medical Research Council (NHMRC grants 241944, 389875, 389891, 389892, 389938, 442915, 442981, 496739, 496688 and 552485), the U.S. National Institute of Health (grants AA07535, AA10248, AA014041, AA13320, AA13321, AA13326 and DA12854) and the Australian Research Council (ARC grant DP0770096).
Footnotes
AUTHOR CONTRIBUTIONS
P.M.V. and M.E.G. designed the study. J.Y. performed statistical analyses. B.B., B.P.M., A.K.H., D.R.N. and S.G. performed quality control analyses and prepared data. D.R.N., P.A.M., A.C.H. and N.G.M. contributed genotype and phenotype data. J.Y., G.W.M., M.E.G. and P.M.V. contributed to writing the paper.
References
- 1.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–31. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- 3.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 4.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 6.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–37. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johannes F, Colot V, Jansen RC. Epigenome dynamics: a quantitative genetics perspective. Nat Rev Genet. 2008;9:883–90. doi: 10.1038/nrg2467. [DOI] [PubMed] [Google Scholar]
- 8.Johannes F, et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009;5:e1000530. doi: 10.1371/journal.pgen.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Trans Roy Soc Edin. 1918;52:399–433. [Google Scholar]
- 10.Galton F. Hereditary stature. Nature. 1886;33:295–298. [Google Scholar]
- 11.Macgregor S, Cornes BK, Martin NG, Visscher PM. Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum Genet. 2006;120:571–80. doi: 10.1007/s00439-006-0240-z. [DOI] [PubMed] [Google Scholar]
- 12.Silventoinen K, et al. Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. 2003;6:399–408. doi: 10.1375/136905203770326402. [DOI] [PubMed] [Google Scholar]
- 13.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9:255–66. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 14.Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 15.Shiang R, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 16.Gudbjartsson DF, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–15. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- 17.Lettre G, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–91. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weedon MN, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Visscher PM. Sizing up human height variation. Nat Genet. 2008;40:489–90. doi: 10.1038/ng0508-489. [DOI] [PubMed] [Google Scholar]
- 20.Hayes BJ, Visscher PM, Goddard ME. Increased accuracy of artificial selection by using the realized relationship matrix. Genet Res. 2009;91:47–60. doi: 10.1017/S0016672308009981. [DOI] [PubMed] [Google Scholar]
- 21.Patterson HD, Thompson R. Recovery of inter-block information when block sizes are unequal. Biometrika. 1971;58:545–554. [Google Scholar]
- 22.Zhang XS, Hill WG. Predictions of patterns of response to artificial selection in lines derived from natural populations. Genetics. 2005;169:411–25. doi: 10.1534/genetics.104.032573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haseman JK, Elston RC. The investigation of linkage between a quantitative trait and a marker locus. Behav Genet. 1972;2:2–19. doi: 10.1007/BF01066731. [DOI] [PubMed] [Google Scholar]
- 24.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9:255–66. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 25.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–50. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–91. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- 28.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–8. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meuwissen TH, Solberg TR, Shepherd R, Woolliams JA. A fast algorithm for BayesB type of prediction of genome-wide estimates of genetic value. Genet Sel Evol. 2009;41:2. doi: 10.1186/1297-9686-41-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEvoy BP, et al. Geographical structure and differential natural selection among North European populations. Genome Res. 2009;19:804–14. doi: 10.1101/gr.083394.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price AL, et al. Principal components analysis corrects for stratification in genome- wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 32.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon WJ. Simplified Estimation from Censored Normal Samples. Ann Math Stat. 1960;31:385–391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.