

The crystallization and preliminary X-ray diffraction analysis of Val57 mutants of human cystatin C, designed to assess the influence of changes in the properties of loop L1 on dimerization propensity, are reported.
Keywords: cysteine protease inhibitors, human cystatin C, single-point mutations
Abstract
Human cystatin C (hCC) is a low-molecular-mass protein (120 amino-acid residues, 13 343 Da) found in all nucleated cells. Its main physiological role is regulation of the activity of cysteine proteases. Biologically active hCC is a monomeric protein, but all crystallization efforts have resulted in a dimeric domain-swapped structure. Recently, two monomeric structures were reported for cystatin C variants. In one of them stabilization was achieved by abolishing the possibility of domain swapping by the introduction of an additional disulfide bridge connecting the two protein domains (Cys47–Cys69). In the second structure, reported by this group, the monomeric hCC fold was preserved by stabilization of the conformationally constrained loop (L1) by a single-amino-acid substitution (V57N). To further assess the influence of changes in the sequence and properties of loop L1 on the dimerization propensity of cystatin C, two additional hCC mutants were obtained: one with a residue favoured in β-turns (V57D) and another with proline (V57P), a residue that is known to be a structural element that can rigidify but also broaden turns. Here, the expression, purification and crystallization of V57D and V57P variants of recombinant human cystatin C are described. Crystals were grown by the vapour-diffusion method. Several diffraction data sets were collected using a synchrotron source at the Advanced Photon Source, Argonne National Laboratory, Chicago, USA.
1. Introduction
Human cystatin C (hCC) is a natural inhibitor of cysteine proteases, which are widely distributed in animals, plants and microorganisms (Grubb, 2000 ▶). This low-molecular-mass protein (13 343 Da) is composed of 120 amino-acid residues constituting a single polypeptide chain with two disulfide bonds in its C-terminal part (Grubb & Löfberg, 1982 ▶). Under physiological conditions wild-type hCC is a monomeric protein, but in all crystal structures reported to date it exists in the form of a symmetric domain-swapped dimer (Fig. 1 ▶ a; Janowski et al., 2001 ▶, 2004 ▶, 2005 ▶). Until very recently, the monomeric fold of cystatin C could only be approximated on the basis of the available structure of its chicken analogue (Bode et al., 1988 ▶) or the structures of family 1 cystatins either alone (Martin et al., 1995 ▶) or in complex with an enzyme (Jenko et al., 2003 ▶). Last year two papers were published in which the monomeric fold of hCC was finally revealed; however, this was not for the wild type but for a mutated protein. Kołodziejczyk and coworkers presented an hCC variant in which two additional Cys residues were introduced by mutagenesis at positions 47 and 69 to form a new disulfide bridge which was intended to hold together the protein domains undergoing the swapping process (Kołodziejczyk et al., 2010 ▶). The second paper came from our group and presented the monomeric structure of an hCC mutant in which the dimerization propensity was diminished by a single point mutation in loop L1 (Orlikowska et al., 2011 ▶). This structural element of cystatin C, encompassing residues 55–59 (QIVAG), connects the protein subdomains that are involved in the exchange process and is the only element that undergoes significant conformational changes during dimerization (a loop–extended β-strand transition). Experimental (Szymańska et al., 2009 ▶) and theoretical (Dehouck et al., 2003; Rodziewicz-Motowidło et al., 2009 ▶) studies confirmed the important role of loop L1 in the stabilization of the monomeric fold of cystatin C. Conformational tensions in this structural element, which are mostly attributed to unfavourable values of the ψ angles of the Val57 residue, which is located near the top of the loop, have been proposed to facilitate the ‘opening’ of the molecule and subsequent domain swapping (Rodziewicz-Motowidło et al., 2009 ▶). Stabilization of loop L1, accomplished by exchanging the Val57 residue for a β-turn-promoting asparagine, occurred to a sufficient extent to preserve the human cystatin C molecule in a monomeric form (Fig. 1 ▶ b).
Figure 1.

Crystal structures of (a) dimeric wild-type hCC (PDB entry 1g96; Janowski et al., 2001 ▶) and (b) monomeric V57N hCC (PDB entry 3nx0; Orlikowska et al., 2011 ▶). Residue Asn57 is marked in red.
In order to confirm the importance of the type of amino acid at position 57 to the oligomeric state of the protein molecule, we have enlarged our pool of hCC variants by obtaining two other single point mutants: V57D and V57P. The introduced substitutions had an impact on the dimerization propensity of hCC that was either similar (aspartic acid) or opposite (proline) to that of asparagine (Szymańska et al., 2009 ▶). To better understand the structural basis of this result, we initiated studies aiming at obtaining the crystal structures of these two hCC variants. Here, we describe our efforts to express, purify and crystallize both hCC mutants, as well as initial X-ray diffraction data collection.
2. Materials and methods
2.1. Protein expression and purification
The hCC variants were obtained by introduction of the appropriate point mutation (V57D or V57P) into the hCC gene engineered into the expression vector pHD313 (Abrahamson et al., 1988 ▶) using the PCR-based oligonucleotide-directed mutagenesis approach with the aid of thermostable Pfu DNA polymerase (Fermentas) according to the manufacturer’s protocol. The changes in the DNA sequences were confirmed by sequencing. The proteins were then overexpressed in Escherichia coli C41 (DE3) strain and the soluble fractions were isolated from bacterial periplasmic space using a modified protocol elaborated in our laboratory (Szymańska et al., 2009 ▶). Briefly, the harvested bacteria were first resuspended in a buffer consisting of 20 mM Tris pH 7.5, 10%(v/v) glycerol and then subjected to repeated freeze–thaw treatment followed by the classic cold osmotic shock procedure (Neu & Heppel, 1965 ▶). Appropriate fractions were collected and the proteins were purified to homogeneity by a two-step protocol. Firstly, crude proteins were loaded onto ion-exchange resin (S-Sepharose), from which hCC mutants were eluted with a linearly increasing salt-concentration gradient (0–0.5 M NaCl in 20 mM Tris, 1 mM benzamidinium chloride pH 7.5). Fractions containing the hCC mutant were pooled, extensively dialyzed against 10 mM ammonium bicarbonate pH 8.0 and lyophilized. The lyophilized protein was dissolved in 20 mM ammonium bicarbonate buffer pH 8.0 and further purified on Superdex 75 PC 10/300 in the same buffer. Pure protein fractions were lyophilized and stored at 253 K. The purity of the protein was confirmed using SDS–PAGE electrophoresis, gel filtration and mass spectrometry.
2.2. Crystallization
Preliminary crystallization trials for the V57D mutant were performed using the commercially available screen Classics Suite (Qiagen) and were further optimized using reagents generated in-house. Crystals were grown at 293 K using the hanging-drop vapour-diffusion method with a 10 mg ml−1 protein solution diluted in a 1:1 ratio with well solution. The crystals used for the diffraction experiment were grown from 0.1 M imidazole pH 6.5, 1.0 M sodium acetate (cubic form; Fig. 2 ▶ a) and from 0.1 M sodium acetate pH 4.6, 6% PEG 4000 (tetragonal form; Fig. 2 ▶ b). The crystals were soaked in reservoir solution containing glycerol to a final concentration of 30% before cryocooling in liquid nitrogen.
Figure 2.

Representative crystals of hCC mutants. (a) V57D (0.1 M imidazole pH 6.5, 1.0 M sodium acetate), (b) V57D (0.1 M sodium acetate pH 4.6, 6% PEG 4000), (c) V57P (0.1 M imidazole pH 6.5, 1.0 M sodium acetate, additive 2.0 M NDSB-211), (d) V57P (0.1 M imidazole pH 6.5, 1.0 M sodium acetate, additive 2,5-hexanediol).
Initial crystallization conditions for the V57P mutant were investigated by high-throughput techniques with a Phoenix robot using the commercially available screens Classics Suite, PACT Suite, JCSG+ Suite (Qiagen) and Index Suite (Hampton Research). All assays were carried out using the sitting-drop vapour-diffusion method at 293 K in a 96-well plate by mixing 200 nl protein solution with 200 nl precipitant solution and equilibrating against 50 µl well solution. Many conditions produced crystals and these conditions were further optimized using solutions generated in-house, as well as by using Additive Screen (Hampton Research) and the sitting-drop method. Crystallization was performed by mixing 1 µl protein solution (10 mg ml−1) with 1 µl precipitant solution and equilibrating against 500 µl well solution in EasyXtal plates (Qiagen). Crystals of hCC V57P were observed under various conditions, but those used for diffraction experiments were grown from 0.1 M imidazole pH 6.5, 1.0 M sodium acetate with NDSB-211 as an additive (Fig. 2 ▶ c). Before cryocooling in liquid nitrogen, the crystals were soaked in reservoir solution containing PEG 400 at a final concentration of 30%.
2.3. Data collection and processing
Diffraction data for the cubic form of hCC V57D were collected at 100 K using synchrotron radiation on beamline 19BM at the Advanced Photon Source (APS), Argonne National Laboratory, Chicago at a wavelength of 0.97923 Å with an ADSC Quantum Q210r CCD detector. 180 frames were collected at a crystal-to-detector distance of 290 mm using an oscillation range of 0.5°. The resolution of 3.0 Å was obtained by measuring the intensities of 110 993 reflections. The final merged data set of 5422 unique reflections was 99.9% complete (100% in the last resolution shell) and was characterized by an R merge of 0.138 and an 〈I/σ(I)〉 of 27.4.
For the tetragonal form of hCC V57D, data were collected at 100 K using synchrotron radiation on beamline 19BM of APS at a wavelength of 0.97923 Å with an ADSC Quantum Q210r CCD detector. 120 frames were collected at a crystal-to-detector distance of 230 mm using an oscillation range of 1.0°. 328 951 reflections were measured to 2.5 Å resolution. After merging, they were reduced to a unique data set consisting of 1302 reflections. This data set was 99.8% complete (100% in the last resolution shell) and was characterized by an R merge of 0.132 and an 〈I/σ(I)〉 of 27.3.
Single-wavelength X-ray diffraction data for the cubic crystals of hCC V57P were measured at 100 K using synchrotron radiation on beamline 19ID at APS at a wavelength of 0.97921 Å with an ADSC Quantum Q210r CCD detector. 240 frames were collected at a crystal-to-detector distance of 320 mm using an oscillation range of 0.5°. A total of 256 497 reflections were measured and reduced to 11 322 unique reflections extending to 2.25 Å resolution. This data set was 100% complete (100% in the last resolution shell) and was characterized by an R merge of 0.126 and an 〈I/σ(I)〉 of 47.0.
The images were indexed, integrated and scaled using the HKL-2000 or HKL-3000 package (Otwinowski & Minor, 1997 ▶; Minor et al., 2006 ▶). Data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection statistics for the mutants of human cystatin C.
Values in parentheses are for the highest resolution shell.
| V57D | V57D | V57P | V57P | |
|---|---|---|---|---|
| Space group | I432 | P4212 | I432 | P622 |
| Unit-cell parameters (Å) | a = 140.14 | a = 141.36, c = 96.25 | a = 140.38 | a = 246.83, c = 375.04 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Wavelength (Å) | 0.97923 | 0.97923 | 0.97921 | 0.97929 |
| Resolution (Å) | 3.0 (3.06–3.00) | 2.50 (2.54–2.50) | 2.25 (2.29–2.25) | 4.66 (4.75–4.66) |
| Unique reflections | 5422 | 1302 | 11322 | 40704 |
| Rmerge† (%) | 0.138 (N/A) | 0.096 (N/A) | 0.126 (N/A) | 0.236 (N/A) |
| 〈I/σ(I)〉 | 27.4 (2.7) | 21.9 (2.06) | 47.0 (2.0) | 7.19 (2.26) |
| Completeness (%) | 99.9 (100) | 99.8 (100) | 100 (100) | 99.8 (100) |
| Multiplicity | 20.5 (21.3) | 9.7 (9.6) | 22.7 (11.6) | 7.9 (8.1) |
R
merge = 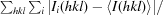
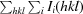 , where I
i(hkl) is the intensity of observation i of reflection hkl. If R
merge exceeds 1.0, SCALEPACK does not report its value because it is uninformative. Instead, the 〈I/σ(I)〉 criterion is used to define the resolution cutoff.
, where I
i(hkl) is the intensity of observation i of reflection hkl. If R
merge exceeds 1.0, SCALEPACK does not report its value because it is uninformative. Instead, the 〈I/σ(I)〉 criterion is used to define the resolution cutoff.
3. Results and discussion
Cubic crystals of hCC V57D (Fig. 2 ▶ a) appeared within four weeks of equilibration against 0.1 M imidazole pH 6.5, 1.0 M sodium acetate. These crystals diffracted to a resolution of 3.0 Å and fulfilled the parameters describing space group I432. The Matthews coefficient was 2.14 Å3 Da−1, corresponding to 42.55% solvent content and two molecules per asymmetric unit. Tetragonal crystals of hCC V57D (Fig. 2 ▶ b) grew in 0.1 M sodium acetate pH 4.6 with 6% PEG 4000 at a protein concentration of 10 mg ml−1 within four weeks. The crystals diffracted to 2.5 Å resolution and belonged to space group P4212. The Matthews coefficient was 2.26 Å3 Da−1, corresponding to 45.60% solvent content and eight molecules per asymmetric unit. Most of the V57P crystals (Fig. 2 ▶ d) did not diffract well (resolution not better than 5 Å). Data indexing resulted in the high-symmetry space group P622 with large unit-cell parameters: a = 246, b = 246, c = 375 Å. In the asymmetric unit of the tetragonal form of hCC V57P, between 40 (Matthews coefficient 3.10 Å3 Da−1) and 46 (Matthews coefficient 2.70 Å3 Da−1) independent copies of the molecule may be present. The propensity of hCC to crystallize with multiple copies of the molecule in the asymmetric unit, in combination with the additional possibilities offered by the point-symmetry elements of the unit cell, may be indicative of the tendency of the protein towards self-association and subsequent oligomerization. Good-quality data were only obtained for the crystals grown in 0.1 M imidazole pH 6.5, 1.0 M sodium acetate with 2.0 M NDSB-211 as an additive. The Matthews coefficient for these crystals was 2.12 Å3 Da−1, corresponding to 42.02% solvent content and two molecules per asymmetric unit.
Structure determination of hCC mutants will be pursued by molecular replacement using the coordinates of hCC V57N, the structure of which has already been deposited (PDB entry 3nx0; Orlikowska et al., 2011 ▶). Analysis of the V57D and V57P mutant structures should provide the opportunity to identify the structural features of loop L1 and to assess the impact of the type of amino acid at position 57 on the oligomeric state of the protein. The results of these efforts will be reported shortly.
Acknowledgments
This work was supported by grant No. 2739/B/H03/2010/38 from the Polish Ministry of Science and Higher Education and by grant DS/8440-4-0172-1.
References
- Abrahamson, M., Dalbøge, H., Olafsson, I., Carlsen, S. & Grubb, A. (1988). FEBS Lett. 236, 14–18. [DOI] [PubMed]
- Bode, W., Engh, R., Musil, D., Thiele, U., Huber, R., Karshikov, A., Brzin, J., Kos, J. & Turk, V. (1988). EMBO J. 7, 2593–2599. [DOI] [PMC free article] [PubMed]
- Grubb, A. (2000). Adv. Clin. Chem. 35, 63–99. [DOI] [PMC free article] [PubMed]
- Grubb, A. & Löfberg, H. (1982). Proc. Natl Acad. Sci. USA, 79, 3024–3027. [DOI] [PMC free article] [PubMed]
- Janowski, R., Abrahamson, M., Grubb, A. & Jaskolski, M. (2004). J. Mol. Biol. 341, 151–160. [DOI] [PubMed]
- Janowski, R., Kozak, M., Abrahamson, M., Grubb, A. & Jaskolski, M. (2005). Proteins, 61, 570–578. [DOI] [PubMed]
- Janowski, R., Kozak, M., Jankowska, E., Grzonka, Z., Grubb, A., Abrahamson, M. & Jaskolski, M. (2001). Nature Struct. Biol. 8, 316–320. [DOI] [PubMed]
- Jenko, S., Dolenc, I., Guncar, G., Dobersek, A., Podobnik, M. & Turk, D. (2003). J. Mol. Biol. 326, 875–885. [DOI] [PubMed]
- Kołodziejczyk, R., Michalska, K., Hernandez-Santoyo, A., Wahlbom, M., Grubb, A. & Jaskolski, M. (2010). FEBS J. 277, 1726–1737. [DOI] [PubMed]
- Martin, J. R., Craven, C. J., Jerala, R., Kroon-Zitko, L., Zerovnik, E., Turk, V. & Waltho, J. P. (1995). J. Mol. Biol. 246, 331–343. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Neu, H. C. & Heppel, L. A. (1965). J. Biol. Chem. 249, 3685–3692. [PubMed]
- Orlikowska, M., Jankowska, E., Kołodziejczyk, R., Jaskólski, M. & Szymańska, A. (2011). J. Struct. Biol. 173, 406–413. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rodziewicz-Motowidło, S., Iwaszkiewicz, J., Sosnowska, R., Czaplewska, P., Sobolewski, E., Szymańska, A., Stachowiak, K. & Liwo, A. (2009). Biopolymers, 91, 373–383. [DOI] [PubMed]
- Szymańska, A., Radulska, A., Czaplewska, P., Grubb, A., Grzonka, Z. & Rodziewicz-Motowidło, S. (2009). Acta Biochim. Pol. 56, 455–463. [PubMed]