

SpFcsU is a fucose mutarotase from S. pneumoniae that links the harvesting of fucose from glycans by glycoside hydrolases to processing of the fucose monosaccharide by subsequent enzymes in a fucose-utilization pathway. Here, the decameric structure of SpFcsU in complex with fucose is described.
Keywords: Streptococcus pneumoniae, mutarotases, fucose metabolism
Abstract
Streptococcus pneumoniae relies on a variety of carbohydrate-utilization pathways for both colonization of its human host and full virulence during the development of invasive disease. One such pathway is the fucose-utilization pathway, a component of which is fucose mutarotase (SpFcsU), an enzyme that performs the interconversion between α-l-fucose and β-l-fucose. This protein was crystallized and its three-dimensional structure was solved in complex with l-fucose. The structure shows a complex decameric quaternary structure with a high overall degree of structural identity to Escherichia coli FcsU (EcFcsU). Furthermore, the active-site architecture of SpFcsU is highly similar to that of EcFcsU. When considered in the context of the fucose-utilization pathway found in S. pneumoniae, SpFcsU appears to link the two halves of the pathway by enhancing the rate of conversion of the product of the final glycoside hydrolysis step, β-fucose, into the substrate for the fucose isomerase, α-fucose.
1. Introduction
Carbohydrate metabolism is a fundamental mode of energy acquisition and generation across all taxonomic kingdoms. The most well established and central carbohydrate-utilization pathway is glycolysis, which converts glucose into pyruvate through a number of intermediates. Other hexose sugars as well as pentose sugars have specific metabolic pathways that produce intermediates that feed into glycolysis. One such example is demonstrated by the utilization of fucose in Escherichia coli, in which free l-fucose is converted into lactaldehyde and dihydroxyacetone phosphate (Fig. 1 ▶), the latter of which is a glycolytic intermediate (Chen et al., 1987 ▶). The first biochemical modification in the pathway requires fucose isomerase (FcsI) to convert the α-anomer of fucose into its ketose form, fuculose. However, upstream of FcsI is a fucose mutarotase (FcsU; EC 5.1.3.n2), which is present to ensure that as the α-fucose is depleted by FcsI the comparatively slow rate of natural mutarotation between the α-fucose and β-fucose anomers is enzymatically enhanced, thus maintaining the pool of α-fucose substrate for FcsI (Seemann & Schulz, 1997 ▶; Lee et al., 2009 ▶). Following FcsI, the fuculose kinase (FcsK) then uses ATP to phosphorylate fuculose, creating a fuculose 1-phosphate substrate (Heath & Ghalambor, 1962 ▶) for fuculose 1-phosphate aldolase (FcsA), which performs the final cleavage to produce lactaldehyde and dihydroxyacetone phosphate (Ghalambor & Heath, 1962 ▶).
Figure 1.
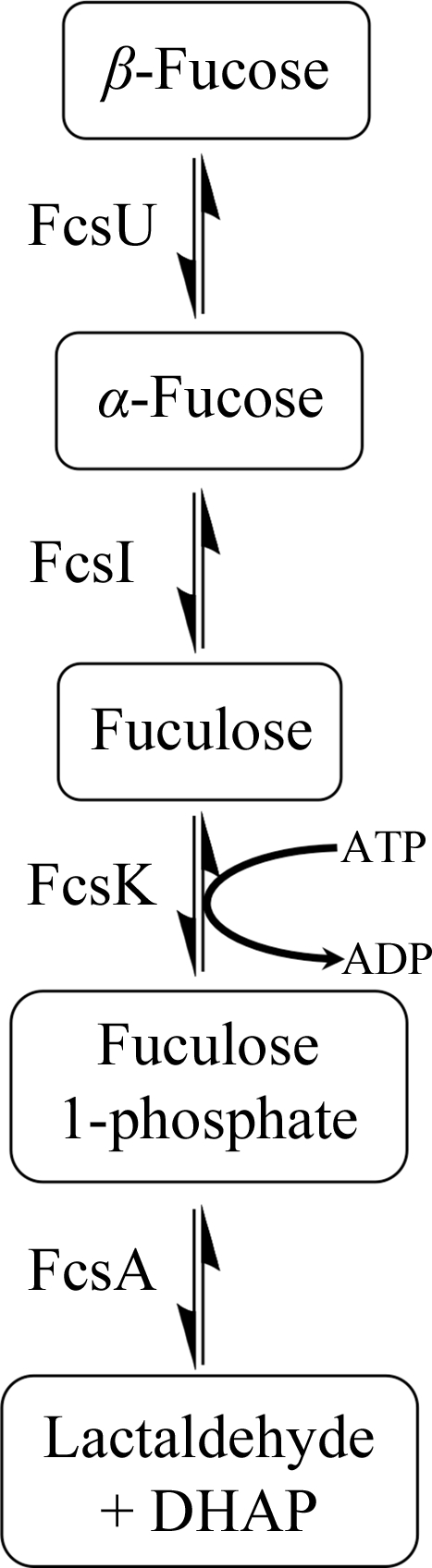
General scheme of fucose processing by bacterial enzymes.
The genome of Streptococcus pneumoniae contains a large number of genes that encode proteins dedicated to carbohydrate metabolism and, remarkably, a high proportion of these enzymes have been shown to be virulence factors in the TIGR4 strain (Polissi et al., 1998 ▶; Hava & Camilli, 2002 ▶). Notable among these identified virulence factors were components of a fucose-utilization operon. This operon is more complex than the E. coli operon and, together with the fucose-processing enzymes (FcsU, FcsI, FcsK and FcsA), contained two putative glycoside hydrolases (GH98 and GH95) and a putative PTS (phosphoenolpyruvate:carbohydrate phosphotransferase system) carbohydrate-transporter system (EIIA, EIIB, EIIC and EIID). This fucose-utilization pathway allows S. pneumoniae to harvest the LewisY blood-group antigen from host cells (Higgins et al., 2009 ▶) and a strain lacking the operon renders the bacterium nearly avirulent in a mouse model of pneumonia (Embry et al., 2007 ▶), showing the importance of this pathway in pathogenesis. Surprisingly, S. pneumoniae is unable to use fucose as a sole carbon source (Chan et al., 2003 ▶), presenting questions about the function of this operon. With the exception of GH98, details of the individual functions of the enzymes comprising the S. pneumoniae fucose-utilization pathway are presently lacking.
The predicted S. pneumoniae fucose mutarotase (SpFcsU) has an amino-acid identity of 43% to E. coli FcsU (EcFcsU) and thus is thought to have activity interconverting the α and β forms of l-fucose. Here, we report the three-dimensional crystal structure of the fucose mutarotase from S. pneumoniae TIGR4 in complex with l-fucose and discuss its possible role with regard to blood-group antigen processing by S. pneumoniae.
2. Materials and methods
2.1. Expression and purification
The fcsU gene (SP_2165) was amplified from S. pneumoniae TIGR4 genomic DNA (American Type Culture Collection BAA-334D) using primers FcsU-Fwd (GGCAGCCATAGTAAAACATATACCGAAA) and FcsU-Rev (GTGGTGCTCGAGTTATTGAACATTTTCTCTTTC) with engineered NdeI and XhoI restriction sites, respectively. This DNA fragment was subsequently cloned into pET-28a using these specific restriction sites to yield pFcsU, the sequence fidelity of which was confirmed by bidirectional DNA sequencing. The construct encoded the desired FcsU protein preceded by the amino-acid sequence MGSSHHHHHHSSGLVPRGSH, which comprises a thrombin-cleavable N-terminal six-histidine tag.
Protein production and purification were carried out using previously described procedures (Higgins et al., 2009 ▶). Briefly, E. coli BL21 Star (DE3) cells (Novagen) were transformed with the pFcsU plasmid. Cultures of the transformed E. coli harbouring pFcsU were initially grown at 310 K in LB broth supplemented with 50 µg ml−1 kanamycin until they reached an absorbance at 600 nm (A 600) of about 0.6. Gene expression was then induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside followed by overnight incubation at 298 K with shaking. After centrifugation, the cells were ruptured via chemical lysis (Robb et al., 2010 ▶). Initial purification of the target protein was carried out by Ni2+-immobilized metal-affinity chromatography (Amersham) followed by thrombin cleavage to remove the polyhistidine tag. After a final size-exclusion chromatography step using a Sephacryl S-200 column (GE Healthcare), the purified protein was concentrated for crystallization screening.
Protein concentration was determined by measuring the absorbance at 280 nm (A 280) and using a calculated molar extinction coefficient of 0.017545 cm−1 µM −1 (Wilkins et al., 1999 ▶).
2.2. Crystallization
Crystallization was carried out using hanging-drop vapour diffusion at 291 K and a protein concentration of 20 mg ml−1. Optimization of the initial crystallization conditions yielded the best crystals in 20% PEG 3350, 0.1 M HEPES pH 8.0, 0.2 M KCl. Crystals were soaked with crystallization solution supplemented with a molar excess of l-fucose to obtain a structure in complex with this sugar.
2.3. Data collection, structure determination and refinement
Crystals of SpFcsU proved difficult to cryoprotect, thus a protocol was used in which powdered fucose was added directly to the crystallization drop and the drops were exposed to air until evaporation was sufficient to result in cryoprotecting conditions, at which point the crystals were mounted straight onto the detector in an N2 cryo-stream at 113 K. Diffraction data were collected using a Rigaku R-AXIS IV++ area detector coupled to a MicroMax-002 X-ray generator with Osmic Blue optics and an Oxford Cryostream 700.
Using a pentamer of Bacillus subtilis RbsD (PDB entry 1ogc; Kim et al., 2003 ▶) as a search model, a molecular-replacement solution comprising four pentamers was found with the program Phaser (McCoy et al., 2005 ▶). Manual correction of the 20 monomers in the asymmetric unit was performed using Coot (Emsley et al., 2010 ▶) and was followed by refinement using REFMAC (Murshudov et al., 2011 ▶). Finally, 20 β-l-fucose molecules were manually placed in the active sites and water molecules were found using ‘Find Waters’ in Coot and manually inspected after refinement. Refinement procedures were monitored by flagging 5% of all observation as ‘free’ (Brünger, 1992 ▶). Model validation was performed with RAMPAGE (Lovell et al., 2003 ▶) and MolProbity (Chen et al., 2010 ▶). Data-processing and refinement statistics are given in Table 1 ▶. Coordinates and structure factors have been deposited in the Protein Data Bank as PDB entry 4a34.
Table 1. Data-collection and refinement statistics for SpFcsU.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength | 1.5419 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 64.3, b = 144.1, c = 165.4, β = 97.51 |
| Resolution (Å) | 20.00–2.50 (2.59–2.50) |
| Rmerge† | 0.087 (0.400) |
| 〈I/σ(I)〉 | 8.4 (2.4) |
| Completeness (%) | 99.7 (98.9) |
| Multiplicity | 4.0 (3.8) |
| Mosaicity (°) | 0.43 |
| No. of crystals | 1 |
| Temperature (K) | 113 |
| Crystal-to-detector distance (mm) | 110 |
| Detector | R-AXIS IV++ |
| Refinement | |
| Resolution (Å) | 2.5 |
| No. of reflections | 97486 |
| Rwork/Rfree | 0.225/0.292 |
| No. of atoms | |
| Protein | 21695 |
| Ligand | 220 |
| Water | 850 |
| B factors (Å2) | |
| Protein | 38.9 |
| Ligand | 40.3 |
| Water | 31.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.0084 |
| Bond angles (°) | 1.20 |
| Ramachandran statistics‡ | |
| Preferred (%) | 96.1 |
| Generously allowed (%) | 3.1 |
| Disallowed (%) | 0.8 |
R
merge = 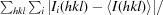
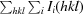 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Ramachandran statistics from RAMPAGE (Lovell et al., 2003 ▶).
3. Results
3.1. S. pneumoniae fucose mutarotase structure
The refined structure of SpFcsU, determined to a resolution of 2.5 Å, revealed 20 monomeric SpFcsU subunits in the asymmetric unit that were arranged as two decamers, with each decamer comprising a dimer of pentamers. This is consistent with the observation that SpFcsU behaves as a high-molecular-weight multimer in solution as assessed by size-exclusion chromatography. The SpFcsU monomer consists of a central β-sheet packed between three α-helices on one side of the β-sheet and two α-helices on the opposing side (Fig. 2 ▶ a). The root-mean-square deviations (r.m.s.d.s) of the monomers making up the two decamers in the asymmetric unit varied between 0.23 and 0.58 Å, with a mean of ∼0.31 Å. Each monomer was generally well ordered and could be completely traced, although the loop between residues 77 and 82 did display some disorder and elevated B factors. The pentamer of the FcsU monomers forms a ring-like structure in which the diameters of the inner and outer rings are approximately 40 and 90 Å, respectively (Fig. 2 ▶ b). The decameric structure, which has a height of approximately 56 Å, is formed by two pentamers that stack on top of one another and interact via the same faces of the pentamer ring (Fig. 2 ▶ c). The pentamer rings, however, are rotated by about 36° relative to one another, resulting in a slightly offset association of the pentamer rings (Fig. 2 ▶ c).
Figure 2.

Structure of SpFcsU. (a) Stereo image of a cartoon representation of an SpFcsU monomer. (b) Top and (c) side views of the decamer organization of SpFcsU in complex with l-fucose. Each monomer is displayed in a different colour, with the potassium ions shown as magenta spheres and the fucose residues as red stick representations.
The decameric quaternary structure of SpFcsU is stabilized by three molecular interfaces labelled (i), (ii) and (iii) in Fig. 3 ▶(a). The first and likely most significant, interface (i), occurs between adjacent monomers, referred to as monomers A and B, in the opposing pentameric rings. The extensive interface of 985.4 Å2 is made by identical surfaces donated by each monomer, reflecting the twofold symmetry that relates the two monomers, and is created by the pairing of the N-terminal α-helices (α1, residues 11–17) from each monomer, while the arms created by residues 2–10 reach over to embrace the neighbouring monomer (Figs. 3 ▶ a and 3 ▶ b). Four hydrogen bonds are formed at this interface with additional extensive van der Waals interactions, notably including the parallel stacking of the side-chain rings of His4 and Tyr59 from separate monomers (Fig. 3 ▶ b). Interface (ii) occurs between monomers B and C within the same pentamer ring. The interacting surface area of this interface is 441.9 Å2 and includes nine hydrogen bonds (Figs. 3 ▶ a and 3 ▶ c). Interface (iii) has an even smaller surface area at 258.2 Å2 and no polar interactions (Figs. 3 ▶ a and 3 ▶ d). Notably, PISA predictions suggest that interfaces (ii) and (iii) are energetically insignificant and do not contribute to stabilization of the quaternary structure, whereas interface (i) is predicted to play an essential role in complex formation. Thus, the decameric organization of FcsU is more accurately described as a pentamer of dimers in which the dimers are of the A/B monomer type. These apparently stable dimers associate via interfaces (ii) and (iii), which together may provide a sufficient driving force for the assembly of the decamer. However, in addition to the protein–protein interactions at the monomer interfaces, metal ions were also identified at each interface (iii) (Figs. 3 ▶ a and 3 ▶ d). These ions were modelled as potassium on the basis of the presence of KCl in the crystallization conditions, coordination geometry, bond lengths and analysis of the refined B factors (average B factor of 47 Å2, which was in accordance with neighbouring atoms). This potassium ion was in the same position in all cases and was pentacoordinated by the main-chain O atoms of two Arg44 residues, one each from monomers A and C (Fig. 3 ▶ d, inset). Two Asp46 residues, again originating from the same A and C monomers, help coordinate two of the water molecules around the potassium ion. Accepting that the building block of the decamer is A/B-type dimers, the coordination of the K ion at interface (iii) is then likely to aid in stabilizing decamer formation by promoting the interaction between separate A/B-type dimers at interface (iii).
Figure 3.

The interfaces of SpFcsU monomers that contribute to decamer organization. (a) Side view of the decamer showing the three monomers involved in interfaces (i), (ii) and (iii). Monomer A is shown in yellow, monomer B is shown in green and monomer C is shown in orange. Stick representations of amino acids involved in (b) interface (i), (c) interface (ii) and (d) interface (iii) are shown with an inlay showing the coordination of the potassium ion by the main-chain O atoms of two Arg44 residues and three water molecules. Side-chain N atoms and O atoms are shown in blue and red, respectively, while potassium ions and water molecules are shown as magenta and blue spheres, respectively.
3.2. Recognition of fucose
The crystals of SpFcsU were soaked in an excess of fucose, and electron density that could be easily modelled as β-l-fucose was found in each of the active sites (Fig. 4 ▶), which, on the basis of the B factors for these residues, were completely occupied. The active site resides at interface (ii) (Fig. 3 ▶ a), thus giving five active sites per pentamer ring and ten active sites in the entire decamer. The fucose monomer is coordinated by amino-acid side chains donated by both SpFcsU monomers that contribute to interface (ii), although the C-type monomer contributes the majority of the residues (Fig. 4 ▶). Specificity appears to be conferred by hydrogen bonds from Asn132 and Asp30 to the axial hydroxyl groups of the fucose C2 and C3. The ring of Tyr130 packs against the apolar surface created by the plane of C3–C4–C5 in the fucose residue, selecting for the axial hydroxyl on C4. His22 (from the adjacent monomer) and Tyr112 hydrogen bond to O1 and O5, respectively, and also make up part of the catalytic machinery (see below).
Figure 4.

The active site of SpFcsU shown as a stereo image with the side chains of the catalytic and/or interacting residues from monomer B in green and those from monomer C in orange, while the fucose molecule is shown in yellow with the side-chain O atoms, N atoms and S atoms in red, blue and yellow, respectively. The blue mesh shows the maximum-likelihood/σA-weighted F obs − F calc maps for fucose contoured at 0.25 e Å−3.
3.3. Similarities among fucose mutarotases
The SpFcsU monomer has amino-acid identities of 43, 50 and 27% to and r.m.s.d.s of 1.25, 1.01 and 1.72 Å with corresponding monomers from EcFcsU (PDB entry 2wcv; Lee et al., 2009 ▶), MmFcsU (PDB entry 2wcu; Lee et al., 2009 ▶) and BsRbsD (PDB entry 1ogd; Kim et al., 2003 ▶), respectively (Figs. 5 ▶ a and 5 ▶ b). Although all four of these structures are very similar, as revealed by the r.m.s.d. values, only the prokaryote structures share the same decamer organization (Fig. 5 ▶ c), while the mammalian fucose mutarotase is a homodimer (Kim et al., 2003 ▶; Lee et al., 2009 ▶). Not surprisingly, the dimensions of the decamers formed by the prokaryotic enzymes are similar, as in general are many characteristics of the interfaces that result in the formation of the quaternary structures. A somewhat distinctive feature of SpFcsU, however, is the potassium ion found in interface (iii). Notably, there are no ions present in the EcFcsU structure, although the pocket that accommodates the potassium in SpFcsU is conserved in EcFcsU. The BsRbsD structure has well ordered chloride ions which are present at the equivalent of interface (i). The role of metal ions in the quaternary structure of these enzymes is unclear and indeed their observation in the structures of SpFcsU and BsRbsD may be artifacts of crystallization. However, it is also plausible that these ions stabilize the quaternary structures in a manner that compensates for the slight species-specific differences at the molecular interfaces of the protein monomers.
Figure 5.

Comparison of SpFcsU. (a) Alignment of SpFcsU with other fucose mutarotases from both prokaryotes and eukaryotes and the paralogue RbsD. Black stars and triangles denote the catalytic residues and fucose-interacting residues, respectively. Cartoon-representation overlay of the (b) monomers and (c) decamers of SpFcsU (green) and EcFcsU (blue). (d) A stick representation of the catalytic residues of SpFcsU and EcFcsU. Side-chain O atoms and N atoms are shown in red and blue, respectively.
The residues in the catalytic site of SpFcsU are highly conserved compared with those of EcFcsU (Fig. 5 ▶ a). Residues Arg108, Tyr112, His22′ and Asp64′ of SpFcsU overlap extremely well with those of EcFcsU (Fig. 5 ▶ d; Kim et al., 2003 ▶), while these residues are also highly conserved among related family members. This high level of identity in the active site, together with the observed ability of SpFcsU to accommodate fucose in its active site, supports the contention that SpFcsU is indeed a fucose mutarotase.
4. Discussion
In prokaryotes, the assimilation of fucose is initiated by fucose isomerase (FcsI) catalyzing the isomerization of fucose to fuculose, which is subsequently processed in two additional steps to dihydroxyacetone phosphate and lactaldehyde. Like d-xylose isomerase, fucose isomerase is thought to be specific for the α-anomer of fucose (Seemann & Schulz, 1997 ▶). In solution, however, the anomeric equilibrium of fucose favours β-l-fucose over α-l-fucose by roughly 2.5:1 (Park et al., 2007 ▶), providing a potential bottleneck for fucose metabolism. This limitation is compounded by inherently slow rates of spontaneous mutarotation, which, for example, can be of the order of minutes for glucose (Livingstone et al., 1977 ▶). The specificity of FcsI for the less abundant anomer of fucose and the potential limits that this imposes on flux through the fucose-assimilation pathway provides a rationale for incorporation of FcsU into fucose-utilization pathways. As FcsI depletes the α-l-fucose, the more abundant β-l-fucose is converted into α-l-fucose by FcsU at a rate that is more consistent with the needs of the downstream enzymes, thus increasing the rate of fucose conversion. Indeed, as an analogous example, it has been shown that an E. coli strain lacking the yiiL gene, which encodes an l-rhamnose mutarotase, displays a longer lag phase and reduced maximal growth compared with the wild type when grown on a minimal medium with a low concentration of rhamnose (Ryu et al., 2005 ▶).
The importance of FcsU in controlling the rate of fucose metabolism is clearly demonstrated in S. pneumoniae. Although this bacterium is unable to use exogenously supplied fucose as a sole carbon source (Chan et al., 2003 ▶; Embry et al., 2007 ▶), it does possess a fucose-utilization operon. Indeed, two types of mutually exclusive variations of the fucose operon, called type 1 and type 2, are found in a strain-dependent manner in S. pneumoniae (Higgins et al., 2009 ▶). A common feature of both operon-encoded pathways are the fucose-processing enzymes FcsI, FcsK and FcsA. The pathways differ, however, in their type of carbohydrate transporter and, importantly, their complement of glycan-processing enzymes that harvest fucose-containing glycans from host tissue and subsequently break them down into monosaccharide components.
Based on available genome sequences, the majority of S. pneumoniae strains possess the type 1 fucose-utilization pathway. This pathway is initiated by the action of an extracellular family 98 glycoside hydrolase that cleaves host LewisY antigen, yielding the H-disaccharide (Higgins et al., 2009 ▶). Once transported into the bacterial cell, it has been suggested that a putative intracellular GH95 hydrolyzes the H-disaccharide with the inverting mechanism demonstrated for the Bifidobacterium bifidum α-1,2-l-fucosidase homologue (Katayama et al., 2004 ▶). This ultimately releases the β-anomer of l-fucose. As the α-anomer is the supposed substrate for the fucose isomerase, this pathway employs SpFcsU (studied here) to speed the conversion of β-l-fucose to α-l-fucose and thus increase the flux through the fucose-conversion steps of the pathway (Fig. 6 ▶). In contrast, the type 2 fucose-utilization pathway is dedicated to harvesting and depolymerizing the A and B blood-group antigens (Higgins et al., 2009 ▶). These sugars share similarities with the LewisY antigen, including the α-1,2-fucose appended to a galactose. In this type 2 pathway the fucose is predicted to be liberated from the blood-group antigens by a family 29 glycoside hydrolase; previously characterized GH29 enzymes have been found to be retaining α-l-fucosidases (Berteau et al., 2002 ▶, 2004 ▶), suggesting that the S. pneumoniae GH29 liberates α-l-fucose from its substrate (Fig. 6 ▶). Unlike the type 1 pathway, therefore, the type 2 pathway releases α-l-fucose, which can be efficiently processed by FcsI without the need for SpFcsU.
Figure 6.

Schematic representation of the link between glycan degradation and fucose processing in S. pneumoniae by both putative fucose-metabolic pathways: the type 1 and type 2 operons. The type I operon harvests the LewisY blood-group antigen and encodes a GH95 and SpFcsU to release fucose for further processing by FcsI, while the type 2 operon harvests the A/B blood-group antigens and encodes a GH29 that does not contain SpFcsU for further fucose processing. Inverting and retaining refer to the catalytic mechanism of the glycoside hydrolases.
FcsU is commonly found in pathogenic bacteria such as E. coli and Clostridium perfringens and is often the first step in fucose-utilization pathways, in which it converts excess free β-l-fucose to α-l-fucose and thus creates substrate for FcsI, which is the next enzyme in the pathway. In S. pneumoniae, however, the initiation of the fucose pathway does not involve FcsU acting on free fucose; instead it starts with a series of glycoside hydrolases releasing fucose from host complex carbohydrates (Higgins et al., 2009 ▶). One such glycoside hydrolase, GH95, releases β-l-fucose, and thus SpFcsU acts as a link between glycoside hydrolase activity on host carbohydrates and bacterial carbohydrate metabolism.
Supplementary Material
PDB reference: fucose mutarotase, 4a34
Acknowledgments
This work was supported by a Canadian Institute of Health Research grant (FRN 86610). MAH is supported by doctoral fellowships from the Natural Sciences and Engineering Research Council of Canada and the Michael Smith Foundation for Health Research (MSFHR). ABB is a Canada Research Chair in Molecular Interactions and an MSFHR Scholar.
References
- Berteau, O., Bielicki, J., Kilonda, A., Machy, D., Anson, D. S. & Kenne, L. (2004). Biochemistry, 43, 7881–7891. [DOI] [PubMed]
- Berteau, O., McCort, I., Goasdoué, N., Tissot, B. & Daniel, R. (2002). Glycobiology, 12, 273–282. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Chan, P. F., O’Dwyer, K. M., Palmer, L. M., Ambrad, J. D., Ingraham, K. A., So, C., Lonetto, M. A., Biswas, S., Rosenberg, M., Holmes, D. J. & Zalacain, M. (2003). J. Bacteriol. 185, 2051–2058. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Chen, Y. M., Zhu, Y. & Lin, E. C. (1987). Mol. Gen. Genet. 210, 331–337. [DOI] [PubMed]
- Embry, A., Hinojosa, E. & Orihuela, C. J. (2007). BMC Microbiol. 7, 80. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Ghalambor, M. A. & Heath, E. C. (1962). J. Biol. Chem. 237, 2427–2433. [PubMed]
- Hava, D. L. & Camilli, A. (2002). Mol. Microbiol. 45, 1389–1406. [PMC free article] [PubMed]
- Heath, E. C. & Ghalambor, M. A. (1962). J. Biol. Chem. 237, 2423–2426. [PubMed]
- Higgins, M. A., Whitworth, G. E., El Warry, N., Randriantsoa, M., Samain, E., Burke, R. D., Vocadlo, D. J. & Boraston, A. B. (2009). J. Biol. Chem. 284, 26161–26173. [DOI] [PMC free article] [PubMed]
- Katayama, T., Sakuma, A., Kimura, T., Makimura, Y., Hiratake, J., Sakata, K., Yamanoi, T., Kumagai, H. & Yamamoto, K. (2004). J. Bacteriol. 186, 4885–4893. [DOI] [PMC free article] [PubMed]
- Kim, M.-S., Shin, J., Lee, W., Lee, H.-S. & Oh, B.-H. (2003). J. Biol. Chem. 278, 28173–28180. [DOI] [PubMed]
- Lee, K.-H., Ryu, K.-S., Kim, M.-S., Suh, H.-Y., Ku, B., Song, Y.-L., Ko, S., Lee, W. & Oh, B.-H. (2009). J. Mol. Biol. 391, 178–191. [DOI] [PubMed]
- Livingstone, G., Franks, F. & Aspinall, L. J. (1977). J. Solution Chem. 6, 203–216.
- Lovell, S. C., Davis, I. W., Arendall, W. B., de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Park, D., Ryu, K.-S., Choi, D., Kwak, J. & Park, C. (2007). Glycobiology, 17, 955–962. [DOI] [PubMed]
- Polissi, A., Pontiggia, A., Feger, G., Altieri, M., Mottl, H., Ferrari, L. & Simon, D. (1998). Infect. Immun. 66, 5620–5629. [DOI] [PMC free article] [PubMed]
- Robb, C. S., Nano, F. E. & Boraston, A. B. (2010). Acta Cryst. F66, 1596–1598. [DOI] [PMC free article] [PubMed]
- Ryu, K.-S., Kim, J.-I., Cho, S.-J., Park, D., Park, C., Cheong, H.-K., Lee, J.-O. & Choi, B.-S. (2005). J. Mol. Biol. 349, 153–162. [DOI] [PubMed]
- Seemann, J. E. & Schulz, G. E. (1997). J. Mol. Biol. 273, 256–268. [DOI] [PubMed]
- Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D. & Hochstrasser, D. F. (1999). Methods Mol. Biol. 112, 531–552. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: fucose mutarotase, 4a34