

Abstract
Subgroups of patients with oral pre-malignant lesions (OPLs) are at extremely high risk for developing invasive cancer in spite of surgical excision. The objective of this study was to evaluate the utility of specific genes and their associated centromeres as markers to stratify OPLs for their cancer risk. Samples used in this study included 35 oral dysplasia with known outcome and 20 normal oral mucosa. Of the dysplasias, 20 were from an ongoing longitudinal study showing progression. The remaining 15 cases (2 of which progressed) were chosen from the population-based, provincial BC Oral Biopsy Service (OBS). Copy number alterations at EGFR, CEP7, CCND1, and CEP11 were evaluated by fluorescent in situ hybridization (FISH). There was no significant difference in demographics between progressors and non-progressors. Specific FISH profiles at these genes and their corresponding centromeres were associated with progression. High gene gain of CCND1 was associated with an 8-fold elevated risk of progression compared with those with no gain in time-to-progression analysis. Numerical alterations of EGFR and CCND1 and their centromeres might be an effective means for identifying OPLs at risk. Future studies will expand on this analysis and set the stage for application of this approach in routine clinical practice.
Keywords: oral dysplasia, progression, EGFR, CCND1, fluorescent in situ hybridization, chromosomal instability
Introduction
Oral squamous cell carcinoma (SCC) is the 6th most common cancer in the world (Warnakulasuriya, 2009). It is believed to progress through various stages of oral pre-malignant lesions (OPL), with or without dysplasia, to invasive cancer. Once cancer has developed, prognosis is poor, with 5-year survival rates of ~ 50% (Epstein et al., 2008). Even when OPLs are successfully treated, survivors frequently have serious cosmetic and/or functional compromise. These dismal mortality rates accentuate the need to prevent the formation of invasive lesions through early diagnosis and effective treatment of OPLs.
The current ‘gold standard’ in OPL risk assessment is the presence and degree of dysplasia. However, the determination of progression risk in OPLs with no or low-grade (mild/moderate) dysplasia remains challenging, since the majority of these lesions will not progress (Waldron and Shafer, 1975; Schepman et al., 1998). This inability to define aggressive behavior in OPLs underscores the need for markers predicting this behavior. Advances in molecular technologies have provided useful tools in developing markers to characterize "high-risk" OPLs (Mao et al., 1994; Rosin et al., 2000; Garnis et al., 2009; Tsui et al., 2009).
Previously, we applied whole-genome analysis to a panel of high-grade lesions and identified chromosomal loci 7p11.2 and 11q13 as the most frequently occurring regions of amplification (Tsui et al., 2009). The 7p11.2 region spans EGFR, the well-characterized tyrosine kinase that has been found to be over-expressed in a number of cancers, including oral cancers (Garnis et al., 2004, 2009; Temam et al., 2007; Ryott et al., 2009; Sheu et al., 2009; Tsui et al., 2009). EGFR is currently viewed as a promising molecular target for cancer therapy and chemoprevention, with multiple inhibitory strategies being developed that will target either the receptor itself or its downstream signaling pathway (Langer, 2008; Egloff and Grandis, 2009). Over-expression of EGFR has been found in a wide variety of solid tumors and may relate to poor prognosis. Over-expression of Cyclin D1 (CCND1) – which maps to the chromosome 11q13 region – has been well-documented in oral cancer and has been reported to be associated with disease progression and poor prognosis (Michalides et al., 1997; Kuo et al., 1999; Shintani et al., 2002). Whole-genome analysis of tumors also reported high frequency of amplification for both EGFR and CCND1 in oral cancers, indicating the ongoing importance of these alterations in later-stage disease (Garnis et al., 2004). Numerical aberration of the CCND1 gene has been linked to failure of regional control with neck metastasis in early-staged oral SCC (Myo et al., 2005).
Chromosomal instability (CIN), resulting in gains and losses of parts of or whole chromosomes, has been recognized as one of the critical driving forces for carcinogenesis (Weaver and Cleveland, 2006). Imbalance of chromosome copy number is responsible for large-scale gene dysregulation and is observed in various cancers, including oral cancers (Squire et al., 2002). In tumors, FISH-detected chromosomal instability in cells at resection margins has been reported as a strong indicator of recurrence (Bergshoeff et al., 2008).
In this study, we investigated the chromosome imbalances and the numerical alterations of specific genes in OPLs and the association of these changes with disease progression following surgical intervention. Analyses were undertaken on a set of invaluable OPLs with extensive clinical follow-up, including rigorously collected temporal clinicopathological data. Alterations identified as significantly associated with aggressiveness by this analysis could have immediate clinical utility, since they were evaluated by an established clinical technique.
Materials & Methods
Sample Selection
Fifty-five archival tissue blocks from patients were collected from the province-wide population-based BCOBS, including 35 dysplasia with no prior cancer history and 20 normal oral mucosa. The histological diagnoses were determined by study pathologists (LZ, KWB) using criteria from the World Health Organization (Axéll et al., 1996). Among patients seen in the ongoing longitudinal study in 2007, 20 cases progressed into carcinoma in situ or invasive squamous cell carcinoma. The initial biopsies showing dysplasia that had enough tissue left on the tissue block were used for analysis. The remaining 15 dysplasia cases were randomly chosen from the BCOBS to match the former group by the year of the initial biopsy. When cross-checked with the pathology database and the BC Cancer Agency Registry, where all cancer cases in the province are documented, two of these cases were identified as showing progression. As a control, we also included 20 normal oral mucosa samples (amalgam tattoo with minimal inflammation). This study was approved by the Research Ethics Board of the University of British Columbia.
Fluorescent in situ Hybridization (FISH) and Scoring
The protocol used was a modification of Romeo et al. (2003) with a lower concentration of pepsin (0.032%) and a longer digestion time (80-90 min) (Tsui et al., 2009). In brief, 4-µm sections on charged slides were incubated in 2X SSC at 37°C for 30 min, dehydrated in ethanol series, and incubated in 70% glacial acetic acid for 1 min. The specimens were then digested in 0.032% pepsin at 37°C for 80 to 90 min, and fixed in 1% neutral buffered formaldehyde at room temperature for 10 min. Regions for analysis were selected based on our previous analysis of genomic alterations in oral tumors and a review of the literature, which indicated that these are significantly altered regions in oral cancer (Garnis et al., 2004). Two sets of dual-colored probes (Vysis, Downers Grove, IL, USA) were performed on the sequential sections according to the manufacturer’s instructions, which included the pairs of CEP7 (7p11.1-q11.1, SpectrumGreen)/EGFR (7p12, SpectrumOrange) and CEP11 (centromere 11p11.11-q11, SpectrumGreen)/CCND1 (11q13, SpectrumOrange). The probe set was applied, and the hybridization area was sealed and co-denatured at 80°C for 8 min, followed by incubation at 37°C for 24 hrs in a humidified chamber. Post-hybridization washes were performed consecutively in 50% formamide/2X SSC, 2X SSC, and 2X SSC/0.1% NP-40, each at 46°C for 6 min. DAPI in Vectashield antifade was applied as chromatin counterstain.
Signals were captured and imaged with Olympus BX61 and ImagePro Plus 5.1. At least 200 non-overlapping intact nuclei were scored. Samples with > 90% nuclei showing signals were considered informative.
The sample was scored and classified according to the frequency of nuclei with specific numbers of copies of the chromosome centromeres or genes, in a protocol modified from Hirsch et al. (2003) and Cappuzzo et al. (2005): for gene dosage change, (1) no gain, ≤ 2 copies of gene in >90% of nuclei, (2) low gain, 3 copies of gene in ≥ 10% and ≥ 4 copies of gene in < 10% of nuclei, and (3) high gain, ≥ 4 copies of gene in > 10% of nuclei, a ratio of gene to centromere ≥ 2, or ≥ 15 copies of gene per nuclei in ≥ 10% of nuclei analyzed; and for numerical change of centromere, (1) disomy, ≤ 2 copies in > 90% of nuclei), (2) trisomy, 3 copies in > 10% and ≥ 4 copies < 10% of nuclei, and (3) polysomy, ≥ 4 copies in > 10% of nuclei analyzed.
Statistical Analyses
Differences and associations between progressors (N = 22) and non-progressors (N = 13) to carcinoma in situ or invasive squamous cell carcinoma and clinical parameters were examined by either Fisher’s exact test for categorical variables or unpaired t tests for continuous variables. Correspondence analyses were used to produce 2-dimensional displays of similarities of relative frequency among groups, by SAS9.1.2. Time-to-progression curves were estimated by the Kaplan-Meier method, and comparisons were performed by log-rank tests. Relative risks were determined by Cox regression analysis. All tests were two-sided. Any p < 0.05 was considered to be statistically significant.
Results
Characteristics of Progressors and Non-progressors
There were no differences in gender, age, and smoking status between the 2 groups (Appendix Table 1). Lesions that progressed were more commonly located at high-risk anatomical sites (i.e., those from the lateroventral tongue, floor of mouth, or soft palate, p = 0.05). The average time to outcome or last follow-up was 36 ± 19 mos. When follow-up times were compared based on progression status, non-progressors had twice the length of follow-up time compared with progressors (52 ± 11 and 26 ± 15 mos, p < 0.0001).
Gene Dosage Change and Clinical Outcome
While absence of EGFR gain was more commonly associated with non-progressors (84%) and normal oral tissue (100%), 82% of progressors showed either low or high gene gain (p < 0.0001). A similar trend was observed for CCND1 (p < 0.0001). The increases in EGFR and CCND1 gene dosage (i.e., low or high gene gain) were significantly associated with progression (p = 0.0006 and 0.0002, respectively). The frequency of high gene gain of CCND1 was significantly higher in progressors than in non-progressors (64% vs. 15%, p = 0.01). Any level of EGFR gain was strongly associated with progressors (p = 0.0002). One progressor in each group showed clumps of gene amplifications for EGFR and CCND1, with ~20-30 gene signals clumped together (see Fig. 1).
Figure 1.

Example of histology and FISH analysis on a progressor. (a) The histomicrograph shows a moderate epithelial dysplasia with a band-like lymphohistiocytic inflammatory cell infiltrate in the subtending connective tissue (H&E, original magnification, 200X). (b) FISH analysis with dual-color probe set (Vysis, Downers Grove, IL, USA) consisting of a centromeric probe for chromosome 7 (CEP7; SpectrumGreen) and a locus-specific probe for EGFR (7p12; SpectrumOrange). The blue line represents the basement membrane separating the epithelium (superior left half) and connective tissue (inferior right half) (original magnification, 600X). Clustered high gene gain signals (orange clumps) with normal CEP7 disomy (green signals) were noted in the epithelium [higher magnification of a dysplastic nucleus in (c), 1000X], and 2:2 signals of EGFR gene (orange signals) and CEP7 (green signals) were noted in the connective tissue [higher magnification of an inflammatory cell in (d), 1000X].
The association of gene dosage change and clinical aggressive behavior can be easily visualized by correspondence analysis. High gene gain of CCND1 was located very close to the progressors in a 2-dimensional display, demonstrating the similarity of their relative frequencies (Fig. 2a). Any level of EGFR gain was even more strongly tied to the progressors (Fig. 2b).
Figure 2.

Correspondence analysis of gene gains of CCND1 and EGFR among progressor, non-progressor, and normal groups. (a) A 2-dimensional display indicated that high gene gain of CCND1 showed a close distance to 3, the progressors, indicating the similarity of their patterns of relative frequencies. (b) A 2-dimensional display indicated that any gene gain of EGFR showed an even closer distance to 3, the progressors (3): (1) normal, (2) non-progressors, (3) progressors; no gain = ≤ 2 copies of gene in > 90% of nuclei; low gain = 3 copies of gene in ≥ 10% and ≥ 4 copies of gene in < 10% of nuclei; high gain = ≥ 4 copies of gene in > 10% of nuclei, a ratio of gene to centromere ≥ 2, or ≥ 15 copies of gene per nuclei in ≥ 10% of nuclei scored.
Chromosomal Instability with the Presence of Trisomy and Polysomy and Outcome
When centromere number was analyzed, trisomy or polysomy was more frequently observed in the progressors (p < 0.0001 in CEP7 and CEP11, Table) than in the non-progressors and normal mucosa. When the diploid status and outcome were considered, the non-progressors were more commonly disomy for both CEP7 (84%) and CEP11 (77%) compared with the progressors (32%, p = 0.005; 23%, p = 0.004, respectively). This lends further weight to the idea that chromosomal instability might be one of the driving forces for oral carcinogenesis.
Table.
| Progressor | Non-progressor | Normal | p | |||||
|---|---|---|---|---|---|---|---|---|
| Case number | 22 | 13 | 20 | All groups | Progressor vs. Non-progressor | Progressor vs. Normal | Non-progressor vs. Normal | |
| EGFR | ||||||||
| No gain (%) | 4 (18%) | 11 (84%) | 20 (100%) | < 0.0001 | 0.0006 | < 0.0001 | 0.19 | |
| Low gain (%) | 13 (59%) | 1 (8%) | 0 | |||||
| High gain (%) | 5 (23%) | 1 (8%) | 0 | |||||
| CCND1 | ||||||||
| No gain (%) | 1 (4%) | 9 (70%) | 20 (100%) | < 0.0001 | 0.0002 | < 0.0001 | 0.03 | |
| Low gain (%) | 7 (32%) | 2 (15%) | 0 | |||||
| High gain (%) | 14 (64%) | 2 (15%) | 0 | |||||
| CEP7 | ||||||||
| Disomy (%) | 7 (32%) | 11 (84%) | 20 (100%) | < 0.0001 | 0.009 | < 0.0001 | 0.19 | |
| Trisomy (%) | 12 (54%) | 1 (8%) | 0 | |||||
| Polysomy (%) | 3 (14%) | 1 (8%) | 0 | |||||
| CEP11 | ||||||||
| Disomy (%) | 5 (23%) | 10 (77%) | 20 (100%) | < 0.0001 | 0.007 | < 0.0001 | 0.08 | |
| Trisomy (%) | 9 (41%) | 1 (8%) | 0 | |||||
| Polysomy (%) | 8 (36%) | 2 (15%) | 0 | |||||
No gain: gene signal ≤ 2 in > 90% of nuclei counted; low gain: gene signal ≥ 3 in ≥ 10% nuclei counted and gene signal ≥ 4 in < 10% nuclei counted; high gain: gene signal ≥ 4 in ≥ 10% of nuclei counted or Gene/CEP ratio ≥ 2 or tight clusters of gene signal ≥ 15 in ≥ 10% of nuclei counted.
Disomy: centermere signal ≤ 2 in > 90% of nuclei counted; Trisomy: centremere signal =3 in ≥ 10% and centremere signal ≥ 4 in < 10% nuclei counted; Polysomy: centremere signal ≥ 4 in ≥ 10% of nuclei counted.
Time-to-Progression Risk
Specific numerical change in gene dosage for EGFR and CCND1, and the centromeres of chromosomes 7 and 11, were examined for their association with disease progression by the Kaplan-Meier method. Time-to progression curves were plotted as a function of EGFR gain, CCND1 gain, and the frequency of disomy, trisomy, and polysomy of CEP7 and CEP11. When these 4 markers were considered independently, gene gains were significantly associated with time to progression for EGFR (p = 0.004) and CCND1 (p = 0.002; Fig. 3), but not with changes in CEP7 and CEP11. When EGFR and CCND1 were considered together in a Cox proportional hazards model, CCND1 appeared to be significantly associated with time to progression (p = 0.05, adjusted for EGFR) but not EGFR (p = 0.2, adjusted for CCND1). This is possibly due to some association/interaction between CCND1 and EGFR. Using a logistic regression model to evaluate the performance of gene gain at CCDN1, EGFR, CCDN1+EGFR, and the interactive term of CCDN1+EGFR for their sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and the area under curve (AUC), we found that all models performed well according to these criteria (Appendix Table 2). The model with the interaction term of CCND1 and EGFR as predictor fitted the data best (AUC = 0.95). Furthermore, the cancer risk of high gene gain of CCND1 is estimated to be eight times that of no gain, and the cancer risk of low gene gain of CCND1 is estimated to be three times that of no gain, both adjusted for EGFR. The frequencies of disomy, trisomy, and polysomy of CEP7 and CEP11 did not seem to be significantly associated with time to progression.
Figure 3.
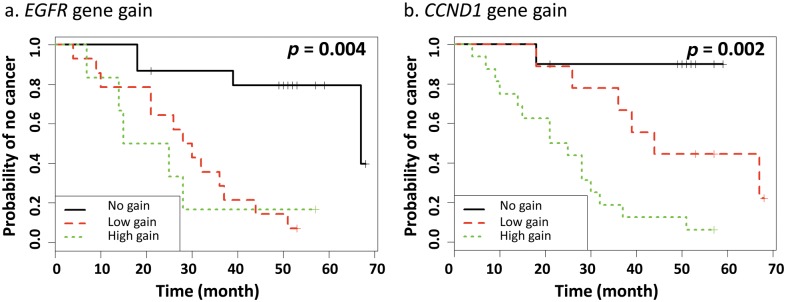
Probability of cancer progression according to gene gains of EGFR (a) and CCND1 (b). Time-to-progression curves were estimated by the Kaplan-Meier method, and the comparisons were performed by log-rank tests (black line for no gain, ≤ 2 copies of gene in > 90% of nuclei; red dash line for low gain, 3 copies of gene in ≥ 10% and ≥ 4 copies of gene in < 10% of nuclei; and green dotted line for high gain, ≥ 4 copies of gene in > 10% of nuclei, a ratio of gene to centromere ≥ 2, or ≥ 15 copies of gene per nuclei in ≥ 10% of nuclei analyzed).
Discussion
There are two main challenges for the development of effective markers for risk prediction in OPLs. One challenge surrounds the rarity of acquiring lesions for analysis, particularly lesions for which outcome information has been accrued. The other challenge has been deriving biomarkers that can be easily adopted for use in a standard histology laboratory.
In British Columbia, we have addressed the first challenge by making use of cases included in an ongoing longitudinal study since 1998. This unique cohort is made possible by our centralized population-based OBS (~ 60,000 archived cases in over 30 years’ history).
To address the second challenge described above, we have restricted our molecular assays to those already in routine use in the clinic. Our application of FISH in the present work could be readily translated for use in a clinical lab setting, since this approach has already been standardized for other genes and in other tissues at many pathology laboratories. Probes used to detect EGFR/CEP7 and CCND1/CEP11 are currently commercially available.
Amplifications of EGFR and CCND1 Are Associated with Aggressive Clinical Behavior
EGFR activation has previously been implicated in several cancer types, including oral cancer (Grandis, 2006; Karamouzis et al., 2007). It has been demonstrated to play a role in gene transcription, cell proliferation, angiogenesis, apoptosis inhibition, and tissue invasion. Activation of CCND1 has also been implicated in tumorigenesis and contributes to oral tumor phenotypes (Todd et al., 2002; Brinkman and Wong, 2006). Analysis of our data indicates that increased gene gain of EGFR and CCND1 is a recurring event in OPLs, confirming findings from earlier whole-genome analyses performed by our group (Garnis et al., 2004; Tsui et al., 2009).
More specifically, we found that any level gains of EGFR and high level of gene gain of CCND1 were strongly associated with progression. High gene gain of CCND1 was associated with an approximate eight-fold elevated risk of progression after adjustment for any gain of EGFR. These findings are intuitive, since both EGFR and CCND1 govern a wide array of critical cancer cell processes.
Chromosomal Instability and Its Associated Outcome
We observed significant differences in numerical changes for CEP7 and CEP11 among progressors, non-progressors, and normal tissues, indicating that chromosomal instability might play a role during oral carcinogenesis. Single chromosomal imbalances have previously been proposed as a mechanism for simultaneous disruption of several genes critical for tumorigenesis (Weaver and Cleveland, 2006). The presence of this kind of broad alteration in lesion cells that linger after surgery – or in histologically ‘normal’ tissue adjacent to resection margins – could serve as a driver for disease recurrence and possibly progression. The finding that greater genomic instability exists in the progressors agrees well with previous reports demonstrating that increased genomic instability is also a marker for progression risk in oral lesions (Garnis et al., 2009; Torres-Rendon et al., 2009).
Taken together, our findings may have immediate clinical impact. FISH-based analysis is a common diagnostic set-up for many pathology laboratories, and tissue requirements are minimal (a 4-µm section); hence our findings could be readily translated into clinical patient care.
In conclusion, we have shown that the presence of polysomy and gene amplification at specific genomic loci is significantly associated with progression in OPLs. Validation of these results in a larger sample set is required and is ongoing. Widespread application of this approach will stratify OPLs, identifying high-risk lesions requiring more aggressive treatment (and justifying the increased toxicity and morbidity risks associated with these treatments).
Supplementary Material
Acknowledgments
The authors acknowledge Dr. J.J. Lee for guidance in data analysis and Dr. Buys for manuscript editing.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
This work was supported by the National Institute of Dental and Craniofacial Research/National Institutes of Health (R01DE17013), Canadian Institutes of Health Research (MOP-77663), and the Canadian Cancer Society (CCS-20336). CFP is supported by a Scholar Award from the Michael Smith Foundation for Health Research.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Axéll T, Pindborg JJ, Smith CJ, van der Waal I. (1996). Oral white lesions with special reference to precancerous and tobacco-related lesions: conclusions of an international symposium held in Uppsala, Sweden, May 18-21 1994. International Collaborative Group on Oral White Lesions. J Oral Pathol Med 25:49-54. [DOI] [PubMed] [Google Scholar]
- Bergshoeff VE, Hopman AH, Zwijnenberg IR, Ramaekers FC, Bot FJ, Kremer B, et al. (2008). Chromosome instability in resection margins predicts recurrence of oral squamous cell carcinoma. J Pathol 215:347-348. [DOI] [PubMed] [Google Scholar]
- Brinkman BM, Wong DT. (2006). Disease mechanism and biomarkers of oral squamous cell carcinoma. Curr Opin Oncol 18:228-233. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F, Varella-Garcia M, Shigematsu H, Domenichini I, Bartolini S, Ceresoli GL, et al. (2005). Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol 23:5007-5018. [DOI] [PubMed] [Google Scholar]
- Egloff AM, Grandis JR. (2009). Improving response rates to EGFR-targeted therapies for head and neck squamous cell carcinoma: candidate predictive biomarkers and combination treatment with Src inhibitors. J Oncol 2009:896407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JB, Gorsky M, Cabay RJ, Day T, Gonsalves W. (2008). Screening for and diagnosis of oral premalignant lesions and oropharyngeal squamous cell carcinoma: role of primary care physicians. Can Fam Physician 54:870-875. [PMC free article] [PubMed] [Google Scholar]
- Garnis C, Campbell J, Zhang L, Rosin MP, Lam WL. (2004). OCGR array: an oral cancer genomic regional array for comparative genomic hybridization analysis. Oral Oncol 40:511-519. [DOI] [PubMed] [Google Scholar]
- Garnis C, Chari R, Buys TP, Zhang L, Ng RT, Rosin MP, et al. (2009). Genomic imbalances in precancerous tissues signal oral cancer risk. Mol Cancer 8:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandis JR. (2006). Prognostic biomarkers in head and neck cancer. Clin Cancer Res 12:5005-5006. [DOI] [PubMed] [Google Scholar]
- Hirsch FR, Varella-Garcia M, Bunn PA, Jr, Di Maria MV, Veve R, Bremmes RM, et al. (2003). Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 21:3798-3807. [DOI] [PubMed] [Google Scholar]
- Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. (2007). Intriguing issues of EGFR targeting in head and neck cancer. Ann Oncol 18:962. [DOI] [PubMed] [Google Scholar]
- Kuo MY, Lin CY, Hahn LJ, Cheng SJ, Chiang CP. (1999). Expression of cyclin D1 is correlated with poor prognosis in patients with areca quid chewing-related oral squamous cell carcinomas in Taiwan. J Oral Pathol Med 28:165-169. [DOI] [PubMed] [Google Scholar]
- Langer CJ. (2008). Targeted therapy in head and neck cancer: state of the art 2007. and review of clinical applications. Cancer 112:2635-2645. [DOI] [PubMed] [Google Scholar]
- Mao L, Lee DJ, Tockman MS, Erozan YS, Askin F, Sidransky D. (1994). Microsatellite alterations as clonal markers for the detection of human cancer. Proc Natl Acad Sci USA 91:9871-9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalides RJ, van Veelen NM, Kristel PM, Hart AA, Loftus BM, Hilgers FJ, et al. (1997). Overexpression of cyclin D1 indicates a poor prognosis in squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg 123:497-502. [DOI] [PubMed] [Google Scholar]
- Myo K, Uzawa N, Miyamoto R, Sonoda I, Yuki Y, Amagasa T. (2005). Cyclin D1 gene numerical aberration is a predictive marker for occult cervical lymph node metastasis in TNM Stage I and II squamous cell carcinoma of the oral cavity. Cancer 104:2709-2716. [DOI] [PubMed] [Google Scholar]
- Romeo MS, Sokolova IA, Morrison LE, Zeng C, Baron AE, Hirsch FR, et al. (2003). Chromosomal abnormalities in non-small cell lung carcinomas and in bronchial epithelia of high-risk smokers detected by multi-target interphase fluorescence in situ hybridization. J Mol Diagn 5:103-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin MP, Cheng X, Poh C, Lam WL, Huang Y, Lovas J, et al. (2000). Use of allelic loss to predict malignant risk for low-grade oral epithelial dysplasia. Clin Cancer Res 6:357-362. [PubMed] [Google Scholar]
- Ryott M, Wangsa D, Heselmeyer-Haddad K, Lindholm J, Elmberger G, Auer G, et al. (2009). EGFR protein overexpression and gene copy number increases in oral tongue squamous cell carcinoma. Eur J Cancer 45:1700-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepman KP, van der Meij EH, Smeele LE, van der Waal I. (1998). Malignant transformation of oral leukoplakia: a follow-up study of a hospital-based population of 166 patients with oral leukoplakia from The Netherlands. Oral Oncol 34:270-275. [PubMed] [Google Scholar]
- Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC, et al. (2009). Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res 69:2568-2576. [DOI] [PubMed] [Google Scholar]
- Shintani S, Mihara M, Nakahara Y, Kiyota A, Ueyama Y, Matsumura T, et al. (2002). Expression of cell cycle control proteins in normal epithelium, premalignant and malignant lesions of oral cavity. Oral Oncol 38:235-243. [DOI] [PubMed] [Google Scholar]
- Squire JA, Bayani J, Luk C, Unwin L, Tokunaga J, MacMillan C, et al. (2002). Molecular cytogenetic analysis of head and neck squamous cell carcinoma: by comparative genomic hybridization, spectral karyotyping, and expression array analysis. Head Neck 24:874-887. [DOI] [PubMed] [Google Scholar]
- Temam S, Kawaguchi H, El-Naggar AK, Jelinek J, Tang H, Liu DD, et al. (2007). Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol 25:2164-2170. [DOI] [PubMed] [Google Scholar]
- Todd R, Hinds PW, Munger K, Rustgi AK, Opitz OG, Suliman Y, et al. (2002). Cell cycle dysregulation in oral cancer. Crit Rev Oral Biol Med 13:51-61. [DOI] [PubMed] [Google Scholar]
- Torres-Rendon A, Stewart R, Craig GT, Wells M, Speight PM. (2009). DNA ploidy analysis by image cytometry helps to identify oral epithelial dysplasias with a high risk of malignant progression. Oral Oncol 45:468-473. [DOI] [PubMed] [Google Scholar]
- Tsui IF, Poh CF, Garnis C, Rosin MP, Zhang L, Lam WL. (2009). Multiple pathways in the FGF signaling network are frequently deregulated by gene amplification in oral dysplasias. Int J Cancer 125:2219-2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron CA, Shafer WG. (1975). Leukoplakia revisited. A clinicopathologic study [of] 3256 oral leukoplakias. Cancer 36:1386-1392. [DOI] [PubMed] [Google Scholar]
- Warnakulasuriya S. (2009). Global epidemiology of oral and oropharyngeal cancer. Oral Oncol 45:309-316. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. (2006). Does aneuploidy cause cancer? Curr Opin Cell Biol 18:658-667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.