

Abstract
A new high performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS/MS) assay for cediranib, a tyrosine kinase inhibitor for VEGFRs, was developed and validated, for the determination of plasma and brain levels of cediranib in small specimen volumes. Tyrphostin (AG1478) was used as internal standard. Mouse plasma and brain homogenate samples were prepared using liquid-liquid extraction. The assay was validated for a 2.5–2500 ng/mL concentration range for plasma, and for 1–2000 ng/mL range for brain homogenate. For these calibration ranges, within-assay variabilities were 1.1–14.3% for plasma and 1.5–9.4% for brain homogenate; between-assay variabilities were 2.4–9.2% for plasma, and 4.9–10.2% for brain homogenate. Overall accuracy ranged from 101.5 to 107.0% for plasma and 96.5 to 100.2% for brain homogenate, for all target concentrations. The developed assay has been successfully applied for a mouse brain distribution study in mice at an oral dose of 5 mg/kg.
Keywords: Cediranib, Liquid–liquid extraction, Liquid chromatography mass spectrometry, Pharmacokinetics
1. Introduction
The vascular endothelial growth factor (VEGF) and receptors (VEGFR-1, -2, -3) have been recognized as one of the critical mediators of pathological angiogenesis and are involved in the regulation of vascular endothelial cell proliferation, survival, migration, and tumor cell invasion. Numerous solid tumors overexpress VEGF, the role of which in tumor angiogenesis is via stimulation of VEGFR. VEGFRs are also highly expressed in many types of human solid tumors, including glioma, lung, breast, renal, ovarian and gastrointestinal tract carcinomas [1]. Inhibition of the VEGFR-mediated signal transduction pathways stabilizes the progression of tumors by disrupting angiogenesis - the formation of new blood vessels that tumors need to grow and spread. There have been at least two potential therapeutic strategies for cancers via blocking VEGFR signaling, which have been clinically validated with FDA-approval: (i) anti-VEGF monoclonal antibodies (e.g., bevacizumab) and (ii) tyrosine kinase inhibitors (TKI, e.g., sorafenib and sunitinib). TKIs are a class of small molecules blocking intracellular signals involved in growth and angiogenesis.
Cediranib, 4-[(4-Fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-pyrrolidin-1-yl)propoxy]q uinazoline (RECENTIN™, AZD2171) (Fig. 1a), is an orally active pan-VEGFR inhibitor with additional activity against platelet-derived growth factor β and c-Kit [2]. Cediranib has exhibited the antitumor and antiangiogenic activity in various cell lines [3–5] and xenografts including colon, lung, prostate, breast and ovary [5,6]. Its pharmacological effect has been evaluated in Phase II/III clinical trials for advanced non-small cell lung cancer, advanced colorectal cancer, metastatic renal cell carcinoma, and recurrent glioblastoma [7]. However, due to the physiological structures of the brain capillary endothelium, i.e., blood-brain barrier (BBB), the brain delivery of cediranib may be limited and lead to limited pharmacological effects when used as a CNS anti-tumor agent.
Fig. 1.
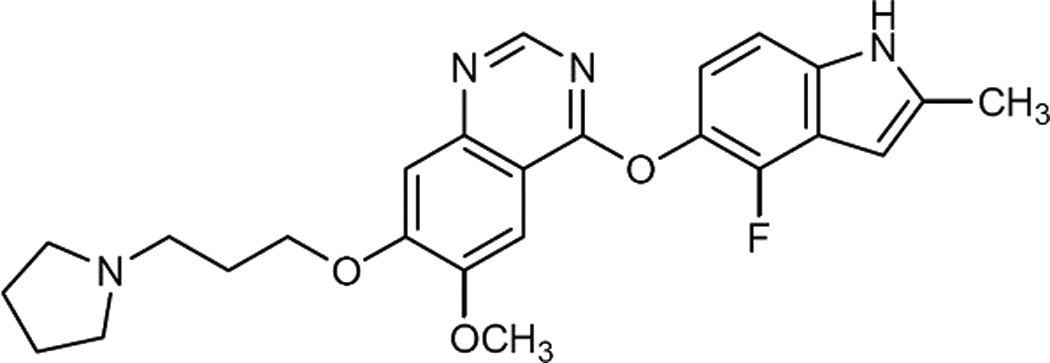
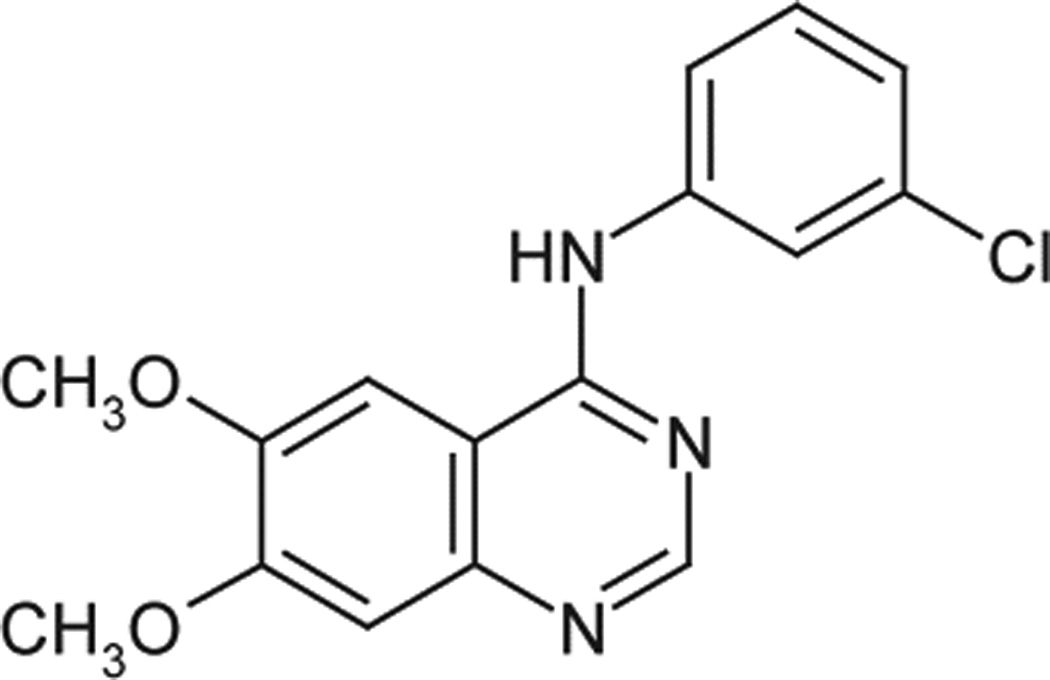
Chemical structures of cediranib and the ISTD. a, cediranib; b, AG1478.
To comprehensively characterize the preclinical pharmacokinetics (PK) of cediranib and the brain disposition of cediranib, a highly sensitive, accurate and reproducible quantification assay is required. To date there are no published methods for the analysis of cediranib in biological samples, except for one study using HPLC with radiochemical detection (HPLC-RAD), UV (HPLC-UV) and mass spectrometric (HPLC–MSn) to characterise [14C]-cediranib and its biosynthetic N+-glucuronide metabolite in hepatocytes from human and preclinical species [8]. The method described here quantifies cediranib in mouse plasma and brain homogenate utilizing high performance liquid chromatography coupled with mass spectrometry (HPLC-MS/MS) equipped with electrospray positive ionization interface following a liquid-liquid extraction. This method allows the evaluation of compound concentrations in both the plasma and the whole brain at a range of time points in mouse specimens, and employs tyrphostin (AG1478, 4-(3-Chloroanilino)-6,7-dimethoxyquinazoline, as shown in Fig. 1b), a tyrosine kinase inhibitor, as the internal standard (ISTD).
2. Experimental
2.1. Chemicals and reagents
Cediranib (98% purity) and internal standard tyrphostin AG1478 (free base, >99% purity) were purchased from Selleck Chemicals LLC (Houston, TX, USA) and LC Laboratories (Woburn, MA, USA), respectively. HPLC grade acetonitrile (ACN), methanol (MeOH), ethyl acetate and dimethyl sulfoxide (DMSO) were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Ammonium formate and formic acid were of analytical grade and supplied from Sigma-Aldrich (St. Louis, MO, USA). Bovine serum albumin (BSA; ≥96% pure from agarose gel electrophoresis) was also obtained from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals used were HPLC or reagent grade. Drug-free mouse plasma was obtained from Valley Biomedical (Catalog# AP3054, Winchester, VA, USA). Deionized water was obtained from a Milli-Q-UF system (Millipore, Milford, MA, USA) and used throughout. The mobile phase was vacuum-filtered through a 0.45 µm filter (Millpore, Milford, MA, USA).
2.2. Preparation of stock and working solutions, calibration standards, and quality controls
The stock solution of cediranib at 1 mg/mL was prepared by dissolution of 1.14 mg cediranib in 1.14 mL DMSO. Sub-stock solutions were prepared by dilution of the stock solution into 100, 10, and 1 µg/mL in MeOH. The stock solutions were stored at −20°C in glass vials, with caps tightly wrapped with Parafilm®. Working solutions were diluted with MeOH from the stock and sub-stock solutions as indicated in Table 1. 25 µL of each concentration of working solutions of cediranib and 10 µL of the working solution of AG1478 were aliquoted, dried under nitrogen, and then reconstituted in blank mouse plasma or brain homogenate on each day of analysis to provide nine calibration standards containing cediranib for plasma samples at the following concentrations: 1, 2.5, 5, 10, 25, 50, 500, 1000, 2500 ng/mL, and for brain homogenate samples at 1, 2.5, 5, 10, 25, 50, 500, 1000, 2000 ng/mL.
Table 1.
Sub-stock solutions were diluted with MeOH from the stock solutions and used to prepare calibration standards and QC samples as follows.
| Target concentration (µg/mL) |
Initial concentration (µg/mL) |
Solution (mL) |
Final volume (mL) |
Final concentration (µg/mL) |
|
|---|---|---|---|---|---|
| Substock 1 | 100 | 1000 | 0.5 | 5 | 100 |
| Substock 2 | 10 | 100 | 0.5 | 5 | 10 |
| Substock 3 | 1 | 10 | 0.5 | 5 | 1 |
Quality control (QC) samples were prepared independently from sub-stock solutions in MeOH at four different concentrations, for plasma, 2.5 ng/mL, the lower limit of quantitation (LLOQ); 15 ng/mL, the low QC; 200 ng/mL, the medium QC; and 800 ng/mL, the high QC; for brain homogenate, 1 ng/mL, the LLOQ; 5 ng/mL, the low QC; 50 ng/mL, the medium QC; and 200 ng/mL, the high QC. The QC samples were stored at −80°C until used.
The ISTD compound (AG1478) was dissolved in MeOH to a concentration of 400 ng/mL.
2.3. Sample pretreatment
Prior to drug extraction, frozen samples were thawed in a water bath at ambient temperature. Brain tissues were homogenized with a tissue homogenizer (Power Gen 125, Fisher Scientific, Pittsburgh, PA) in 3 volumes of ice-cold 5% (w/v) BSA in phosphate-buffered saline (PBS) solution. A 50 µL aliquot of plasma and a 100 µL aliquot of brain homogenate samples were dispensed into disposable borosilicate glass culture tubes (13mm×100 mm) containing AG1478 (400 ng/mL in 10 µL MeOH) and were vigorously mixed for 5 s on a vortex-mixer. The liquid-liquid extraction procedures were as follows: 800 µL ethyl acetate was added to each tube and vortexed vigorously for 30 s then centrifuged at 3000 rpm for 10 min at 4°C (SORVALL® LEGEND RT, Kendro). A volume of 600 µL of the top organic layer was transferred to a glass culture tube and dried under a gentle stream of nitrogen. The samples were reconstituted in 75 µL mobile phase and transferred to autosampler vials for injection. A volume of 10 µL was injected at 10°C using a temperature-controlled autosampling device.
2.4. Chromatographic and mass-spectrometric conditions
HPLC analysis was performed using an Agilent Model 1200 separation system (Santa Clara, CA, USA). Separation was achieved on a ZORBAX Eclipse XDB-C18 RRHT threaded column (4.6 × 50 mm, 1.8 µm; Agilent Technologies). Column temperature was set to be 30°C. The mobile phase was composed of 20mM ammonium formate containing 0.1% formic acid: acetonitrile (62:38 v/v). The flow rate was maintained at 0.25 mL/min and the chromatographic run time was 10 minutes.
The HPLC system was interfaced to a TSQ® Quantum 1.5 triple quadrupole mass spectrometer (Thermo Finnigan, San Jose, CA, USA) equipped with an electrospray ionization (ESI) source. The samples were analyzed using an electrospray probe in the positive ionization mode operating at an ion spray voltage of 4000V for both cediranib and the ISTD. Selected reaction monitoring (SRM) was employed for mass spectrometric quantification. Data acquisition and analysis was controlled by the Xcalibur version 2.0.7 data system. The spectrometer was programmed to allow the [MH]+ ion of cediranib at m/z 451.7 and that of ISTD at m/z 317 to pass through the first quadrupole (Q1) and into the collision cell (Q2). The collision gas was argon (1.5 mTorr) and the collision energy was set at 17 volts for cediranib and 16 volts for AG1478. The transitions monitored were m/z 451.7 -> 112.2 for cediranib, m/z 317->301 for the ISTD, and monitored through the third quadrupole (Q3). The scan width for the two product ions was set 1.5 unit with 0.5 second of scan time.
2.5. Calibration curve
Calibration curves for cediranib were computed with the peak area ratios of analyte and ISTD using weighted quadratic regression, with a weighting factor of 1/concentration. Parameters of each calibration curve were used to compute concentrations for the QC samples and unknown samples by interpolation.
2.6. Method validation
2.6.1. Accuracy and precision
The method developed for cediranib quantification in mouse plasma and brain homogenate was validated for 5 different days by repeated analysis (n=5) of QC samples. The low, median and high QC levels correspond to the drug levels anticipated in most mouse samples. Precision and accuracy for the three quality controls were calculated by a single factor analysis of variance (ANOVA) to determine accuracy as well as within- and between-assay variability. The within-assay and between-assay precision was expressed as the relative standard deviation (RSD%), and the total accuracy as the percentage of mean measured (back-calculated) to nominal concentration.
2.6.2. Limit of quantification
Replicate analysis of the lower limits of quantification (LLOQ, i.e., the lower calibration level) samples was also performed. The assay LLOQ was determined following the criteria that the accuracy and precision were less than 20% with the ratio of signal/noise greater than 10, according to the FDA (US Food and Drug Administration, 2001) guidance for bioanalytical method validation. Calculations of the signal/noise ratio were based on peak areas. The peak area for background noise was measured in the plasma blank and the brain homogenate blank at the corresponding retention time window.
2.6.3. Evaluation of matrix effect
The effect of matrix was determined post extraction in both the plasma and brain homogenate. 100 µL of plasma or brain homogenate in triplicate was extracted as previously mentioned, using 800 µL of ethyl acetate. 600 µL of the supernatant was dried under nitrogen. Then 75 µL of the stock solution (10 ng/mL, 100 ng/mL and 1000 ng/mL) in methanol and 400 ng/mL ISTD was added and dried under nitrogen. The samples were then reconstituted in 75 µL of mobile phase and 10 µL was injected for analysis. The matrix effect was determined by [the ratio of the peak area of the analyte in the post-extraction matrix versus the peak area of the non-extracted sample in the reconstituted solvent (mobile phase) − 1]×100%.
2.6.4. Recovery
The plasma and homogenate samples were extracted in triplicate according to the liquid-liquid extraction procedure described previously. The extraction efficiency was determined by comparing the absolute peak areas of drug extracted from biomatrices with those of non-extracted samples in the reconstitution solvent (mobile phase). The extraction efficiency of the ISTD was assessed with an identical procedure. Analyses were performed in triplicate with three levels of cediranib (10, 100 and 1000 ng/mL) and 40 ng/mL ISTD.
2.6.5. Analyte stability
The stability over five freeze-thaw cycles was evaluated for the QC samples stored at −80°C for plasma and brain homogenate. The freeze-thaw stability was expressed as a percentage of the drug concentration measured after five freeze-thaw cycles compared to the concentration on day one. Frozen samples were thawed at a 37°C water bath and refrozen each day at −80°C. The long-term stability of the stock solution of cediranib in methanol in −80C was determined by comparing the absolute peak area of the analyte in triplicate when freshly prepared to the absolute peak area of the analyte after 29 weeks in −80C.
2.7. Method application
The described analytical method has been applied to the brain and plasma pharmacokinetic study of cediranib in mice. 28 wild-type FVB mice were randomly assigned to seven groups. Mice in each group were dosed via oral gavage with 5mg/mL cediranib suspended in 10% (w/v) Tween 80 aqueous solution as vehicle. At various time points (0.25, 0.5, 1, 2, 4, 8, 20 h postdose), mice (n=4) were euthanized. Blood samples (~400 µL) were immediately harvested into plastic tubes containing 20 uL of 100 unit/mL heparinized saline via cardiac puncture and whole brain was collected within 3 min and rinsed with ice-cold saline to remove extraneous blood. At the end of the experiment, plasma was separated from blood by centrifugation at 3500 rpm for 10 min at 4°C. Plasma and whole brain samples were stored at −80°C until analysis. All studies were approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Pharmacokinetic data analysis was performed using Phoenix WinNonlin 6.1® (Pharsight, CA).
3. Results and discussion
3.1. Chromatography
Some TKIs have similar physicochemical properties, hence we chose AG1478 as the ISTD for cediranib since deuterated analogues of cediranib were not available. Optimal resolution of analyte and ISTD peaks from one another and from the void volume was achieved with 62% aqueous mobile phase when run isocratically at 0.25 mL/min. With the chromatographic conditions described above, retention time ranges were 3.8–4.0 min for cediranib and 6.3–6.9 min for the ISTD AG1478. Typical chromatograms of the highest QC sample and LLOQ in plasma and brain homogenate are shown in Fig. 2. The low background from the biological matrix and the sharp and symmetrical resolution of the peaks show good selectivity for cediranib and AG1478.
Fig. 2.
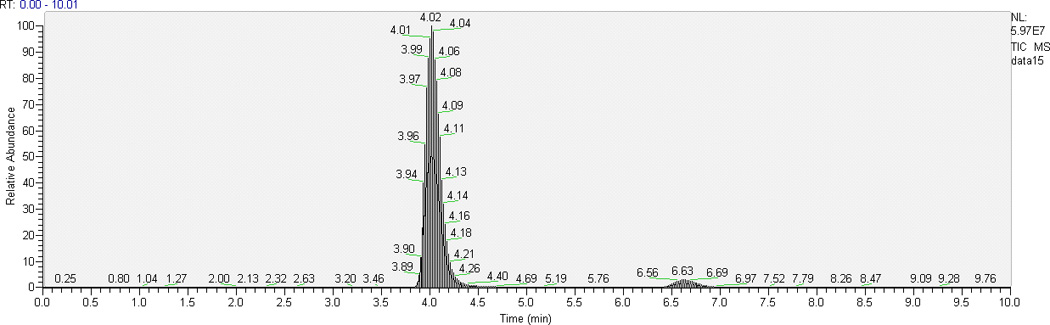
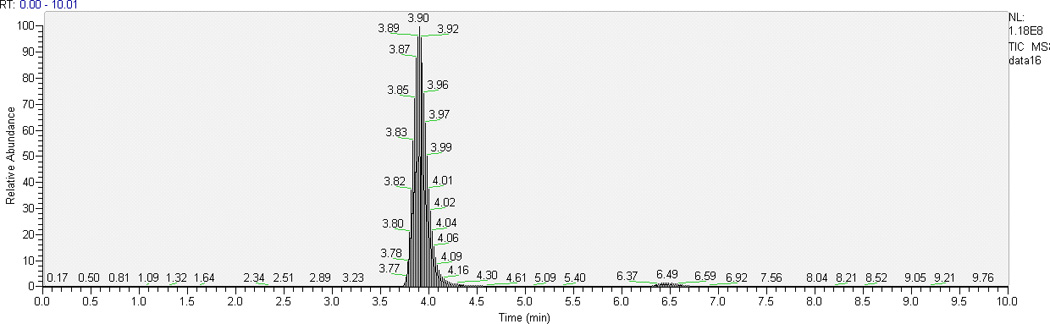
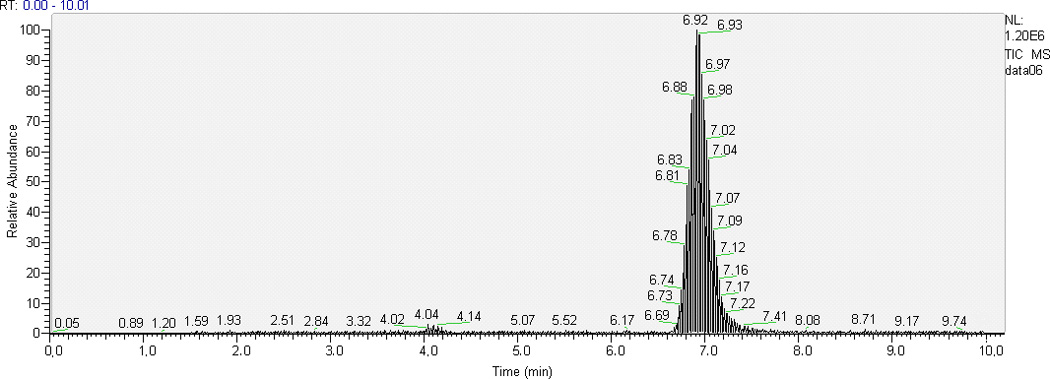
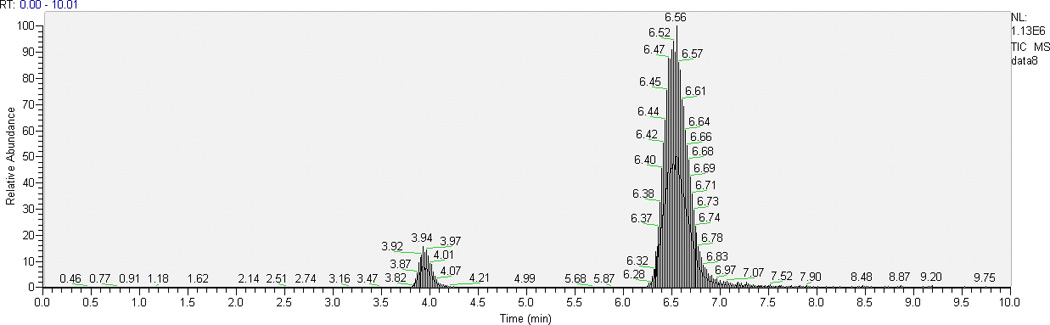
Representative HPLC-MS/MS chromatograms of plasma and brain homogenate extracts: (a) chromatogram of the highest level of calibrator samples in ; 1, plasma; 2, brain homogenate; (b) chromatogram of LLOQ (the lowest level of calibrator samples) in 1, plasma; 2, brain homogenate.
3.2. Linearity, accuracy, precision and sensitivity
Calibration curves over the entire ranges of concentrations (2.5–2500 ng/mL for plasma samples; 1–2000 ng/mL for brain samples) were adequately described by 1/concentration weighted quadratic regression of the peak-area ratios of cediranib to its ISTD and the nominal cediranib concentrations, with regression coefficient r2 always greater than 0.9990 in all analytical runs. The weighting factor was selected based on evaluation of the r2 value and the deviation of back-calculated calibrators from nominal values (% Diff). The assay LLOQ was determined in five aliquots to be 2.5 ng/mL in plasma and 1 ng/mL in brain homogenate. The ANOVA performed on the three QCs from 5×5 validation runs resulted in twenty and four degrees of freedom for within- and between-assay comparisons, respectively. The levels of QCs were selected to reflect the range of concentrations found in mouse plasma and brain homogenate after typical dosing. Accuracy and precision (RSD%) assessments are shown in Table 2. For plasma samples, within-assay variabilities ranged from 1.1 to 1.3% and between-assay variabilities ranged from 2.4 to 6.0%. Additionally, for brain homogenate samples, within-assay variabilities ranged from 1.5 to 3.7% and between-assay variabilities ranged from 4.9 to 6.0%. Overall accuracy was described by the percentage of the grand mean of each calculated concentration to the nominal concentration, and ranged from 102.1 to 107.0% for plasma and 96.5 to 99.8% for brain homogenate, respectively, for all target concentrations. Both within-assay and between-assay variabilities were within ±10% for QCs and within ±20% for LLOQs. These results satisfied the acceptance criteria in the FDA guidance and the assay was suitable in terms of accuracy and precision.
Table 2.
Within-assay and between-assay variabilities and accuracy of the HPLC-MS/MS assay for cediranib in mouse plasma and brain homogenate.
| Matrix | Nominal (ng/mL) | RSD (%) | Accuracy (%) |
|
|---|---|---|---|---|
| Within-assay | Between-assay | |||
| Plasma | 2.5 (LLOQ) | 14.3 | 9.2 | 101.5 |
| 15 (Low QC) | 1.3 | 6.0 | 107.0 | |
| 200 (Medium QC) | 1.1 | 4.7 | 104.0 | |
| 800 (High QC) | 1.2 | 2.4 | 102.1 | |
| Brain | 1 (LLOQ) | 9.4 | 10.2 | 100.2 |
| 5 (Low QC) | 3.7 | 4.9 | 96.5 | |
| 50 (Medium QC) | 1.5 | 6.0 | 99.8 | |
| 200 (High QC) | 1.7 | 5.5 | 98.2 | |
3.3. Matrix effect and extraction efficiency
The bio-matrix effect on analyte ionization was assessed under the utilized chromatographic and extraction conditions for three concentrations of cediranib and one concentration of AG1478. As shown in Table 3a and 3b, the absolute plasma effect was −38% ~ −49% for cediranib and −53% for ISTD; the absolute brain homogenate effect was −36 ~ −53% for cediranib and −45% for ISTD. These results indicated that the plasma and brain extract caused ionization suppression for the compounds.
Table 3.
Matrix effect on ionization suppression of cediranib and ISTD.
| a. Matrix effect on cediranib (Mean ± SD) | ||
| ng/mL | Plasma | Brain Homogenate |
| 10 | −39.1% ± 4.2% | −35.6% ± 4.9% |
| 100 | −38.1% ± 2.0% | −46.8% ± 0.8% |
| 1000 | −48.6% ± 1.3% | −53.0% ± 1.4% |
| b. Matrix effect on AG1478 (Mean ± SD) | ||
| ng/mL | Plasma | Brain Homogenate |
| 40 | −52.6% ± 8.5% | −44.8% ± 4.1% |
The recovery of cediranib from spiked plasma samples and brain homogenate samples were calculated by comparing the peak area of extracted samples at 10, 100 and 1000 ng/mL, using the extraction conditions described above, with those from corresponding unextracted samples in mobile phase. Extraction efficiency for ISTD at 40 ng/mL was also assessed. High and similar recoveries using this extraction method were obtained for both compounds. Average recoveries for cediranib at all three evaluated concentration levels and the ISTD were above 80% in both plasma and brain homogenate. These recoveries allowed quantification of cediranib to as low as to 1 ng/mL from a sample volume of 100µL. The recovery results are summarized in Table 4.
Table 4.
Absolute recoveries for cediranib and ISTD in the mouse plasma and brain homogenate.
| a. Cediranib recovery (Mean ± SD) | ||
| ng/mL | Recovery in Plasma | Recovery in Brain Homogenate |
| 10 | 83.2% ± 1.1% | 101.6% ± 2.0% |
| 100 | 79.0 % ± 0.7% | 80.0% ± 0.6% |
| 1000 | 83.5% ± 2.4% | 89.5% ± 3.4% |
| b. AG1478 recovery (Mean ± SD) | ||
| ng/mL | Recovery in Plasma | Recovery in Brain Homogenate |
| 40 | 85.7% ± 3.6% | 81.7% ± 0.6% |
3.4. Stability
For freeze-thaw stability, some QC samples (n=5 for each concentration level) were extracted on day one and the rest of them were frozen at −80°C, then thawed, extracted and refrozen each day for further studies. Cediranib concentrations at −80°C undergoing four freeze-thaw cycles in mouse plasma and brain homogenate were all within ± 15% of the starting concentrations, indicating that cediranib can be considered stable in stored plasma and homogenate samples at all three concentration levels and that there was no concentration-dependent degradation observed. Percentage recoveries for cediranib at each level and in both biological matrices are summarized in Table 5a and 5b. Evaluation of more than one freeze-thaw cycle was performed in case that occasionally samples will be analyzed more than once.
Table 5.
Freeze - thaw stability of the plasma and brain homogenate samples. a. cediarnib stability in mouse plasma; b. cediarnib stability in mouse brain homogenate.
| a. | |||||
| ng/mL | 1st freeze-thaw cycle % recovery |
2nd freeze-thaw cycle % recovery |
3rd freeze-thaw cycle % recovery |
4th freeze-thaw cycle % recovery |
5th freeze-thaw cycle % recovery |
| 15 | 100.0% | 103.8% | 108.2% | 99.1% | 114.4% |
| 200 | 100.0% | 103.6% | 108.9% | 98.3% | 97.1% |
| 800 | 100.0% | 97.8% | 103.8% | 98.8% | 99.1% |
| b. | |||||
| ng/mL | 1st freeze-thaw cycle % recovery |
2nd freeze-thaw cycle % recovery |
3rd freeze-thaw cycle % recovery |
4th freeze-thaw cycle % recovery |
5th freeze-thaw cycle % recovery |
| 5 | 100.0% | 93.8% | 98.9% | 103.0% | 110.4% |
| 50 | 100.0% | 92.5% | 93.7% | 106.7% | 102.3% |
| 200 | 100.0% | 89.1% | 93.6% | 99.7% | 102.0% |
Evaluation of bench-top stability at room temperature was not deemed necessary as all samples were kept on the bench for less than 15 minutes. Long-term stability of cediranib stock solution in MeOH was evaluated under −80°C. The results are shown in Table 6. The long-term stability of low, median, and high levels of cediranib stock solution was all within ± 30% for up to 29 weeks.
Table 6.
Long - term stability of cediranib stock solution in methanol.
| Cediranib (ng/mL) |
Long-term Stability (Mean ± SD) |
|---|---|
| 10 | 86% ± 1.8% |
| 100 | 122% ± 2.3% |
| 1000 | 130% ± 4.6% |
3.5. Method application
The established HPLC-MS/MS assay was applied to study the brain and plasma pharmacokinetics of cediranib in FVB mice following a single oral dose of 5 mg/kg cediranib suspension. The assay was found to be sufficiently sensitive and accurate for determining cediranib in plasma and brain samples in order to characterize its pre-clinical pharmacokinetics and brain distribution in mice. Fig. 3 shows the plasma and brain concentration-time profiles (means±S.D.) at 0.25, 0.5, 1, 2, 4, 8, 20 hours post-dose. All measured concentrations were above the LLOQ. The maximum plasma concentration (Cmax) achieved was 510 ± 205 ng/mL and was reached at 4 hour after oral dosing; the apparent oral clearance and terminal half-life of cediranib in the plasma were determined to be 28.8 mL/hour and 3.5 hours, respectively. However, the brain delivery of cediranib was limited. The brain area under the curve from time zero to the last time point (AUC) was 849 ± 80 hour×ng/mL, and only 21.8% compared with that in the plasma, which was 3892 ± 434 hour×ng/mL.
Fig. 3.
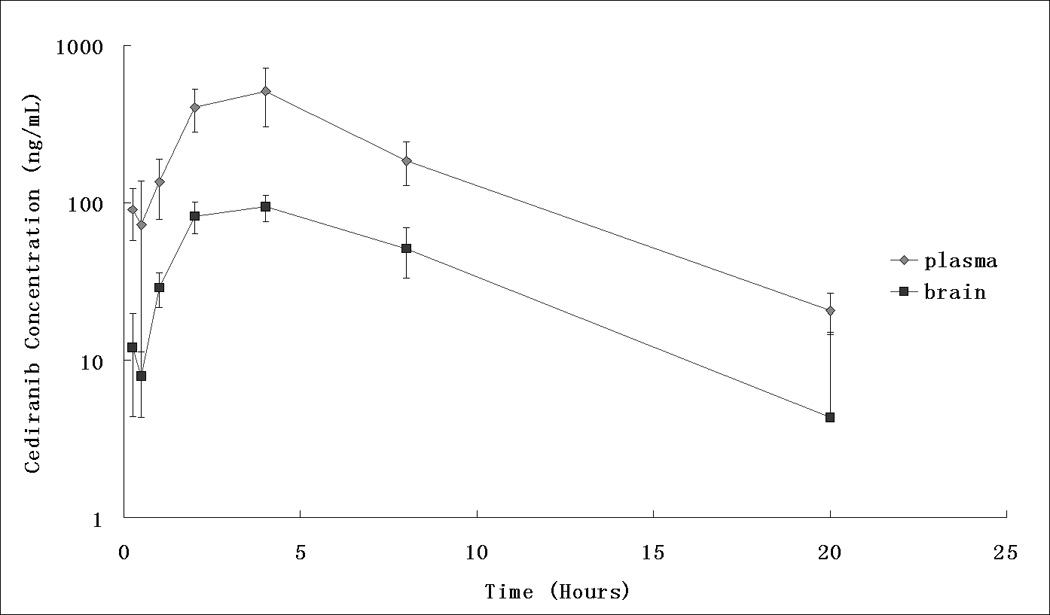
Mouse plasma ( ) and brain (
) and brain ( ) pharmacokinetic profiles of cediranib following 5 mg/kg oral administration.
) pharmacokinetic profiles of cediranib following 5 mg/kg oral administration.
4. Conclusion
In conclusion, a robust, sensitive and reproducible HPLC-MS/MS assay for quantification of cediranib in mouse plasma and brain homogenate samples has been developed and validated. This is the first report for quantification of cediranib in two biomatrices with high accuracy and precision. The liquid-liquid extraction procedure gives a high extraction yield and is simple to apply. The quantification range of 2.5–2500 ng/mL is sufficient to allow both plasma and brain tissue pharmacokinetic studies. Furthermore, given the sensitivity and the relatively small sample volume (50 µL for plasma and 100µL for brain homogenate), the present assay is also feasible for and has been applied in our laboratory to in vitro studies of cediranib (e.g., intracellular uptake studies). It may help fill the current knowledge gaps in preclinical pharmacokinetic studies of cediranib in small specimens.
Highlights.
-
-
This is the first study to describe a sensitive and precise LC-MS assay for cediranib.
-
-
This assay allows small specimen volumes and adequate detectability to use in tissue distribution studies.
-
-
We have employed the assay in a brain distribution study in the mouse, to examine different mechanisms that limit the CNS distribution of cediranib.
-
-
The assay is generally applicable to a variety of pharmacokinetic and in vitro cell culture experiments
Acknowledgements
We thank James Fisher (Clinical Pharmacology Analytical Services Laboratory, Department of Experimental and Clinical Pharmacology, University of Minnesota) for his assistance with the method development. This work was supported by National Institutes of Health grant R01 CA 138437.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sharma PS, Sharma R, Tyagi T. Curr Cancer Drug Targets. 2011 Nov;:624. doi: 10.2174/156800911795655985. [DOI] [PubMed] [Google Scholar]
- 2.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J, Hochberg FH, Benner T, Louis DN, Cohen KS, Chea H, Exarhopoulos A, Loeffler JS, Moses MA, Ivy P, Sorensen AG, Wen PY, Jain RK. J Clin Oncol. 2010;28:2817. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeda M, Arao T, Yokote H, Komatsu T, Yanagihara K, Sasaki H, Yamada Y, Tamura T, Fukuoka K, Kimura H, Saijo N, Nishio K. Clin Cancer Res. 2007;13:3051. doi: 10.1158/1078-0432.CCR-06-2743. [DOI] [PubMed] [Google Scholar]
- 4.Siemann DW, Brazelle WD, Jurgensmeier JM. Int J Radiat Oncol Biol Phys. 2009;73:897. doi: 10.1016/j.ijrobp.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S, Richmond GH, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S, Jurgensmeier JM, Ogilvie DJ. Cancer Res. 2005;65:4389. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 6.Medinger M, Esser N, Zirrgiebel U, Ryan A, Jurgensmeier JM, Drevs J. Anticancer Res. 2009;29:5065. [PubMed] [Google Scholar]
- 7.Dietrich J, Wang D, Batchelor TT. Expert Opin Investig Drugs. 2009;18:1549. doi: 10.1517/13543780903183528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenz EM, Spear M, Drake C, Pollard CR, Ward M, Schulz-Utermoehl T, Harrison M. J Pharm Biomed Anal. 2010;53:526. doi: 10.1016/j.jpba.2010.03.023. [DOI] [PubMed] [Google Scholar]