

Abstract
Congenital disorders of glycosylation (CDG) are a group of clinically heterogeneous inborn errors of metabolism. At present, treatment is available for only one CDG, but potential treatments for the other CDG are on the horizon. It will be vitally important in clinical trials of such agents to have a clear understanding of both the natural history of CDG and the corresponding burden of disability suffered by patients. To date, no multicentre studies have attempted to document the natural history of CDG. This is in part due to the lack of a reliable assessment tool to score CDG’s diverse clinical spectrum. Based on our earlier experience evaluating disease progression in disorders of oxidative phosphorylation, we developed a practical and semi-quantitative rating scale for children with CDG. The Nijmegen Paediatric CDG Rating Scale (NPCRS) has been validated in 12 children, offering a tool to objectively monitor disease progression. We undertook a successful trial of the NPCRS with a collaboration of nine experienced physicians, using video records of physical and neurological examination of patients. The use of NPCRS can facilitate both longitudinal and natural history studies that will be essential for future interventions.
Electronic supplementary material
The online version of this article (doi:10.1007/s10545-011-9325-5) contains supplementary material, which is available to authorized users.
Introduction
Since the first report of patients diagnosed with congenital disorder of glycosylation (CDG) in 1980 (Jaeken et al. 1980), a large number of diverse CDG has been reported. CDG are multisystem disorders with highly variable phenotypes and consequently the clinical spectrum associated continues to expand. Almost any organ system can be affected in CDG, though the expression and severity of signs and symptoms can differ widely (Grünewald et al. 2009; Morava et al. 2009).
Due to the high variability in the phenotype, CDG can mimic other diseases such as mitochondrial disorders (Briones et al. 2001; Chi et al. 2010; Morava et al. 2006). Prediction of disease prognosis and assessment of potential therapeutic interventions depend on a comprehensive understanding of the natural history of a disease. To date, no multicentre study has attempted to prospectively document the natural history of CDG. This is in part due to the lack of a reliable assessment tool to score the diverse clinical spectrum. Several tools are available aimed at specific aspects of progressive diseases (WeeFIM for functional independence, PEDI and ICF surveys for disability, or PedSQL for quality of life), not however including multisystem aspects of the disease. Based on our earlier experience of evaluating disease progression in disorders of oxidative phosphorylation, we developed a practical and semi-quantitative rating scale for children with CDG .
A novel rating scale was developed for use in patients with CDG, to be able to monitor the natural history of CDG and the corresponding burden of disability suffered by patients. The overlapping clinical spectrum of CDG and mitochondrial diseases allowed adaptation of the mitochondrial disease rating scale, which has already been validated and successfully used in three metabolic paediatric centres (Newcastle, Nijmegen and Rome) (Phoenix et al. 2006). The Nijmegen Paediatric CDG Rating Scale (NPCRS) has been developed to offer a tool to objectively follow the clinical disease progression. The score avoids the diagnostic aspects and is designed to evaluate easily recognizable clinical signs, assessable by clinical examination, that occur frequently in CDG. The use of NPCRS will facilitate longitudinal natural history studies and will also be helpful in assessing the impact of future therapeutic interventions.
Methods
Development of NPCRS
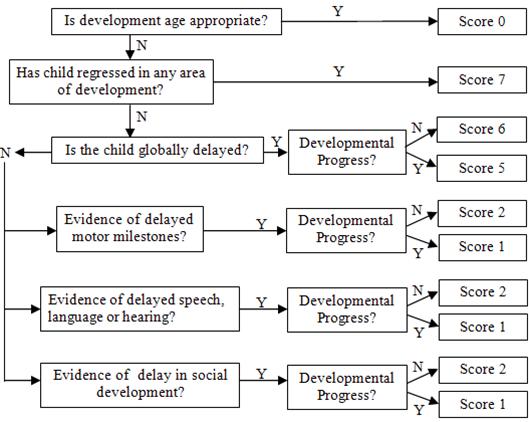
The Nijmegen Paediatric CDG Rating Scale (NPCRS) comprises a domain structure identical to the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS), which has already been successfully validated (Phoenix et al. 2006). Like NPMDS, the NPCRS is based around three domains, with each domain containing specific items relevant to the assessment of CDG patients. The three domains are as follows: Section I (Current Function) is a domain consisting of five questions regarding five general functions (vision, hearing, communication, feeding and mobility); Section II (System Specific Involvement) includes questions directly assessing CNS, blood, gastrointestinal, endocrine, respiratory, cardiovascular, renal and liver function over the preceding 6 months; Section III (Current Clinical Assessment) includes questions related to status. For each item, there are four possible responses reflecting normal (0), mild (1), moderate (2) and severe (3) impairment, with one exception in the form of a flow chart to assess developmental progress for which a score of 0–7 is possible (see Fig. 1 in the Electronic Supplementary Material). The NPCRS is subdivided into three age ranges, according to developmental phases, including infancy and early childhood (0–24 months), middle childhood (2–11 years) and adolescence (12–18 years).
Patients
We included 12 patients (mean age 8.7 years, SD: 5.6) who were regularly followed in our clinical centre. Patients’ parents or guardians provided informed consent for participation. Subsequently we collected biochemical and genetic information of these patients, and the findings are shown in Table 1.
Table 1.
Clinical characteristics, biochemical and genetic data on patients 1–12
| Patient (M/F) | Age (years) | Clinical features | Diagnosis | Mutations |
|---|---|---|---|---|
| 1 M (Drijvers et al. 2010) | 0.8 | Psychomotor retardation, failure to thrive, protein-losing enteropathy, epilepsy | ALG6-CDG | p.A333V (c.998 C > T); del exon 3 (c.257 + 5 G > A) |
| 2 F | 1 | Psychomotor retardation, failure to thrive, hypoglycemia, ataxia | PMM2-CDG | hom. p.F119L (c.357 C > A) |
| 3 F (Morava et al. 2010a) | 5 | Glaucoma, psychomotor retardation, abnormal liverfunction, hypotonia, inverted nipples | SRD5A3-CDG | p.S10x (c.29 C > A) |
| 4 F (Morava et al. 2008) | 10 | Cutis laxa, failure to thrive, psychomotor retardation, myopathy | ATP6V0A2-CDG | hom. p.R63x (c.187 C > T) |
| 5 F (Morava et al. 2009) | 11 | Psychomotor retardation, failure to thrive, diarrhea, CMP, ataxia, hypotonia | PMM2-CDG | p.R141H (c.422 G > A); p.F119 L (c.357 C > A) |
| 6 M | 5 | Positive family history, elevated transaminases, cholestasis | CDG-IIx | Unknown |
| 7 F | 8 | Liver failure, psychomotor retardation, clotting abnormalities | CDG-IIx | Unknown |
| 8 F | 3 | Strabismus, hypotonia, psychomotor retardation | PMM2-CDG | p.F119L (c.357 C > A); p.E151G (c.452A > G) |
| 9 M | 18 | Delayed motor development | CDG-Ix | Unknown |
| 10 M | 16 | Short stature, diarrhoea, hypoglycemia | CDG-Ix | Unknown |
| 11 M | 15 | Psychomotor retardation, ataxia | PMM2-CDG | p.R123G (c.324 G > A); p.R162W (c.484 T > C) |
| 12 F (Hutchagowder 2009) | 11 | Cutis laxa, growth delay, psychomotor retardation | ATP6V0A2-CDG | p.P405L (c.31908 C > T); p.R5101 (c.32579 G > T) |
Each patient was examined independently by four different investigators, on the same day in the same hospital, or in different settings within a 1 week period. The investigators rated the patients current clinical status by performing a physical examination.
Collaborative trial
We further trialed the use of the NPCRS with the collaboration of nine experienced physicians (European Metabolic Group conference, Lissabon, 2010). Clinical history, detailed biochemical information and video records of the physical examination of two patients with SRD5A3-CDG (Morava et al. 2010b) demonstrated neurological involvement (mental retardation, hypotonia, ataxia, dystonia, and visual loss) but no internal organ involvement. A 15 min video of the clinical presentation and neurological examination was reviewed independently after which the participants were allowed to request additional information, as would occur during a clinical visit. The age-appropriate version of NPCRS was used to assess the disease severity in both patients and the results discussed at the workshop.
Statistics, validation/inter-rater validation
The Kappa test of agreement was used for the measure of agreement (excess of agreement on categorical data, such as mild, moderate and severe, is expected owing to chance and sample size). This was calculated with the observed agreement (ObsA) and the agreement expected by chance (ExpA) using the following formula: [ObsA − ExpA/1 − ExpA ]. Kappa scores were classified as follows: <0.20 (poor), 0.21–0.40 (fair), 0.41–0.60 (moderate), 0.61–0.80 (good) and 0.81–1.00 (very good).
Results
Development of the NPCRS
We included only the most common symptoms of CDG patients in our rating scale corresponding with the literature. We excluded rare and unique symptoms due to their lack of discriminatory power of ‘severity’. We have made the assumption that in performing a detailed examination of all 12 patients we have not missed any clinical features. We found application of the scale to be straightforward, taking approximately 15 min for each patient. Parents and patients (where possible) found the questions to be clear, and the investigators did not encounter any particular difficulties in applying the rating scale to patients.
Patients
Our patient group consisted of five male and seven female patients ranging from 8 months to 19 years of age. There was a great diversity in the phenotypic spectrum of the different patients (see Table 1). Some of the patients have been described earlier (Morava et al. 2008, 2010b; Drijvers et al. 2010; Hutchagowder et al. 2009)
Inter-rater variation
The total scores assigned by each clinician to each of the 12 patients are shown in Table 1 in the Electronic Supplementary Material. These scores are used to scale patients into a mild (0–14), moderate (15–25) and severe ( >26) category. There was very little inter-rater variation regarding the categories into which the nine experienced clinicians placed the patients, based on video observation (see domain III in Table 2 in the Electronic Supplementary Material). All inter-rater comparisons were good or very good. In domains I and II there were no differences between the rates (Kappa value very good), and in domain III the Kappa values were either good or very good. There was no significant inter-rater variation and no significant difference regarding the final score.
Discussion
Most of the CDG types commonly present with a severe clinical phenotype in the first years of life, affecting multiple organ systems including the central nervous system. Those who survive this crucial period will stabilise and show a relatively steady clinical picture by puberty (Truin et al. 2008; Morava et al. 2008; Cantagrel et al. 2010; Drijvers et al. 2010). One of the most important aspects of CDG relates to how the disease will affect the patients and their families in the future. We have been unable to accurately answer questions regarding prognosis due to lack of a comprehensive and effective tool to objectively document the natural history of the disease.
A similar problem was faced by clinicians managing patients diagnosed with mitochondrial disease, a group that shares many clinical features with CDG patients. Based on our positive experience of using the recently developed Newcastle Paediatric Mitochondrial Disease Scale (NPMDS, Newcastle, Rome and Nijmegen) in regular clinical practice (Morava et al. 2010a; Koene et al. 2009), we decided to adapt this existing scale and to validate it in this group of CDG patients. Therefore, we have collaborated with multiple experienced clinicians who regularly follow patients with CDG.
One of the major challenges in designing a scale for CDG patients has been addressing the severity of clinical features, especially neurological symptoms. Another aspect is the need to be comprehensive, e.g., many of the clinical features are either not apparent at diagnosis or are found in only a minority of patients with that particular type of disorder. A good example is sensorineural deafness, a very rare symptom, also difficult to assess in the clinical outpatient setting and follow progression based on physical examination in CDG patients (Jaeken et al. 2009).
We additionally reviewed all clinical features of the patients included in the validation process, and we have not found any clinical symptoms not included on our clinical scale. Although our patient cohort was small, their phenotype was representative for the various types of CDG. We concluded that the scale is a comprehensive tool for assessing the disease progression in CDG patients. In addition to being comprehensive the scale had to be easy and rapid to use and outpatient clinic–friendly.
Similar to the original NPMDS (Phoenix et al. 2006), the NPCRS has three key domains which comprise key questions relating to functional ability. These clinical criteria are sometimes difficult to define without performing additional studies (objective measurements of strength, EMG, nerve conduction studies), especially in the case of a mild to moderate myopathy or neuropathy. This problem was also encountered during the validation and use of the mitochondrial disease scale.
The NPCRS is designed to be applied at the outpatient visits each 6 months or at least annually throughout the follow-up period for individual CDG patients. During this period patients may be assessed by a number of different paediatricians and therefore the consistency of the scoring system is very important. In the current validation process we found no substantial inter-rater variability, but a very good level of inter-rater agreement in our relatively small, but phenotypically heterogeneous, cohort of paediatric patients with CDG (Morava et al. 2008; Hucthagowder et al. 2009).
Our collaborative study showed no significant inter-rater variability and no differences between the total scores recorded, further demonstrating the reliability of the CDG rating scale. Based on those findings we recommend applying the NPCRS both to facilitate disease severity assessment and to study longitudinal natural history. In addition, we believe that the NPCRS will be an important tool in assessing the clinical impact of future therapeutic interventions.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
NPCRS total scores (including sections I, II and III) of examiners (A–D) for each patient (DOC 16 kb)
Sub-scoring of examiners (A–I) for all symptoms within section III in one of the patients with homozygous SRD5A3 mutations based on video recording of the physical examination (patient 1 in Morava et al. 2010a) (DOC 18 kb)
{kind=link}
Developmental flowchart (JPEG 43 kb)
0–24 months (DOC 98 kb)
2–11 years (DOC 91.5 kb)
12–18 years (DOC 90.5 kb)
Acknowledgements
The authors are thankful for the subsidy granted to the corresponding author in 2010/2011 by “Metakids”.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Competing interest: None declared.
S. Achouitar and M. Mohamed contributed equally.
References
- Briones P, Vilaseca MA, Garcia-Silva MT, et al. Congenital disorders of glycosylation (CDG) may be underdiagnosed when mimicking mitochondrial disease. Eur J Paediatr Neurol. 2001;5:127–131. doi: 10.1053/ejpn.2001.0483. [DOI] [PubMed] [Google Scholar]
- Cantagrel V, Lefeber DJ, Ng BG, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142:203–17. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi C-S, Lee H-F, Tsai C-R, et al. Clinical manifestations in children with mitochondrial diseases. Pediatr Neurol. 2010;43:183–189. doi: 10.1016/j.pediatrneurol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Drijvers JM, Lefeber DJ, de Munnik SA, et al. Skeletal dysplasia with brachytelephalangy in a patient with a congenital disorder of glycosylation due to ALG6 gene mutations. Clin Genet. 2010;77:507–9. doi: 10.1111/j.1399-0004.2009.01349.x. [DOI] [PubMed] [Google Scholar]
- Grünewald S. The clinical spectrum of phosphomannomutase 2 deficiency. Biochim Biophys Acta. 2009;1792:827–834. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hucthagowder V, Morava E, Kornak U, et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–65. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken J, Vanderschueren-Lodewyckx M, Caeaer P, et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG deficiency, increased serum arysulphatase A and increased CSF protein: a new syndrome? Pediatr Res. 1980;14:179. doi: 10.1203/00006450-198002000-00117. [DOI] [Google Scholar]
- Jaeken J, Vleugels W, Régal L, et al. (2009) RFT1-CDG: deafness as a novel feature of congenital disorders of glycosylation. J Inherit Metab Dis. doi:10.1007/s10545-009-1297-3 [DOI] [PubMed]
- Koene S, Kozicz TL, Rodenburg RJ, et al. Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J Affect Disord. 2009;114:327–32. doi: 10.1016/j.jad.2008.06.023. [DOI] [PubMed] [Google Scholar]
- Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–6. doi: 10.1212/01.wnl.0000244435.27645.54. [DOI] [PubMed] [Google Scholar]
- Morava E, Lefeber DJ, Urban Z, et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur J Hum Genet. 2008;16:28–35. doi: 10.1038/sj.ejhg.5201947. [DOI] [PubMed] [Google Scholar]
- Morava E, Wosik HN, Sykut-Cegielska J, Adamowicz M, et al. Ophthalmological abnormalities in children with congenital disorders of glycosylation type I. Br J Ophthalmol. 2009;93:350–4. doi: 10.1136/bjo.2008.145359. [DOI] [PubMed] [Google Scholar]
- Morava E, Gardeitchik T, Kozicz T, et al. Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion. 2010;10:528–33. doi: 10.1016/j.mito.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Morava E, Wevers RA, Cantagrel V, et al. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010;133(11):3210–20. doi: 10.1093/brain/awq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix C, Schaefer AM, Morava E, et al. A scale to monitor progression and treatment of mitochondrial disease in children. Neuromuscul Dis. 2006;16:814–820. doi: 10.1016/j.nmd.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Truin G, Guillard M, Lefeber DJ, et al. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab. 2008;94:481–4. doi: 10.1016/j.ymgme.2008.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NPCRS total scores (including sections I, II and III) of examiners (A–D) for each patient (DOC 16 kb)
Sub-scoring of examiners (A–I) for all symptoms within section III in one of the patients with homozygous SRD5A3 mutations based on video recording of the physical examination (patient 1 in Morava et al. 2010a) (DOC 18 kb)
Developmental flowchart (JPEG 43 kb)
0–24 months (DOC 98 kb)
2–11 years (DOC 91.5 kb)
12–18 years (DOC 90.5 kb)