

Abstract
This study investigated the immunomodulatory effects of sunitinib in order to rationally design combinational platforms with immunotherapies for the treatment of solid tumors. Using a mouse model, we studied the effects of sunitinib given for 4 weeks at concentrations comparable to 37.5–50 mg/day in humans, followed by 2 weeks off the drug (sunitinib 4/2). We assessed the effect of differently timed combinations of sunitinib and a poxvirus-based vaccine encoding carcinoembryonic antigen (CEA) plus 3 costimulatory molecules on immune responses in CEA-transgenic (CEA-Tg) mice. Antitumor studies were performed in CEA-Tg mice bearing CEA-transfected MC38 murine colon carcinomas (MC38-CEA), treated either concurrently or sequentially with sunitinib and vaccine. In vitro, sunitinib inhibited PDGFR phosphorylation on MC38-CEA cells at concentrations similar to those biologically available during human treatment. In vivo, one cycle of sunitinib 4/2 caused bimodal immune effects: (a) decreased regulatory cells during the 4 weeks of treatment and (b) an immune-suppression rebound during the 2 weeks of treatment interruption. In a model using CEA-Tg mice bearing CEA+ tumors, continuous sunitinib followed by vaccine increased intratumoral infiltration of antigen-specific T lymphocytes, decreased immunosuppressant T regulatory cells and myeloid-derived suppressor cells, reduced tumor volumes, and increased survival. The immunomodulatory activity of continuous sunitinib administration can create a more immune-permissive environment. In combination with immunotherapies, sunitinib treatment should precede vaccine, in order to precondition the immune system, to maximize the response to vaccine-mediated immune enhancement.
Keywords: sunitinib, vaccine, immunotherapy, TKI, cancer
Introduction
Sunitinib is an orally available inhibitor of multiple tyrosine kinase receptors (TKI). It was approved by the FDA in 2006 for the treatment of advanced renal cell carcinoma 1 and imatinib-resistant gastrointestinal stromal tumors (GIST) 2. Sunitinib is currently being investigated as a treatment for other solid and hematologic malignancies in numerous clinical trials, including nearly 150 sponsored by the National Cancer Institute3.
Tyrosine kinase receptors (TKRs) targeted by sunitinib, such as VEGF and PDGF receptors, are widely expressed in many tumor cell types 4, tumor vasculature 5, and as growth factor receptors and protectors of apoptosis, allowing sunitinib to act directly against tumor cells 6 and tumor stroma 4. Sunitinib also targets TKRs expressed on myeloid-derived suppressor cells (MDSCs), such as c-KIT and VEGFR-1, making it a promising immunomodulator 7–8. Sunitinib has been shown to exert powerful immunomodulatory effects in cancer patients, as evidenced by a shift from a Th2 to a Th1 immune response and inhibition of immune suppressor cells 9–10, making this TKI an attractive candidate for combination with immunotherapies 11. In this study, we show that a sunitinib treatment regimen of 4 weeks on/2 weeks off (sunitinib 4/2) has a bimodal effect on the immune system: a decrease in regulatory cells during the 4-week treatment period, followed by an immune-suppressant rebound during the 2-week interruption. We therefore investigated whether continuous daily administration of sunitinib would maintain an environment permissive to the induction of immune response. In a combination regimen, sunitinib given before vaccine increased intratumoral infiltration of antigen-specific T lymphocytes, decreased Tregs and MDSCs, reduced tumor volume, and increased survival.
Materials and Methods
Drug preparation
For in vitro experiments, sunitinib malate salt > 99% (LC Laboratories, Woburn, MA) was dissolved in DMSO at 40 mg/mL and kept at −20° C as stock solution. A working solution of sunitinib was obtained by dissolving stock solution in PBS at 10 μg/mL. For in vivo studies, sunitinib malate salt > 99% was admixed to Open Standard Diet formulated as 20% Kcal proteins, 15% fat, and 65% Kcal carbohydrates at 228.5 mg/kg of diet (Research Diets Inc., New Brunswick, NJ). Open Standard Diet without sunitinib was used as control.
Flow cytometry
Multicolor cytometric analyses were performed using a FACSCalibur cytometer, with blue 488-nm and red 635-nm lasers, or an LSR-II supporting UV 355-nm, violet 405-nm, blue 488-nm, and red 633-nm lasers (BD Biosciences, San Diego, CA). CD140a (PDGFR-α), CD140b (PDGFR-β), and FoxP3 monoclonal antibodies (mAbs), along with appropriate isotypes, were purchased from eBioscience, San Diego, CA. Anti-CEA mAb COL-1 (IgG2a) and the negative control murine myeloma mAb UPC-10 (IgG2a) (Cappel, Organon Teknika Corp., West Chester, PA) were used as previously described 12. H-2Db-restricted CEA572–579, CEA526–532 13, and HIV-Gag tetramer peptides were purchased from Beckman Coulter, Fullerton, CA. All remaining mAbs for flow cytometry were purchased from BD Biosciences.
Animals
Eight- to 12-week-old female C57BL/6 mice were obtained from the National Cancer Institute’s Frederick Cancer Research Facility, Frederick, MD. A breedingpair of CEA-transgenic (CEA-Tg) mice homozygous for expression ofCEA was generously providedby Dr. John Shively (Beckman Research Institute, City of Hope National Medical Center, Duarte, CA) and used as a self-antigen model 14–15. Mice were housed and maintained in microisolator cages under specific pathogen-freeconditions in accordance with Association for Assessmentand Accreditation of Laboratory Animal Care guidelines. Allexperimental studies were approved by the National Cancer Institute’s Intramural Animal Care and Use Committee.
Sunitinib treatment
To model the FDA-approved sunitinib 4/2 treatment, C57BL/6 mice were fed with a sunitinib diet for 4 weeks and then with a control diet for 2 weeks. Mice (5/group) were sacrificed on days 2, 5, 7, 14, 21, 28, 35, and 42. In the day 0 group, 5 mice were fed with the control diet for 42 days. Daily intake of sunitinib, expressed as mg/kg/day, was calculated by the following formula:
Daily food intake, expressed in grams, was calculated by the following formula:
Food and animals were weighed twice/week. As a long-term treatment model, C57BL6 mice (4/group) were treated for 6 months with sunitinib or control diet, and then either splenocytes or cells from compact bone were pooled per treatment, and processed for flow-cytometric analysis.
Sampling techniques and HPLC analysis
On the day of harvest, peripheral blood was collected from the retro-orbital cavity of 3 mice/group. Half of the sample (500 μL) was preserved in 4% sodium citrate and analyzed by a Sysmex XE-2100 hemocytometer (Sysmex America, Mundelein, IL) for complete blood count (CBC). The other 500 μL were centrifuged and sunitinib serum concentration was quantified by the Laboratory of Proteomics and Analytical Technologies, SAIC-Frederick Inc. (NCI-Frederick, MD) via liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS). Mouse spleens were surgically extracted and pressed through a 70-μm cell strainer (BD Biosciences), after which red cells were lysed by ACK Lysing Buffer. After washing, 5×106 cells were stained for flow cytometry.
Clonogenic assays in methylcellulose
Compact bone marrow cells from C57BL6 mice (4/group) were harvested after 6 months of sunitinib or control diet, pooled per treatment, and cultured in triplicate using pre-aliquoted methylcellulose medium with recombinant cytokines and erythropoietin for mouse cells (StemCell Technologies, Vancouver, CA) according to the manufacturer’s protocol.
Suppression assays
Spleens of CEA-Tg mice (5/group) fed for 28 days with the control or sunitinib diet were removed and pressed through a 70-μm cell strainer. CD4+/CD25+ cells from each group were isolated using the CD4+/CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. T regulatory (Treg) suppression assay was performed as previously described 16, with the following minor changes: (a) the CD4+/CD25− fraction from the control group was used as a source of target cells, and (b) 5×104 20 Gy-irradiated syngeneic naïve splenocytes were added to each well as antigen-presenting cells in complete leukocyte medium (RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin) enriched with anti-mouse CD3e (0.5 μg/mL). Treg % suppression of sunitinib group was calculated by the formula:
For the MDSC functional assay, 2 different phenotypes of effector GR1+ splenocytes from CEA-Tg mice (5/group) were enriched, using the Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec), following the manufacturer protocol. In brief, splenocytes underwent gradient centrifugation with histopaque. Ly-6G positive cells were magnetically separated by double positive selection and used as GR1hi MDSCs. The remnant Ly-6 G negative fraction was subjected to a double GR1 positive selection, and the resulting cells were used as GR1dim MDSCs. Both enriched MDSCs populations were determined to be >90% GR1 positive by flow cytometry. As target cells, CD8 T-lymphocytes were negatively selected from spleens of 8 untreated mice, using the CD8a+ T Cells Isolation Kit II (Miltenyi Biotec), following manufacturer instructions. 2×107 CD8 lymphocytes were incubated with 10 μM carboxyfluorescein succinimidyl ester (CFSE) for 20 minutes at 37°C. After 3 washings, 5×104 CFSE-loaded CD8 cells were cocultured in triplicate with an equal number of GR1hi, GR1dim, or a 1:1 mixture of both, in a 96 well plate. Anti-mouse CD3e (1 μg/mL) and 1×105 20 Gy-irradiated syngeneic naïve splenocytes antigen presenting cells (APCs) were added to each well in complete leukocyte medium. CD8 lymphocytes with medium alone were cultured as a non-proliferation control. Cocultures were maintained at 37°C in a 5% CO2 incubator. After 72 hours incubation, cells were harvested and CFSE dilution of CD8 cells was analyzed by flow cytometry. Proliferation data were analyzed using FlowJo 7.6.3 (Treestar, Ashland, OR).
Poxvirus constructs
Recombinant modified vaccinia Ankara (rMVA) and recombinant fowlpox(rF) viruses containing transgenes for the murine costimulatory molecules B7-1, ICAM-1, and LFA-3 (designated TRICOM) in combinationwith the CEA transgene (rMVA/rF-CEA-TRICOM) have been previously described 17. For in vivo studies, subcutaneous rMVA-CEA-TRICOM was administered as prime and rF-CEA-TRICOM as weekly boosts at 1×108 plaque-forming units (PFUs) per mouse 18–19.
Evaluation of CEA-specific T-cell immune responses
To evaluate CEA-specific T-cell immune responses elicited by the combination of sunitinib and vaccine, CEA-Tg mice (5/group) were treated for 28 days as follows: (a) control group: control diet from day 0; (b) sunitinib group: sunitinib from day 0; (c) vaccine group: control diet from day 0, rMVA-CEA-TRICOM prime on day 7 and rF-CEA-TRICOM boost on day 14; (d) coadministration group: sunitinib from day 0, vaccine prime on day 0, and boost on days 7 and 14; and (e) sequential group: sunitinib from day 0, vaccine prime on day 7, and boost on day 14. On day 28, spleens were surgically removed, pooled per group, and pressed through a 70-μm cell strainer. Lymphocytes were separated by gradient centrifugation, followed by CD4 lymphocyte proliferation assay and CEA-tetramer evaluation of CD8 T cells.
CD4 lymphocyte proliferation assay
CD4+ splenocytes were negatively selected by CD4+ T Cell Isolation Kit II (Miltenyi Biotec) and incubated in complete leukocyte medium in the presence of increasing concentrations of CEA protein (AspenBio Pharma, Littleton, CO). Cultures without CEA protein were used as a control response. Cells were cultured for 5 days at 37° C. One μCi [3H] thymidine was added to each well for the last 24 h. Cells were harvested using a Wallac 1205 Betaplate Liquid Scintillation Counter (EG&G Wallac, PerkinElmer, Waltham, MA). The mean cellular proliferation of control response was subtracted from proliferation in response to CEA protein antigens. Data were averaged and reported as CEA-specific CD4 proliferation (cpm).
Analysis of CEA-tetramer+ CD8 T lymphocytes
Bulk cultures from differently treated groups of mice were set as follows: splenocytes were cultured in complete leukocyte medium enriched with 1 μg/μL CEA572–579 peptide for 6 days. Cells were harvested after 24 h of restimulation with fresh medium plus CEA572–579 peptide. After incubation, tetramer staining was performed by incubating cells with antimouse CD8a, CD19, NK1.1 mAbs or their matching isotypes, along with CEA572–579 or HIV-Gag tetramer and acquired by flow cytometry. During analysis, CD8a+/CEA-tetramer+ cells were gated from CD19−/NK1.1− events.
Tumor cell lines
Murine colon carcinoma MC38 cells expressing human CEA (MC38-CEA cells) were generated by retroviral transduction with CEA cDNA, as previously described 20. MC38-CEA cells were cultured in complete medium (DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, 300 μg/mL G418, and 100 μg/mL streptomycin). CEA expression of cultures, checked every 2 weeks by flow cytometric analysis, was > 80%.
Western blot
Inhibition of phosphorylation of PDGFR-α and PDGFR-β was measured after culturing MC38-CEA cells for 24 h in serum-free conditions. Cells were then incubated for 30 min with increasing concentrations of sunitinib (0.1, 1, 10, 100, and 1,000 ng/mL), followed by a 15-min incubation with 2 μL/mL PDGF-BB recombinant protein (GenWay Biotech, San Diego, CA). Cells were lysed in RIPA Lysis Buffer (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Proteins (10 μg) were resolved on SDS-PAGE, transferred onto nitrocellulose membranes, and probed with primary antibodies for PDGFR-α, PDGFR-β, PDGFR-α TYR754, PDGFR-β TYR740-751-771-1009-1021, and PDGFR-α TYR849/PDGFR-β TYR857 (Cell Signaling Technologies, Danvers, MA) at 4° C overnight. Detection was performed with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Experiments were repeated twice, with reproducible results.
Treatment of tumor-bearing mice
MC38-CEA cells (5×105/mouse) were injected subcutaneously in CEA-Tg mice (12/group). Seven days after tumor transplant, groups of mice began treatment as follows: (a) control group: control diet from day 7; (b) sunitinib group: sunitinib from day 7; (c) vaccine group: control diet from day 7, rMVA-CEA-TRICOM prime on day 14, weekly rF-CEA-TRICOM boosts from day 21; (d) sequential group: sunitinib from day 7, rMVA-CEA-TRICOM prime on day 14, weekly rF-CEA-TRICOM boosts from day 21. Where indicated, 2 additional combinations were included in the studies: (e) coadministration: sunitinib from day 7, rMVA-CEA-TRICOM prime on day 7, weekly rF-CEA-TRICOM boosts from day 14; (f) vaccine then sunitinib: sunitinib from day 14, rMVA-CEA-TRICOM prime on day 7, weekly rF-CEA-TRICOM boosts from day 14. Tumor dimensions were measured weekly and tumor volumes were obtained by the formula:
Analysis of intratumoral immune infiltration
To study the immune infiltration of MC38-CEA subcutaneous tumors, CEA-Tg mice (12–17/group from 3 separate experiments) were treated as above and sacrificed on day 21 post-transplant. After surgical resection and enzymatic digestion of tumors, tumor cells were stained for multicolor flow cytometric analysis of immune infiltrates.
Statistical analysis
GraphPad Prism 5® statistical software (GraphPad Software, La Jolla, CA) was used to measure 2-tailed unpaired t-test for differences between groups, one-way ANOVA for differences among groups with Bonferroni’s multiple comparison test, and Wilcoxon tests of survival. CellQuest software (BD Bioscience) was used to measure 2 sample Kolmogorov-Smirnov tests for analysis of 2 populations of pooled cells. P < 0.050 was considered statistically significant.
Results
Effect of one cycle of sunitinib 4/2 on mouse immune system
Studies were first conducted to determine the effect of a treatment cycle of sunitinib 4/2 (4 weeks of sunitinib treatment followed by 2 weeks of therapy interruption) on the immune system of tumor-free mice. Body weights (measured in grams ± SD) were comparable between sunitinib-treated mice (20.5 ± 1.3) and control mice (20.5 ± 1.5) (P = 0.846). Among sunitinib-treated mice, average daily intake of sunitinib (mg/kg/day ± SD) was 28.0 ± 4.7 and average serum concentration of sunitinib (ng/mL ± SD) was 336.5 ± 58.9 (Fig. 1). As shown on Supplemental Fig. 1, the analysis of peripheral blood showed that from day 14 to day 42 there was a slight but significant decrease in red blood count, hemoglobin, and hematocrit, and an increase in mean corpuscular volume (MCV). Platelets decreased slightly at day 7, increased at day 14, and recovered to baseline at all other time points. Lymphocytes decreased from day 0 to day 7, then returned to normal levels at day 14 until day 42. There were no differences in white blood count, polymorphonuclear cells, or basophils during the 6-week cycle. Thereafter, while monocytes remained constant during sunitinib treatment, there was an 8-fold increase at day 35 (one week after treatment interruption), with a return to normal at day 42. Eosinophils remained constant during treatment, but increased 10-fold at day 42. To investigate the cause of this rebound of monocytes and eosinophils in the peripheral blood after sunitinib interruption, flow cytometric analyses of splenocytes were undertaken. As shown in Fig. 1, the number of CD8 T lymphocytes decreased slightly from day 0 to day 2, then returned to normal values from day 5 until the end of the 6-week treatment cycle, while CD4 T lymphocytes decreased during all 4 weeks of sunitinib treatment and returned to baseline after treatment interruption. Analysis of immune-regulatory elements showed that Tregs decreased from day 5 to day 28 and returned to normal levels after treatment interruption (Fig. 1), while MDSCs had a similar decrease during the 4-week treatment and increased 2-fold after treatment interruption (Fig. 1 and 2C).
Fig. 1. Effect of sunitinib 4/2 on mouse immune system.
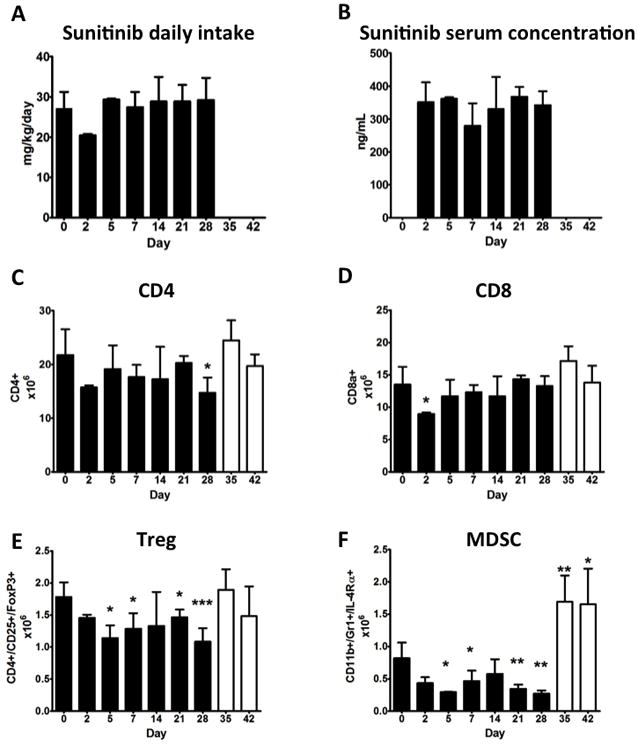
C57BL/6 mice (5/group) were treated with sunitinib corresponding to a human dose of 37.5–50 mg/day for 4 weeks, followed by 2 weeks of rest (sunitinib 4/2). (A) Daily intake of sunitinib was measured as described on materials and methods. (B) Serum concentrations of sunitinib were assessed by HPLC. (C-D) Flow cytometric analysis of CD8 and CD4 splenocytes. (E-F) Flow cytometric analysis of Treg and MDSC. Solid columns: days of sunitinib treatment. Open columns: days of control diet. Bars represent SD. Statistical difference based on unpaired t-test compared to day 0. * = P < 0.050; ** = P < 0.005; *** = P < 0.001.
Fig. 2. Four weeks of sunitinib therapy decreased immune suppression, followed by an immune suppressive rebound after treatment interruption.
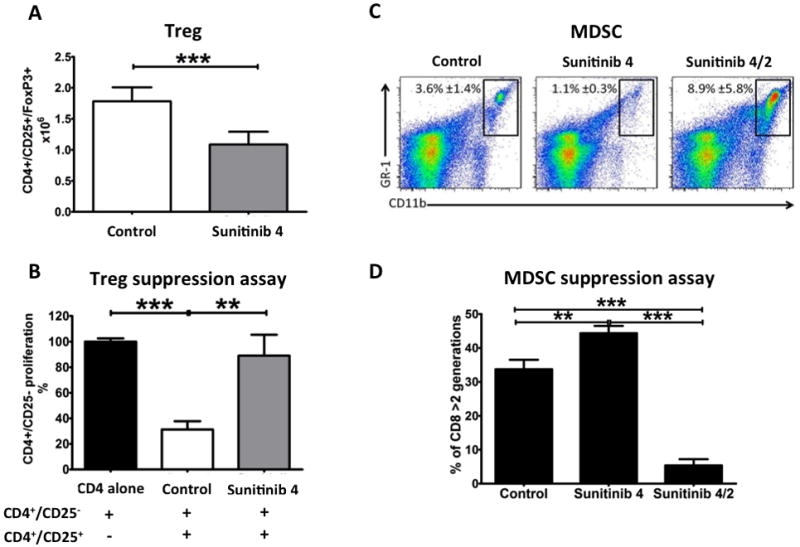
C57BL/6 mice (5/group) were treated with sunitinib corresponding to a human dose of 37.5–50 mg/day for 4 weeks, followed by 2 weeks of rest (sunitinib 4/2). Control: mice fed with control diet for 6 weeks; Sunitinib 4: mice fed with sunitinib diet for 4 weeks; Sunitinib 4/2: mice fed with sunitinib diet for 4 weeks, followed by 2 weeks of control diet. (A) Number of Treg lymphocytes in mouse spleens after 4 weeks of sunitinib or control diet. Statistical difference based on unpaired t-test compared to day 0. (B) Suppression assay of purified Tregs from mice treated for 4 weeks with sunitinib or control diet. Columns indicate proliferation of CD4+/CD25− fraction as measured by 3H release assay. Statistical difference based on one-way ANOVA test with Bonferroni’s multiple comparison tests. (C) Biparametric flow cytometric representation of MDSCs in mouse spleens. Squares indicate GR-1+/CD11b+ MDSCs, with percentage ± SD. (D) Three-day MDSC functional assay of purified CD11b+/GRhi plus CD11b+/GR1dim splenocytes (effectors) cocultured with CFSE-loaded CD8 lymphocytes from control-treated mice (targets). Proliferation was assessed by flow cytometric CFSE dilution assay. Columns represent percentage of CD8 lymphocytes undergone to 3 or more cell division cycle. Statistical difference based on one-way ANOVA test with Bonferroni’s multiple comparison tests. For A, B, and D bars represent SD. * = P < 0.050; ** = P < 0.005; *** = P < 0.001.
Four weeks of sunitinib treatment decreased Treg suppression
We next investigated whether the decreased number of Tregs in the spleen after 28 days of sunitinib treatment (Fig. 1E and 2A) was accompanied by a decreased ability to suppress T-cell proliferation. Treg functional analysis showed that Tregs from the control group suppressed T-lymphocyte proliferation by 69%, while Tregs from the sunitinib diet group suppressed T-lymphocyte proliferation by only 11.0% (Fig. 2B).
Sunitinib interruption increased suppression of MDSC
In order to understand whether the increased number of MDSC after sunitinib interruption was accompanied by changes in their suppressive function, 3-day MDSC functional assays were performed. As shown on Fig. 2C–D, after 4-weeks treatment the decreased number of MDSC was accompanied by decreased ability to suppress the proliferation of CD8 lymphocytes. After 2-weeks of treatment interruption, not only did the number of MDSC increase compared to before treatment initiation but their suppressive function per cell basis increased significantly.
Effects of sunitinib on negative regulatory elements were maintained after 6 months treatment in absence of bone marrow toxicity
In order to evaluate the effect of long-term sunitinib treatment, C57BL/6 mice were treated continuously for 6 months with control diet or sunitinib. After this time, splenocytes and bone marrow (BM) cells from compact bone were harvested, pooled per group, and tested by flow-cytometry. Average splenocyte number was unchanged by sunitinib treatment, being 215 ×106 in control group and 212 ×106 after sunitinib diet. As shown on Supplemental Table 1, in sunitinib-treated mice CD8 T-lymphocytes increased 92% in the spleen and 44% in the BM compared to control. Tregs decreased 41% in the spleen and 7% in the BM. MDSC slightly decreased both in the spleen and BM. As a consequence, the ratio between CD8 and Treg increased 3.3-fold in the spleen and 1.6-fold in BM. Similarly, the CD8:MDSC ratio doubled both in the spleen and BM. To study BM toxicity, 2-week clonogenic assays in cytokine-enriched methylcellulose medium were performed. The number of colonies in control and sunitinib group were comparable (Supplemental Table 1), indicating a functioning erythropoiesis and granulocytopoiesis of murine BM.
Sequential administration of sunitinib and vaccine increased antigen-specific T lymphocytes
To investigate whether sunitinib treatment could enhance vaccine-based immunotherapy, 2 different combinations were studied, as a consequence of the above results: (a) coadministration of a vaccine prime and sunitinib on the same day, and (b) sunitinib followed 7 days later by vaccine (sequential administration). Mice received sunitinib for 4 weeks, rMVA-CEA-TRICOM as a prime and rF-CEA-TRICOM as a boost. As shown in Figure 3A, CEA-dependent CD4 proliferation after coadministration of sunitinib and vaccine was similar to vaccine alone. However, when vaccine followed sunitinib, CD4 proliferation increased significantly compared to control, sunitinib alone, vaccine alone, and coadministration of sunitinib and vaccine. Similarly, the analysis of CEA-tetramer+ CD8 T lymphocytes, when compared to vaccine alone group, increased 75% after sequential therapy, but decreased 55% after coadministration of vaccine and sunitinib (Fig. 3B). Sunitinib alone was not able to increase the CEA-tetramer+ CD8 T lymphocytes compared to control.
Fig. 3. Relation of sunitinib/vaccine sequencing to induction of immune responses.

CEA-Tg mice (5/group) were treated with either concurrent or sequential sunitinib and vaccine for 4 weeks, after which splenocytes were harvested for functional assays. Control group: control diet for 4 weeks. Vaccine group: control diet for 4 weeks, vaccine prime (rMVA-CEA-TRICOM) on day 0, boosts (rF-CEA-TRICOM) on days 7 and 14. Sunitinib group: sunitinib diet for 4 weeks. Sunitinib plus vaccine (coadministration) group: sunitinib for 4 weeks, vaccine prime on day 7, boost on day 14. Sunitinib then vaccine (sequential) group: sunitinib diet for 4 weeks, then vaccine prime on day 7 and boost on day 14. (A) 3H release assay of purified CD4 lymphocytes from splenocytes of differently treated mice cultured in the presence of increasing concentrations of CEA protein. Bars represent SD of triplicate assessments. Statistical difference based on ANOVA. * = P < 0.050; ** = P < 0.005; *** = P < 0.001. (B) Flow cytometric analysis of CEA-specific CD8 lymphocytes from bulk cultures of splenocytes cultured with CEA572–579 peptide or HIV-Gag control peptide. Numbers inside each plot represent percentages of tetramer+ CD8 lymphocytes.
Sunitinib affects MC38-CEA cells through the PDGFR pathway
In order to assess the anti-tumor effect of sunitinib, we first analyzed the effect of sunitinib on murine MC38 colon carcinoma cells transfected with CEA (MC38-CEA). We used flow cytometry and Western blot to measure the in vitro expression of TKRs known to be ligands of the inhibitor: c-KIT, VEGFR-2, VEGFR-3, PDGFR-α, and PDGFR-β. MC38-CEA cells expressed high concentrations of PDGFR-α and moderate concentrations of PDGFR-β (Supplemental Fig. 2A), but did not express the other TKRs (not shown). Phosphorylation of PDGFRs was studied by Western blot at different tyrosine positions of the intracytoplasmic tail of the receptors (PDGFR-α TYR754, PDGFR-β TYR740-751-771-1009-1021, and PDGFR-α TYR849/PDGFR-β TYR857). PDGFR-α TYR849/PDGFR-β TYR857 was the only one to show phosphorylation after in vitro stimulation with PDGF-BB; phosphorylation was inhibited by sunitinib starting at concentrations of 100 ng/mL (Supplemental Fig. 2A). In order to determine whether the inhibition of PDGFR phosphorylation was sufficient to inhibit cell growth, we assessed the effect of 100 ng/mL of sunitinib on in vitro proliferation of MC38-CEA tumor cells. We found that, when compared to DMSO control, sunitinib did not affect the in vitro proliferation of tumor cells up to 72 h cultures either when sunitinib was added only once at 0 hours or when sunitinib was replenished every 24 hours (Supplemental Fig. 2B).
Anti-tumor activity of sequential administration of sunitinib and vaccine
On the basis of the results described above, studies were undertaken to determine if sequential administration of sunitinib followed by vaccine would enhance anti-tumor responses when compared with the use of either agent alone. As shown on Fig. 4 and Supplemental Fig. 3, sunitinib alone led to smaller tumors and longer survival, when compared with control or vaccine alone. Thereafter, the sequential treatment, in which CEA-Tg mice bearing MC38-CEA tumors were treated with sunitinib 4/2 starting 7 days post-transplant and vaccine starting 14 days post-transplant, resulted in significantly smaller tumors compared to control, sunitinib alone, and vaccine alone (Fig. 4A); moreover, 25% of animals in this group were tumor-free 42 days after tumor inoculation. Sequential treatment resulted also in a higher median survival, which was a significant increase over control (P < 0.001), vaccine alone (P < 0.001), and sunitinib alone (P < 0.001) (Fig. 4B). We also tested the coadministration and the sequential therapy in which vaccine preceded sunitinib by one week (Supplemental Fig. 3). Comparing coadministration and vaccine followed by sunitinib, the latter caused smaller tumors than the former, but both were statistically comparable to sunitinib alone. Moreover, similarly to sunitinib alone, coadministration and vaccine followed by sunitinib did not result in tumor-free animals. Finally, when compared to sunitinib alone, the survival was similar both after coadministration with vaccine (p=0.793) and after vaccine followed by sunitinib (p=0.706).
Fig. 4. Sunitinib followed by vaccine decreased tumor volume and improved survival.
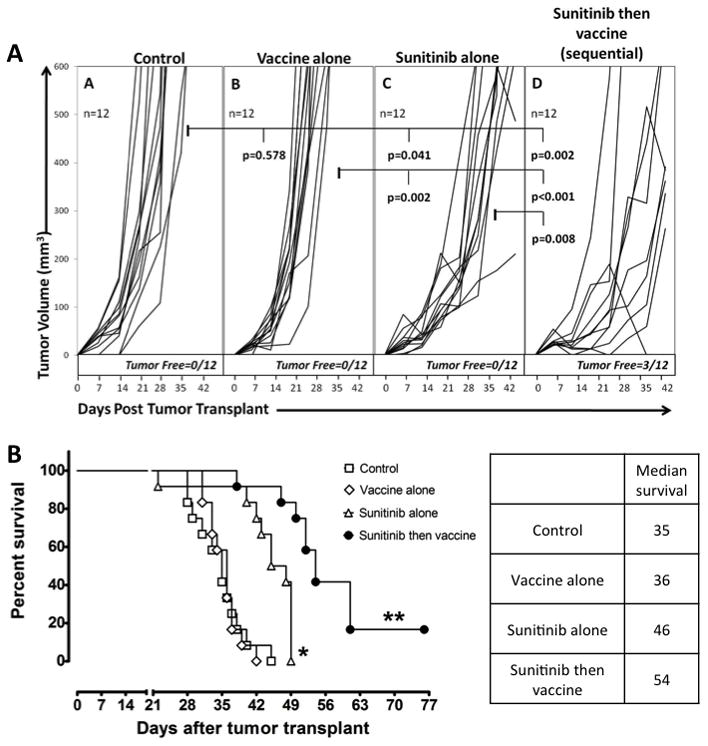
CEA-Tg mice (12/group) bearing subcutaneous MC38-CEA tumors were treated as follows: Control group: continuous control diet starting 7 days after tumor transplant. Vaccine group: prime 14 days after tumor transplant, then weekly boosts. Sunitinib group: continuous sunitinib diet starting 7 days after tumor transplant. Sunitinib then vaccine (sequential) group: continuous sunitinib diet starting 7 days after tumor transplant, vaccine prime on day 14, then weekly boosts. (A) Tumor volumes. Statistical analysis based on unpaired t-test as measured on day 42 post-tumor transplant. Bold values: p < 0.050. (B) Survival analysis. Statistical analysis of survival based on Wilcoxon test. * = P < 0.050 compared to control and vaccine groups. ** = P < 0.050 compared to all other groups. Median survival is indicated.
Sequential administration of sunitinib and vaccine improved intratumoral immune infiltration
To confirm that administering sunitinib before vaccine improved anti-tumor response by triggering favorable immune-balance in the tumor stroma, additional in vivo studies were undertaken in which MC38-CEA tumor-bearing mice were treated for 2 weeks with control diet, sunitinib alone, vaccine alone, or different combinations of both (sunitinib plus vaccine and sunitinib followed by vaccine after 1 week). Outcomes were tumor volumes and intra-tumoral immune-infiltration at the end of the 2-week treatments. As shown on Fig. 5, sunitinib alone treatment resulted in smaller tumors compared to control or vaccine alone, but no animals became tumor free. The sequential treatment gave superior results than sunitinib alone and 32% tumor-free mice. Interestingly, the coadministration of sunitinib and vaccine did not improve the antitumor activity noted with that of sunitinib alone (Supplemental Fig. 4). Flow cytometric analysis showed that, compared to control, changes in the intratumoral immune infiltration were evident within 2 weeks of treatment (Table 1 and Supplemental Table 2). Both sunitinib alone and sunitinib followed by vaccine showed increased CD8 and lower Treg infiltration, but only the sequential therapy led to a significant decrease in MDSC infiltration (Table 1). Analysis of CEA-tetramer+ CD8 cells showed that, compared to untreated mice, tumor-infiltrating CEA-specific CD8 T lymphocytes increased 7-fold after sunitinib alone, 10-fold after vaccine alone, and 12-fold after sequential treatment. As a result, compared to control, the ratio of CEA-specific CD8 cells versus Tregs increased 5-fold in sunitinib alone, 8-fold in vaccine alone, and 15-fold in sequential therapy group. Similarly, the ratio between CEA-specific CD8 T lymphocytes and MDSC increased 5-fold after sunitinib alone, 4-fold after vaccine alone, and 28-fold after sequential treatment over control. The coadministration of sunitinib with vaccine (Supplemental Table 2) showed that this combination failed to decrease tumor-infiltrating MDSC, over that observed for mice treated with sequential therapy. The infiltration of CEA-specific CD8 T lymphocytes after coadministration was greater than that seen in control groups, or groups treated with sunitinib alone or vaccine alone, and similar to what achieved from sequential therapy. However, compared to control, the CEA-tetramer+:Treg ratio increased only 7-fold (compared with a 15-fold after sequential treatment) and the CEA-tetramer+:MDSC ratio increased 8-fold (as compared with a 28-fold increment after sequential therapy).
Fig. 5. Antitumor effect of sunitinib followed by vaccine is evident within 2 weeks of treatment.
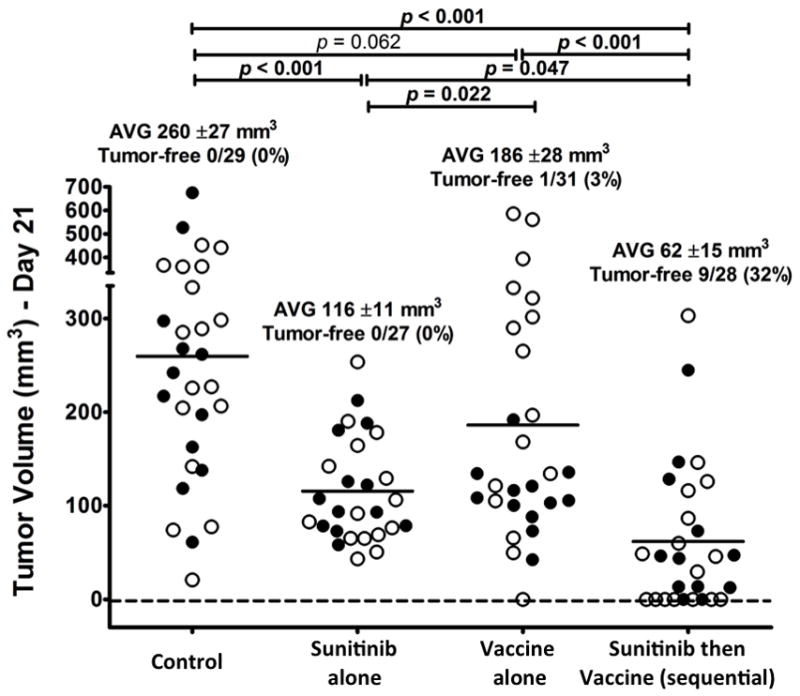
CEA-Tg mice (27–31/group) bearing subcutaneous MC38-CEA tumors were treated as follows: Control group: continuous control diet starting 7 days post-tumor transplant. Vaccine group: vaccine prime 14 days post-tumor transplant. Sunitinib group: continuous sunitinib diet starting 7 days post-tumor transplant. Sunitinib then vaccine (sequential) group: continuous sunitinib diet starting 7 days post-tumor transplant, vaccine prime on day 14. Measurements were taken 21 days after tumor transplant. Closed circles: tumor volumes from experiment on Fig. 4. Open circles: Tumor volumes from 3 additional experiments (n=4–6/group per experiment). All the open circles, if from tumor-bearing mice, were investigated for intratumoral immune-infiltration (Table 1). Statistical difference based on unpaired t-test as measured on day 21 post-tumor transplant. Bold: statistical significance (P < 0.050) based on 2-tailed unpaired t-test.
Table 1.
Vaccinating after Sunitinib improved intratumoral immune infiltration
| Control (±SD) | Sunitinib(a) (±SD) | Vaccine(b) (±SD) | Sunitinib then Vaccine (sequential)(c) (±SD) | |
|---|---|---|---|---|
|
Infiltrates(d)
| ||||
| CD8 % | 0.32 (0.12) | 0.69 (0.22)* | 0.46 (0.22) | 0.66 (0.03)* |
| CD4 % | 3.86 (3.28) | 2.03 (0.75) | 3.41 (2.94) | 1.90 (0.86) |
| Treg % | 0.11 (0.06) | 0.07 (0.02)* | 0.07 (0.02)* | 0.05 (0.02)* |
| MDSC % | 1.48 (0.23) | 1.16 (0.46) | 1.35 (0.99) | 0.48 (0.13)** |
|
| ||||
|
CEA-specific CD8(e)
| ||||
| CEA tetramer % | 0.01 (p<0.001) | 0.07 (p<0.001) | 0.10 (p<0.001) | 0.12 |
| CEA tetramer:Treg | 0.22 | 1.14 | 1.80 | 3.37 |
| CEA tetramer:MDSC | 0.01 | 0.05 | 0.04 | 0.28 |
Flow cytometric analysis of tumor infiltrating immune-cells of CEA-Tg mice bearing MC38-CEA tumors. Measurements taken 21 days after tumor transplant.
Sunitinib started 7 days after tumor transplant.
Vaccine 14 days after tumor transplant.
Sunitinib started 7 days after tumor transplant, then vaccine on day 14.
Data from 3 independent experiments, n=12–17/group (open circles from Fig. 5). Statistical significance based on t-test.
: p<0.050 vs. Control.
: p<0.050 vs. all other groups.
CEA-tetramer+ CD8 lymphocytes were calculated subtracting HIV-GAG tetramer+ control from CEA tetramer+ cells. Data from one experiment (5 mice/group; pooled cells). Statistical analysis based on Kolmogorov-Smrinov (K-S) test vs. Sunitinib then vaccine (sequential) treatment.
Discussion
The study described here was undertaken to investigate the scheduling effects of sunitinib in combination with vaccine to help to rationally design combinational therapy platforms.
The first requirement was to establish an animal model comparable to the human standard-of-care with sunitinib. As previously reported 21–22, 20 to 40 mg/kg/day of sunitinib are necessary in mouse models to achieve plasma concentrations of 50 to 100 ng/mL, which will inhibit VEGFR and PDGFR phosphorylation. This dose corresponds to approximately 0.4 to 0.8 mg/mouse/day. Sunitinib was administered admixed with a balanced purified diet, in order to minimize the stress caused by daily orogastric gavage, that could in turn cause alterations of immune responses 23. Sunitinib administration was 28 mg/kg/day (approximately 0.6 mg/mouse/day), which led to serum concentration of 336 ng/mL (Fig. 1), sufficient to inhibit the same TKRs that are blocked during treatment in humans 21. A preclinical study by Ozao-Choy et al. 24 showed that sunitinib in combination with an immunotherapeutic protocol (IL-12 and 4-1BB activation) improved survival in MCA26 colon tumor-bearing mice by reversal of intratumoral immune suppression. However, the authors utilized a maximum dose of 0.015 mg/mouse/day of sunitinib. In a separate study, we determined that C57BL/6 mice receiving sunitinib for 2 weeks at 0.015 mg/mouse/day had a serum concentration of <5 ng/mL, indicating that the immunomodulatory and anti-tumoral effects described by the authors could be caused by the sole inhibition of c-KIT (IC50 0.5–5 ng/mL) and VEGFR2 (IC50 2 ng/mL) 21. With the sunitinib serum concentrations achieved during human treatment (higher than 50 ng/mL), many additional TKRs are targeted by sunitinib, such as Flt-3, PDGFRs, and CSF-1R. These receptors are known to be crucial to the tumor biology, tumor-stroma organization, and modulation of antitumor immune response. Another recent animal study by Bose et al. 25 showed the benefit of a short-term (7 days) combination of 1 mg/mouse/day of sunitinib with a peptide-pulsed dendritic cell vaccine for the treatment of B16.OVA melanoma. The authors describe that the favorable anti-tumor response depended on increased chemotactic signals that impact antigen-specific CD8 T cell recruitment into the tumor microenvironment. The data presented here confirm and extend these findings with a standard-of-care treatment regimen.
We report here results observed during a cycle of 4 weeks of sunitinib treatment followed by 2 weeks of therapy interruption (sunitinib 4/2). To our knowledge, these are the first data on the dynamics of immunomodulation during an entire cycle of sunitinib 4/2. In the preclinical model described here, as in humans, sunitinib was well tolerated with few hematologic side effects, including moderate anemia and transient thrombocytopenia (Supplemental Fig. 1). Four weeks of sunitinib treatment decreased the number of circulating Tregs and MDSCs (Fig. 1), confirming the finding of Finke et al. of reduced circulating immunosuppressive cells after 4 weeks of treatment in human patients 9–10. Importantly, 4 weeks of sunitinib treatment affected mouse Tregs and MDSC both quantitatively, by decreasing their numbers, and qualitatively, by inhibiting their ability to suppress T-cell proliferation (Fig. 1 and 2). Moreover, we report for the first time an increase of MDSC (Fig. 1 and 2C) along with an increased ability to suppress CD8 proliferation (Fig. 2D) after sunitinib interruption, indicating that interrupting sunitinib treatment triggered immune suppression. This immunosuppressant rebound could play a role in the reported regrowth of metastases and progression of clinical symptoms when sunitinib therapy is discontinued in cancer patients 26–27. In addition, we describe a rebound of monocytes and eosinophils in the CBC after sunitinib interruption (Supplemental Fig. 1). It is possible that these monocytes and granulocytes represent the 2 subpopulations forming the pool of MDSC 28, which are known to directly suppress T lymphocyte proliferation and to activate Tregs 29. These data suggest that to maintain immune-favorable conditions, continuous daily dosing (CDD) with sunitinib is preferable to intermittent dosing.
Extended continuous treatment with sunitinib raises concerns about bone marrow suppression 30–31. However, we found that the improved immune balance favoring effector cells over suppressor cell populations was maintained for up to 6 months of sunitinib treatment, with no bone marrow toxicity (Supplemental Table 1).
Interestingly, while sunitinib inhibited PDGFR phosphorylation on MC38-CEA cells in vitro at concentrations comparable to human therapy (100 ng/mL), the TKI failed to suppress the tumor cell in vitro proliferation at this dose (Supplemental Fig. 1), supporting the assumption that in vivo sunitinib exerts its anti-tumor effect through additional mechanisms, such as the inhibition of tumor stroma 5 and modulation of immune response 9. To sort out whether the in vivo effect of sunitinib alone on tumor shrinkage involves immune modulation, preliminary data of ongoing immunohistochemical studies showed that sunitinib alone affects the intratumoral infiltration of T-lymphocytes by changing intratumoral interstitial pressure (not shown).
To examine the potential additive or synergistic effects of combination therapy of sunitinib and immunotherapies, we designed a therapy regimen where vaccine alone was ineffective (Fig. 4–5 and Supplemental Fig 3–4). In this study, we used a poxviral vaccine platform of recombinant vaccinia prime and recombinant fowlpox boost, bearing CEA antigen and a triad of costimulatory molecules 18–19.
In order to achieve the optimal outcome with the combination of immunotherapy and standard-of-care therapy, three different timings and sequences of administration of vaccine plus the TKI sunitinib are possible: coadministration, sunitinib followed by vaccine, and vaccine followed by sunitinib. The best combination schedule is the result of a robust immune stimulation versus TAAs with an absent or low toxicity versus immune effector cells. We hypothesized that if vaccine was given after sunitinib, the beneficial immune balance created by the TKI would magnify the vaccine-mediated response to TAAs. Indeed, combinatorial studies showed that, while vaccinating after sunitinib initiation improved immune activation, coadministration can affect antigen-specific T-lymphocytes (Fig.3). Based on the transient decrease of T-lymphocytes at day 2 after sunitinib beginning (Fig. 1), vaccinating at the same time of sunitinib treatment could compromise the vaccine-mediated immune response at the time of antigen presentation, decreasing the magnitude of immune response. Furthermore, it has been reported that sunitinib could be able to impair the proliferation and function of T cells 32. If vaccine precedes sunitinib, instead, the first phase of immune activation after vaccination would occur before the sunitinib-dependent inhibition of immune suppressant elements; as a consequence, the T-cell activation would take place in presence of fully active Tregs and MDSCs, which directly inhibit T-cells. Here we show that both vaccine followed by sunitinib and concurrent administration of them showed absence of antitumor benefit (Supplemental Fig. 3) and unchanged intratumoral immune infiltration (Supplemental Table 2) over sunitinib alone. Moreover, sunitinib followed by vaccine (Fig. 4–5 and Supplemental Fig. 3–4), led to greater antitumor activity, longer survival and more efficient intratumoral infiltration of antigen-specific T lymphocytes (Table 1 and Supplemental Table2). Taken altogether, these data indicate that, in order to facilitate immune response, vaccine should follow sunitinib administration.
Recently, a randomized phase III clinical study 33 of the combination of MVA encoding the TAA 5T4 (MVA-5T4; TroVax) with sunitinib in renal cell carcinoma showed no difference in survival between patients receiving sunitinib alone and patients receiving sunitinib with vaccine. In this trial, however, patients were vaccinated before receiving sunitinib which, based on our findings, could explain this result.
This study, showing for the first time the favorable outcomes with sequential administration of continuous daily dosing of sunitinib and recombinant vaccine, could provide a rationale for the design of platforms combining TKIs with immunotherapeutic agents to treat solid tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support: Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH.
The authors acknowledge the excellent editorial assistance of Bonnie L. Casey in the preparation of this manuscript.
References
- 1.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584–90. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 3.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, Dahut WL, Arlen PM, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37. [PubMed] [Google Scholar]
- 5.Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7:225–33. doi: 10.1007/s10456-004-3149-y. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007;17:3–7. doi: 10.1016/j.gde.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen SH. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol. 2011;77:12–9. doi: 10.1016/j.critrevonc.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, Huang B. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One. 2010;5:e8922. doi: 10.1371/journal.pone.0008922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–82. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 10.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 11.Asemissen AM, Brossart P. Vaccination strategies in patients with renal cell carcinoma. Cancer Immunol Immunother. 2009;58:1169–74. doi: 10.1007/s00262-009-0706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hand PH, Robbins PF, Salgaller ML, Poole DJ, Schlom J. Evaluation of human carcinoembryonic-antigen (CEA)-transduced and non-transduced murine tumors as potential targets for anti-CEA therapies. Cancer Immunol Immunother. 1993;36:65–75. doi: 10.1007/BF01754404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehm AL, Higgins J, Franzusoff A, Schlom J, Hodge JW. Concurrent vaccination with two distinct vaccine platforms targeting the same antigen generates phenotypically and functionally distinct T-cell populations. Cancer Immunol Immunother. 2010;59:397–408. doi: 10.1007/s00262-009-0759-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke P, Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58:1469–77. [PubMed] [Google Scholar]
- 15.Schmitz J, Reali E, Hodge JW, Patel A, Davis G, Schlom J, Greiner JW. Identification of an interferon-gamma-inducible carcinoembryonic antigen (CEA) CD8(+) T-cell epitope, which mediates tumor killing in CEA transgenic mice. Cancer Res. 2002;62:5058–64. [PubMed] [Google Scholar]
- 16.Farsaci B, Sabzevari H, Higgins JP, Di Bari MG, Takai S, Schlom J, Hodge JW. Effect of a small molecule BCL-2 inhibitor on immune function and use with a recombinant vaccine. Int J Cancer. 2010;127:1603–13. doi: 10.1002/ijc.25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge JW, Poole DJ, Aarts WM, Gomez Yafal A, Gritz L, Schlom J. Modified vaccinia virus ankara recombinants are as potent as vaccinia recombinants in diversified prime and boost vaccine regimens to elicit therapeutic antitumor responses. Cancer Res. 2003;63:7942–9. [PubMed] [Google Scholar]
- 18.Hodge JW, Sabzevari H, Yafal AG, Gritz L, Lorenz MG, Schlom J. A triad of costimulatory molecules synergize to amplify T-cell activation. Cancer Res. 1999;59:5800–7. [PubMed] [Google Scholar]
- 19.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res. 2003;9:1837–49. [PubMed] [Google Scholar]
- 20.Robbins PF, Kantor JA, Salgaller M, Hand PH, Fernsten PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3657–62. [PubMed] [Google Scholar]
- 21.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–96. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 22.Haznedar JO, Patyna S, Bello CL, Peng GW, Speed W, Yu X, Zhang Q, Sukbuntherng J, Sweeny DJ, Antonian L, Wu EY. Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: comparative plasma kinetics in non-clinical species. Cancer Chemother Pharmacol. 2009;64:691–706. doi: 10.1007/s00280-008-0917-1. [DOI] [PubMed] [Google Scholar]
- 23.Balcombe JP, Barnard ND, Sandusky C. Laboratory routines cause animal stress. Contemp Top Lab Anim Sci. 2004;43:42–51. [PubMed] [Google Scholar]
- 24.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2010 doi: 10.1002/ijc.25863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26:1810–6. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 27.Desar IM, Mulder SF, Stillebroer AB, van Spronsen DJ, van der Graaf WT, Mulders PF, van Herpen CM. The reverse side of the victory: flare up of symptoms after discontinuation of sunitinib or sorafenib in renal cell cancer patients. A report of three cases. Acta Oncol. 2009;48:927–31. doi: 10.1080/02841860902974167. [DOI] [PubMed] [Google Scholar]
- 28.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 29.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhojani N, Jeldres C, Patard JJ, Perrotte P, Suardi N, Hutterer G, Patenaude F, Oudard S, Karakiewicz PI. Toxicities associated with the administration of sorafenib, sunitinib, and temsirolimus and their management in patients with metastatic renal cell carcinoma. Eur Urol. 2008;53:917–30. doi: 10.1016/j.eururo.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 31.van Erp NP, Mathijssen RH, van der Veldt AA, Haanen JB, Reyners AK, Eechoute K, Boven E, Wessels JA, Guchelaar HJ, Gelderblom H. Myelosuppression by sunitinib is flt-3 genotype dependent. Br J Cancer. 2010;103:757–8. doi: 10.1038/sj.bjc.6605813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu Y, Zhao W, Meng F, Qu B, Zhu X, Sun Y, Shu Y, Xu Q. Sunitinib impairs the proliferation and function of human peripheral T cell and prevents T-cell-mediated immune response in mice. Clin Immunol. 2010;135:55–62. doi: 10.1016/j.clim.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Amato RJ, Hawkins RE, Kaufman HL, Thompson JA, Tomczak P, Szczylik C, McDonald M, Eastty S, Shingler WH, de Belin J, Goonewardena M, Naylor S, et al. Vaccination of metastatic renal cancer patients with MVA-5T4: a randomized, double-blind, placebo-controlled phase III study. Clin Cancer Res. 2010;16:5539–47. doi: 10.1158/1078-0432.CCR-10-2082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.