

Abstract
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase involved in cell growth that is often misregulated in cancer. Several recent studies highlight the unique structural mechanisms involved in its regulation. Some elucidate the important role that the juxtamembrane segment and the transmembrane helix play in stabilizing the activating asymmetric kinase dimer, and suggest that its activation mechanism is likely to be conserved amongst the other human EGFR-related receptors. Other studies provide new explanations for two long observed, but poorly understood phenomena, the apparent heterogeneity in ligand binding and the formation of ligand-independent dimers. New insights into the allosteric mechanisms utilized by intracellular regulators of EGFR provide hope that allosteric sites could be used as targets for drug development.
Introduction
The epidermal growth factor (EGF) was discovered in 1962 [1] and since the initial characterization of its interaction with a cell surface receptor in 1975 [2], the epidermal growth factor receptor (EGFR) has been one of the most intensely studied receptor tyrosine kinases. EGFR and its relatives, human EGF receptors 2,3 and 4 (Her2, Her3 and Her4; also known as Erb2, Erb3 and Erb4) play important roles in cell growth and differentiation and are misregulated in numerous cancers [3]. The binding of growth factors such as EGF to these receptors induces the dimerization and activation of their cytosplasmic kinase domains, resulting in the phosphorylation of tyrosine residues in their C-terminal tails and recruitment of downstream effectors [4,5].
The first structural insights into how EGF induces the activation of the receptor, came from crystal structures of the EGFR extracellular domain with and without bound ligand, which showed that ligand binding causes a conformational change that enables dimerization, with the dimer interface mediated entirely by the receptor [6-9]. These findings suggested that EGFR might be activated by a mechanism shared by many other receptor tyrosine kinases, in which ligand-induced dimerization brings the kinase domains close enough together to activate each other through trans-phosphorylation [10,11]. But, unexpectedly, structural and functional studies revealed that the catalytic domain is activated by the formation of an asymmetric dimer in which one kinase domain, the ‘activator’ acts as an allosteric activator for a second ‘receiver’ kinase domain [12] (Figure 1). This activation mechanism resembles the activation of cyclin-dependent kinases by cyclins, with the EGFR kinase domain acting as its own ‘cyclin ’ [11,12]. Formation of an asymmetric dimer explain how Her2, which lacks a known ligand, is activated by Her3, which can bind ligand but has a structurally impaired catalytic site and only residual kinase activity [9].
Figure 1. A simple model for EGFR activation.
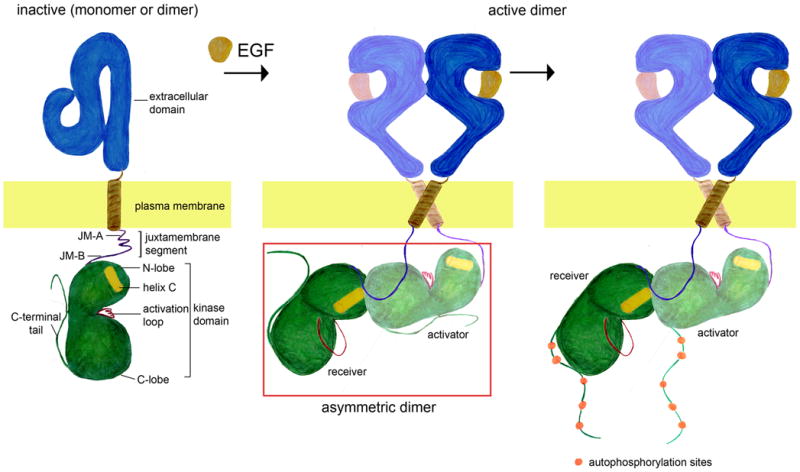
Activation of EGFR by binding of EGF results in the formation of the asymmetric kinase dimer, and phosphorylation of the C-terminal tail of the receptor. Note that in the absence of ligand EGFR is shown as a monomer, but it ligand-independent dimer may also form [20-24].
In this review we discuss several recent studies that have revealed more complexity and nuance in the structural mechanisms that regulate this important family of receptors. New structural and biochemical results explain how the juxtamembrane segment and the transmembrane helix contribute to the activation of the receptor by stabilizing the asymmetric kinase dimer [13-15]. Crystal structures of the Her2, Her3 and Her4 kinase domains suggest that the asymmetric activation mechanism of the kinase is conserved among the other EGFR family members [16-19]. Although earlier studies uncovered the existence of ligand-independent dimers [20-23], a single molecule study provides clearer evidence of their existence and suggests that they could play an important role in the activation of the receptor [24]. New EGF binding studies [25,26] and structures of the Drosophila EGFR extracellular domain [27,28]provide evidence for negative cooperativity in ligand binding. The allosteric nature of the activation of EGFR provides an opportunity for other molecules to use these interactions to either activate or inhibit the receptor, and both activator and inhibitor proteins have been identified and characterized [29,30].
The juxtamembrane segment of EGFR stabilizes the asymmetric dimer of kinase domains
The section of the receptor located between the kinase domain and the membrane spanning transmembrane helix is known as the juxtamembrane segment (residues 645-682, using numbering from human EGFR and not including the signal sequence, Figure 1). The juxtamembrane segment of EGFR was shown to be necessary for its activation [31]. A clue for how this activation occurs came from a crystal structure of the Her4 kinase domain bound to a small molecule inhibitor, in which the C-terminal half of the juxtamembrane segment of the receiver kinase latches the activator kinase to the receiver [32]. Although this aspect of the structure was not commented on in the original report, a similar “juxtamembrane latch” was later shown to be required for full activation of EGFR auto-phosphorylation [13] (Figure 2). Independently, a new crystal structure of the EGFR kinase showed that the juxtamembrane latch does indeed stabilize the asymmetric dimer, and based on this structure the authors provided a rationale for how two oncogenic mutations in the juxtamembrane segment could enhance the activation of the kinase [14]. It is interesting that in both the EGFR [14] and Her4 [32] structures, the kinase domain is actually in an inactive conformation as a result of mutation (EGFR) or inhibitor binding (Her4). Nevertheless, the kinase domains are able to form the asymmetric dimer, reinforcing the important role that the juxtamembrane latch plays in stabilizing this interaction.
Figure 2. Structural model for the activated receptor.
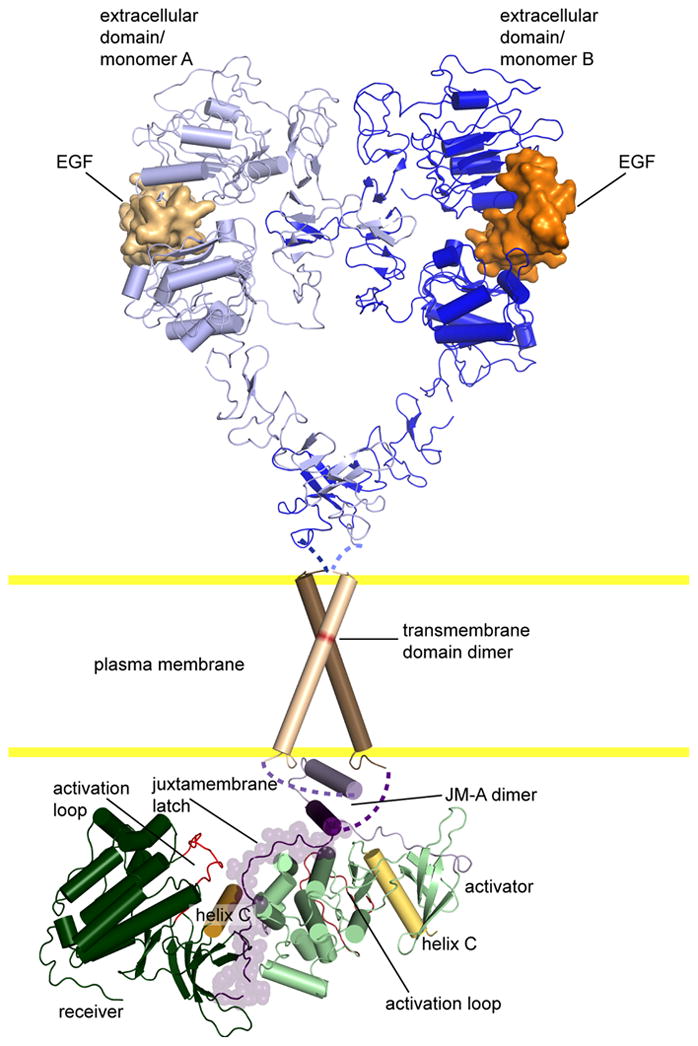
Our current structural model for the activated receptor, based on crystal structures of the extracellular and kinase domains [12-14,32,62]. The transmembrane helices and the anti-parallel coiled-coil are modeled based on NMR studies [13,15]. Note that the extracellular domain is extends upon ligand binding stabilizing an extensive dimer interface. This extended conformation provides the proper spacing for the transmembrane domains to dimerize via the N-terminal GxxxG motif, the JM-A region to forms an anti-parallel coiled-coil, and the active asymmetric kinase dimer to be stabilized by the juxtamembrane latch.
Biochemical studies pointed to an important role for the N-terminal portion of the juxtamembrane in the activation of the kinase [13]. In the EGFR crystal structure [14], this region of the juxtamembrane segment forms an α helix, which protrudes from the kinase and is not involved in stabilizing the dimer interface. This conformation allows the repeating juxtamembrane latch interaction to stabilize a daisy chain of kinase domains in the crystal. Nevertheless, biochemical data support a closed dimer model for kinase activation in which the juxtamembrane segments of the receiver and activator kinases interact with each other [13]. In this model, the juxtamembrane latch only forms on the activator kinase and the N-terminal juxtamembrane segment dimerizes by forming an antiparallel coiled-coil, which has shown to be possible by NMR analysis of an isolated peptide corresponding to the juxtamembrane segment of EGFR [13] (Figure 2).
The role of the transmembrane helices in receptor activation
The importance of the transmembrane helices of EGFR in receptor activation was made clear by the discovery that a single substitution (Val to Glu) in the transmembrane helix of Her2 is associated with cancer [33][34]. Recently, the structure of the transmembrane helix of Her2 was determined in a lipid bilayer environment by NMR [15]. In this structure, two transmembrane helices of Her2 form a symmetric dimer with tight helical packing through an N-terminal GxxxG motif, leaving the C-terminal ends splayed apart. This is reminiscent of the dimer formed by the transmembrane helices of Glycophorin A (GpA), which has become a model system for studying transmembrane helical dimerization [35]. A similar structure is observed in a heterodimer of the EGFR and Her2 transmembrane helices [36]. Although there is currently no structure of the EGFR transmembrane domain, cross-linking and molecular dynamics studies suggest that it also forms a similar dimer [37,38].
The dimerization of the transmembrane helices through their N-terminal GxxxG motifs could, in principle, synergize nicely with the formation of the active asymmetric kinase dimer by providing an optimal separation of the C-terminal ends of the transmembrane helices for the formation of an anti-parallel coiled-coil in the N-terminal portion of the juxtamembrane segment [13] (Figure 2). Cooperation between the transmembrane helices and juxtamembrane segments in the stabilization of the asymmetric kinase dimer could explain why deletion of the extracellular domain can activate the EGFR kinase [39-41]. It could also explain how the oncogenic Val to Glu substitution in the transmembrane helix of Her2, which is hypothesized to stabilize the N-terminal packing of its transmembrane helices [34], leads to constitutive activation of its kinase domains [42].
Surprisingly, extensive mutagenesis of the EGFR transmembrane helices, including mutations analogous to those that activate Her2, did not appear to alter receptor function significantly [37,43]. The free energy of transmembrane dimerization has been determined to be relatively low [44], thus, the weak effect of mutations in this region could be an indication that for EGFR the transmembrane helix may only play a minor role in the stabilization of activated dimer in the context of full length receptor. In addition to the N- terminal dimerization motif in the transmembrane helices, EGFR, Her2 and Her4 also contain a C-terminal GxxxG motif, that contributes to the dimerization of isolated transmembrane segments [45]. While more work is needed to firmly establish a role for this motif in EGFR function, modeling suggests that it could be involved in stabilizing an inactive form of the receptor [46].
Activation of the kinase through asymmetric dimer formation is likely to be conserved throughout the EGFR family
The allosteric mechanism for EGFR kinase activation is likely to be unique among receptor tyrosine kinases [10,11]. However, recent results suggest that this activation mechanism is conserved in all four EGFR family members. Crystal structures of the Her2 [19] and Her 4 [16] kinase domains reveal that these kinases can form an asymmetric dimer interface very similar to that observed for the EGFR kinase domain [12,47]. Biochemical studies have confirmed that activation of these kinases depend on the formation of this interface [17,18,48].
Her3 is regarded as a pseudo kinase due the absence of several catalytically important residues [49]. Her3 conserves the activator surface of the asymmetric dimer interface [12] and thus can activate other EGFR kinases by taking the activator position [17,18,48]. Recent crystal structures of Her3 provide further rationale for why Her3 is defective as a receiver kinase[17,18]. The N-terminal end of helix-αC, a critical component of the receiver portion of the asymmetric dimer interface, is partially unwound and permits hydrophobic interactions that stabilize its inactive conformation. Nevertheless, Her3 can bind ATP and despite having no detectable activity against peptide substrates [17], it has been shown to undergo autophosphorylation at a low rate when concentrated on lipid vesicles [18]. This is an intriguing finding, which along with a recent study demonstrating that the pseudokinase CASK has physiologically relevant kinase activity [50], has forced us to consider that the residual kinase activity of pseudokinases may play a role in signaling. Further studies are needed to see how the ability of Her3 to autophosphorylate itself carries over to its biological function.
Negative Cooperativity in the binding of EGF to EGFR
A long-standing puzzle concerning the activation of EGFR is why EGF binding leads to a “concave-up” Scatchard plot. This result has been interpreted to mean that there are two populations of EGFR, a small population with high affinity, and a larger population with lower affinity [51]. Based on an analysis of EGF binding as a function of receptor concentration, it has recently been shown that the concave-up Scatchard can instead be explained by negative cooperativity, i.e., the binding of one EGF molecule to an EGFR dimer weakens the affinity for EGF at the other binding site in the dimer [25,26]. Negative cooperativity depends on the ability of the extracellular domain to dimerize, and on the presence of the juxtamembrane region (even without the kinase domain) [26].
A surprising finding was that the nature of the concentration dependent binding of EGF to EGFR implies that receptor monomers and unbound receptor dimers have the same affinity for EGF [26]. This is counterintuitive because one would expect that if a ligand induces dimerization of a receptor, then it should have higher affinity for receptor dimers. This is likely to be a property of the phosphorylated receptor because when phosphorylation is prevented by mutations, EGFR dimers appear to bind EGF more tightly then monomers, as would be expected in a ligand-induced dimerization model [25,26]. The full implications of this observation for receptor structure and function have yet to be fully explored, but the data suggest that there could be structural differences between phosphorylated and unphosphorylated receptor that effect ligand binding.
A crystal structure of the extracellular domain of Drosophila EGFR (dEGFR), bound to its ligand Spitz suggested a potential mechanism for negative cooperativity [28] (Figure 3). In this structure, the two extracellular domains in the dimer have different structures. Both have ligand bound, but whereas one resembles the structure of the EGFR-EGF complex [9], the other has the ligand more loosely bound and resembles the extended conformation seen in the unliganded Her2 and dEGFR structures [27]. This asymmetry in binding sites provides an explanation for the negative cooperativity of Spitz binding to the dEGFR extracellular domain. It is tempting to speculate that a similar asymmetry in ligand binding may apply to members of the human EGFR family, and this could explain the observed negative cooperativity of EGF binding to EGFR. We await the results of further studies to probe this connection.
Figure 3. Structural model for Negative Cooperativity.
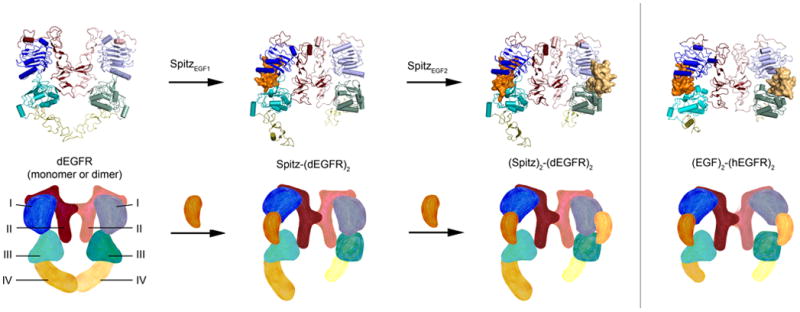
The extracellular domain is shown in four possible states observed in published crystal structures of dEGFR [27,28] and human EGFR [6] extracellular domains. Cartoon diagrams below structures depict conformational rearrangements in each state. In the unliganded structure, as observed for dEGFR and Her2 [27], domain I and domain III are collapsed on each other in both molecules of the dimer, occluding the ligand binding site and bending domain II in a way that would weaken dimerization. When one ligand binds as observed in a structure of dEGFR bound to a mutant of Spitz [28], it creates a wedge between domain I and domain III that allows for extensive contact, while the other molecule of the dimer is in a conformation similar to the unbound state. Note that the asymmetric arrangement in this dimer allows for close packing of domain II, stabilizing the dimer interface. A second ligand can bind in this asymmetric dimer in an apparently weaker interaction as seen in the structure of dEGFR bound to Spitz [28], which does not wedge domains I and III apart, but maintains the asymmetry required for the tight dimer interface. When two ligands are tightly bound as seen in the crystal structure of human EGFR bound to EGF [6], the imposed symmetry of the dimer weakens the dimer interface. Figure was adapted from [28].
The role of ligand-independent dimerization in EGFR activation
Over the past 15 years several studies have demonstrated that EGFR can dimerize in the absence of ligand [20-23], yet the relevance of this phenomenon for regulation has not yet been firmly established. A recent study in which EGFR dimerization was tracked in living cells at the single molecule level, provides new insights into both the dynamics and function of ligand-independent dimerization [24]. Extracellular domain antibody fragments (Fab) were labeled with quantum dots and used to observe the diffusion of endogenous EGFR in a variety of cell types. EGFR was found to transition between states of high and low mobility in the absence of ligand, which the authors interpreted as a transition between monomers and dimers. EGF binding to the receptor led to a dramatic drop in the diffusion constant, possibly due to linkage to signaling complexes or actin.
An important finding of this study was that EGF binding only occurred when the receptor was in a dimeric state, implying that ligand-independent dimerization may play an essential role in EGFR activation by priming the receptor for ligand binding. By examining cell lines with a variety of EGFR expression levels, the authors observe that while the frequency of dimerization is concentration-dependent, the relationship between ligand-independent dimerization and both ligand-binding and activation of the receptor is conserved. This observation suggests that ligand-independent dimerization could still play an important role in receptor function, even when the level of ligand-independent dimerization is low [52].
Although ligand-independent dimerization appears to be a fairly robust phenomenon, there are several unanswered questions about its structure and function. A potential solution to the mystery of how kinase activity is prevented is provided by an inactive kinase domain reported originally in 2009 [53], but also observed in a more recent crystal structure [54]. The dimer is stabilized by the C-terminal tails of the kinase domains, which are both in the inactive conformation. The C-terminal tails in this structure also occlude the docking of the juxtamembrane latch [13]. Although a clear link between this inactive kinase dimer and receptor auto-inhibition has yet to be established, an auto-inhibitory mechanism involving the C-terminal tail could help to explain the oncogenic effect of C-terminal tail deletions [55]. Further complicating our understanding of receptor structure in the plasma membrane is the observation that ligand binding induces the formation of higher order clusters of EGFR (tetramer or higher) [21,56][57]. It remains to be seen whether the formation of these clusters is a result of kinase activity and effector recruitment [57], or a necessary step in receptor activation [21].
Intracellular regulators of EGFR kinase activity may use allosteric mechanisms
Although we have known about intracellular regulators of EGFR activity for years [58,59], recent studies have suggested that these regulators could work by interfering with or promoting allosteric mechanisms of activation [29,30]. A crystal structure of the EGFR kinase domain in complex with the inhibitor Mig6, suggests that this inhibitor works in part by occluding the activator interface and preventing asymmetric dimer formation [30]. Cytohesins were recently demonstrated to be the first known intracellular activators of EGFR activity [29]. Although the structural mechanism for this activation is not yet understood, the authors provide strong evidence that cytohesins enhance the activation of already dimerized receptors, by facilitating conformational rearrangements upon directly binding to the kinase domain or the juxtamembrane segment.
The existence of cytosplamic regulators of EGFR activity suggests that it may be possible to design drugs to modulate EGFR activity via allosteric interfaces. In the EGFR kinase, helix αC helps to form a hydrophobic pocket in the receiver kinase that is critical for the activation of the kinase. A similar hydrophobic pocket is involved in the allosteric regulation of several kinases [11]. Recently, small molecules targeting the PDK1 hydrophobic pocket were discovered that either activate or inhibit the kinase [60,61]. A similar strategy could be used to design inhibitors of EGFR family kinase domains that would block activation of the receptor through the asymmetric dimer interface. Such a compound would create a new class of EGFR family inhibitors that have the potential for increased specificity since they would not target highly conserved active site.
Conclusions
Like a Russian doll, each new insight into the regulation of EGFR opens up new surprises. Instead of a simple two-step model for activation, recent studies suggest that there are multiple layers in the regulation of EGFR activity. In the absence of ligand, EGFR is in a concentration-dependent equilibrium between monomers and dimers. Ligand-independent dimerization may inhibit kinase activity [13] and may also be a prerequisite for EGF binding [24]. In addition to promoting extracellular domain dimerization, EGF binding must also reorient the receptor in such a way that allows the transmembrane, juxtamembrane and kinase domains to form the activating asymmetric dimer [13,14]. This orientation may be sensitive to both the stoichiometry of ligand binding and the phosphorylation state of the receptor [26,28].
The complexity of the EGFR regulation mechanism, though potentially vexing for those who crave elegant simplicity, is likely reflective of the critical role the receptor plays in determining the fate of the cell. Great progress has been made in understanding this mechanism by combining insights gained from structures, with increasingly sophisticated biophysical techniques for monitoring receptor oligomerization and activation. In addition to being intellectually satisfying, these studies have the potential to have important impacts on cancer treatment, because each insight gained into the allosteric regulatory mechanisms of the receptor could provide a new strategy for drug development targeting EGFR or one its family members.
Highlights.
> Review of structural mechanisms regulating EGFR activation.
> New structures show juxtamembrane segment stabilizes activating asymmetric kinase dimer.
> Studies suggest kinase activation mechanism conserved throughout EGFR family.
> New structures suggest mechanism for negative cooperativity in ligand binding.
> New insights into role of ligand-independent dimerization in receptor function.
> Recent findings suggest intracellular regulators use allosteric mechanisms.
Acknowledgments
We thank A. Cantor, T. Chen, Y. Huang and J. Iwig for initial discussion on the content of the review. Work on EGFR in this lab was supported in part by a grant from the National Cancer Institute to J.K. (ROI CA96504-06) and the Susan G. Komen Society (K.E.). N.E. was a fellow of the Leukemia and Lymphoma Society. R.D. is a fellow of the National Science and Engineering Research Council of Canada.
Footnotes
Conflict of interest: The authors have no conflict interest…(should we mention pending patent on drug design using asymmetric dimer interface?)
Submission Declaration: This work is approved by all the authors and will not be published elsewhere.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem. 1962;237:1555–1562. [PubMed] [Google Scholar]
- 2.Carpenter G, Lembach KJ, Morrison MM, Cohen S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J Biol Chem. 1975;250:4297–4304. [PubMed] [Google Scholar]
- 3.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Yarden Y, Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26:1443–1451. doi: 10.1021/bi00379a035. [DOI] [PubMed] [Google Scholar]
- 5.Yarden Y, Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–1442. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]
- 6.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 7.Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 9.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 10.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- **13.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. Biochemical studies show that the C-terminal portion of the juxtamembrane segment stabilizes the activating asymmetric dimer, and the N-terminal portion of the juxtamembrane segment also contributes to activation by forming an anti-parallel helical dimer. A new crystal structure of an inactive EGFR kinase domain also suggests a mechanism for receptor auto-inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **14.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. A new crystal structure of the EGFR kinase domain shows how the juxtamembrane region stabilizes the asymmetric dimer. Biochemical studies support an important role for the juxtamembrane region in the activation of the receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **15.Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, Sobol AG, Chupin VV, Kirpichnikov MP, Efremov RG, Arseniev AS. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283:6950–6956. doi: 10.1074/jbc.M709202200. An NMR structure of the Her2 transmembrane helix in a lipid bicelle shows that it dimerizes via its N-terminal GxxxG motif. [DOI] [PubMed] [Google Scholar]
- 16.Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S, Pal A, Bornmann WG, Lemmon MA, Cole PA, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proceedings of the National Academy of Sciences. 2009;106:21608–21613. doi: 10.1073/pnas.0912101106. A crystal structure of the Her3 kinase domain bound to ATP explains why Her3 cannot take the receiver position in the asymmetric dimer. While Her3 does not have significant catalytic activity when concentrated on lipid vesicles, it can activate an EGFR kinase domain whose activator interface is impaired. A crystallographic dimer is discussed, which could represent and auto-inhibited form of the kinase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. A crystal structure of the Her3 kinase domain bound to an ATP analog is presented. In vitro experiments suggest that the Her3 kinase doman, though generally regarded as a pseudokinase, can bind ATP and can phosphorylate itself when concentrated on lipid vesicles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, et al. Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. J Biol Chem. 2011;286:18756–18765. doi: 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sako Y, Minoghchi S, Yanagida T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat Cell Biol. 2000;2:168–172. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 21.Clayton A, Orchard S, Nice E, Posner R, Burgess A. Predominance of activated EGFR higher-order oligomers on the cell surface. GGRF. 2008;26:316–324. doi: 10.1080/08977190802442187. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Fernandez M, Clarke DT, Tobin MJ, Jones SV, Jones GR. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys J. 2002;82:2415–2427. doi: 10.1016/S0006-3495(02)75585-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadella TW, Jovin TM. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. The Journal of Cell Biology. 1995;129:1543–1558. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **24.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464:783–7. doi: 10.1038/nature08827. Single molecule diffusion rates of endogenously expressed EGFR transition between a fast and slow rate in the absence of ligand, suggesting a monomer to dimer transition. EGF binding to the receptor, leads to a sharp drop in diffusion rate and only occurs when the receptor is in a dimeric state. [DOI] [PubMed] [Google Scholar]
- **25.Macdonald JL, Pike LJ. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc Natl Acad Sci USA. 2008;105:112–117. doi: 10.1073/pnas.0707080105. An analysis of EGF binding to EGFR as a function receptor level, shows that the “concave-up” nature of Scatchard plots is best interpreted as negative cooperativity of ligand binding, in which the binding of the first ligand to an EGFR dimer weakens the affinity of the second ligand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **26.Macdonald-Obermann JL, Pike LJ. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J Biol Chem. 2009;284:13570–13576. doi: 10.1074/jbc.M109.001487. The contribution of the intracellular domain to the binding of EGF to EGFR is examined. The results suggest that the juxtamambrane segment is responsible for the observed negative cooperativity in ligand binding, and that the phosphorylation of the receptor effect the relative affinity of ligand for receptor monomers or dimers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarado D, Klein D, Lemmon M. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature. 2009;461:287–91. doi: 10.1038/nature08297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **28.Alvarado D, Klein DE, Lemmon MA. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell. 2010;142:568–579. doi: 10.1016/j.cell.2010.07.015. A crystal structure of the extracellular domain of Drosophila EGFR bound to its ligand Spitz, is in an asymmetric arrangement that suggest a potential mechanism for the negative cooperativity of ligand binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **29.Bill A, Schmitz A, Albertoni B, Song JN, Heukamp LC, Walrafen D, Thorwirth F, Verveer PJ, Zimmer S, Meffert L, et al. Cytohesins are cytoplasmic ErbB receptor activators. Cell. 2010;143:201–211. doi: 10.1016/j.cell.2010.09.011. Cytohesins are shown to activate EGFR and to bind preferentially to dimerized EGFR. [DOI] [PubMed] [Google Scholar]
- **30.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–744. doi: 10.1038/nature05998. A crystal structure of the EGFR kinase domain bound to the inhibitor Mig6, shows that Mig6 inhibits the EGFR kinase domain by blocking asymmetric dimer formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proceedings of the National Academy of Sciences. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood ER, Shewchuk LM, Ellis B, Brignola P, Brashear RL, Caferro TR, Dickerson SH, Dickson HD, Donaldson KH, Gaul M, et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc Natl Acad Sci USA. 2008;105:2773–2778. doi: 10.1073/pnas.0708281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 34.Sternberg MJ, Gullick WJ. Neu receptor dimerization. Nature. 1989;339:587. doi: 10.1038/339587a0. [DOI] [PubMed] [Google Scholar]
- 35.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 36.Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV, Arseniev AS. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. Journal of Molecular Biology. 2010;400:231–243. doi: 10.1016/j.jmb.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 37.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S, Springer TA. Structural Evidence for Loose Linkage between Ligand Binding and Kinase Activation in the Epidermal Growth Factor Receptor. Molecular and Cellular Biology. 2010;30:5432–5443. doi: 10.1128/MCB.00742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakash A, Janosi L, Doxastakis M. Self-association of models of transmembrane domains of ErbB receptors in a lipid bilayer. Biophys J. 2010;99:3657–3665. doi: 10.1016/j.bpj.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chantry A. The kinase domain and membrane localization determine intracellular interactions between epidermal growth factor receptors. J Biol Chem. 1995;270:3068–3073. [PubMed] [Google Scholar]
- 40.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu HJ, Iaria J, Orchard S, Burgess AW, Walker F. Epidermal Growth Factor Receptor: Association of Extracellular Domain Negatively Regulates Intracellular Kinase Activation in the Absence of Ligand. GGRF. 2003;21:15–30. doi: 10.1080/0897719031000096424. [DOI] [PubMed] [Google Scholar]
- 42.Smith SO, Smith C, Shekar S, Peersen O, Ziliox M, Aimoto S. Transmembrane interactions in the activation of the Neu receptor tyrosine kinase. Biochemistry. 2002;41:9321–9332. doi: 10.1021/bi012117l. [DOI] [PubMed] [Google Scholar]
- 43.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Merzlyakov M, Cohen T, Shai Y, Hristova K. Energetics of ErbB1 Transmembrane Domain Dimerization in Lipid Bilayers. Biophysj. 2009;96:4622–4630. doi: 10.1016/j.bpj.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mendrola JM. The Single Transmembrane Domains of ErbB Receptors Self-associate in Cell Membranes. J Biol Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 46.Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci USA. 2002;99:15937–15940. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stamos J. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 48.Monsey J, Shen W, Schlesinger P, Bose R. Her4 and Her2/neu tyrosine kinase domains dimerize and activate in a reconstituted in vitro system. J Biol Chem. 2010;285:7035–7044. doi: 10.1074/jbc.M109.096032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci USA. 1994;91:8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukherjee K, Sharma M, Urlaub H, Bourenkov GP, Jahn R, Südhof TC, Wahl MC. CASK Functions as a Mg2+-independent neurexin kinase. Cell. 2008;133:328–339. doi: 10.1016/j.cell.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein P, Mattoon D, Lemmon MA, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc Natl Acad Sci USA. 2004;101:929–934. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagy P, Claus J, Jovin TM, Arndt-Jovin DJ. Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc Natl Acad Sci USA. 2010;107:16524–16529. doi: 10.1073/pnas.1002642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol Biol Cell. 2002;13:2547–2557. doi: 10.1091/mbc.01-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fidanze SD, Erickson SA, Wang GT, Mantei R, Clark RF, Sorensen BK, Bamaung NY, Kovar P, Johnson EF, Swinger KK, et al. Imidazo[2,1-b]thiazoles: Multitargeted inhibitors of both the insulin-like growth factor receptor and members of the epidermal growth factor family of receptor tyrosine kinases. Bioorganic & Medicinal Chemistry Letters. 2010;20:2452–2455. doi: 10.1016/j.bmcl.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 55.Pines G, Huang PH, Zwang Y, White FM, Yarden Y. EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene. 2010;29:5850–5860. doi: 10.1038/onc.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saffarian S, Li Y, Elson EL, Pike LJ. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys J. 2007;93:1021–1031. doi: 10.1529/biophysj.107.105494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hofman EG, Bader AN, Voortman J, van den Heuvel DJ, Sigismund S, Verkleij AJ, Gerritsen HC, van Bergen en Henegouwen PMP. Ligand-induced EGF receptor oligomerization is kinase-dependent and enhances internalization. J Biol Chem. 2010;285:39481–39489. doi: 10.1074/jbc.M110.164731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hackel PO, Gishizky M, Ullrich A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 2001;382:1649–1662. doi: 10.1515/BC.2001.200. [DOI] [PubMed] [Google Scholar]
- 59.Segatto O, Anastasi S, Alemà S. Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. Journal of Cell Science. 2011;124:1785–1793. doi: 10.1242/jcs.083303. [DOI] [PubMed] [Google Scholar]
- 60.Hindie V, Stroba A, Zhang H, Lopez-Garcia LA, Idrissova L, Zeuzem S, Hirschberg D, Schaeffer F, Jørgensen TJD, Engel M, et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat Chem Biol. 2009;5:758–764. doi: 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- 61.Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proceedings of the National Academy of Sciences. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mi LZ, Grey MJ, Nishida N, Walz T, Lu C, Springer TA. Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry. 2008;47:10314–10323. doi: 10.1021/bi801006s. [DOI] [PMC free article] [PubMed] [Google Scholar]