

Abstract
Summary: pybedtools is a flexible Python software library for manipulating and exploring genomic datasets in many common formats. It provides an intuitive Python interface that extends upon the popular BEDTools genome arithmetic tools. The library is well documented and efficient, and allows researchers to quickly develop simple, yet powerful scripts that enable complex genomic analyses.
Availability: pybedtools is maintained under the GPL license. Stable versions of pybedtools as well as documentation are available on the Python Package Index at http://pypi.python.org/pypi/pybedtools.
Contact: dalerr@niddk.nih.gov; arq5x@virginia.edu
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Due to advances in DNA sequencing technologies, genomic datasets are rapidly expanding in size and complexity (Stein, 2010). It is now clear that the primary bottleneck in genomics is data analysis and interpretation, not data generation. Therefore, researchers depend upon fast, flexible ‘genome arithmetic’ tools for interrogating and comparing diverse datasets of genome features. For example, genome arithmetic is used to interpret results from whole-genome sequencing, ChIP-seq and RNA-seq experiments by integrating experimental datasets with genes, genetic variation and the wealth of existing genome annotations (1000 Genomes Project Consortium et al., 2010; ENCODE Project Consortium et al., 2011). These analyses are complicated by the fact that they are often done via custom scripts or one-off manipulations that are inefficient and difficult to reproduce and maintain.
Tools designed to manipulate, intersect and annotate these datasets in commonly-used formats greatly facilitate such analyses and provide a consistent framework for reproducible research. Here we introduce pybedtools, which extends the BEDTools (Quinlan and Hall, 2010) genome arithmetic utilities by providing a powerful interface combining the benefits of Python scripting and the BEDTools libraries. Using a simple syntax, it allows researchers to analyze datasets in BED (Kent et al., 2002), VCF (Danacek et al., 2011), GFF, BEDGRAPH (Kent et al., 2002) and SAM/BAM (Li et al., 2009) formats without the need for format conversion.
2 APPROACH
The pybedtools library allows one to manipulate datasets at both the file and individual feature level using the BedTool and Interval classes, respectively. It integrates high-level BEDTools programs through the Python subprocess module, and lower level BEDTools functionality by exposing a subset of BEDTools' libraries. At the core of pybedtools is the BedTool class. Typically, a BedTool is initially created with a file name. BEDTools programs are then accessed as methods of BedTool objects (e.g. BedTool.intersect for the BEDTools program intersectBed) with arguments identical to the user's installed version of BEDTools. However, in addition to passing filenames as in typical BEDTools command line usage, one may also pass collections of Interval objects which can be manipulated in Python on a feature-by-feature basis. Furthermore, BedTool methods return new BedTool instances, allowing users to chain many operations together in a fashion similar to the UNIX command line.
The pybedtools package provides a standardized interface to individual features in diverse genomics datasets, thus allowing one to iterate through datasets while accessing chromosome, start and stop coordinates with identical syntax, regardless of the underlying file format. This abstraction is made possible via Cython (http://cython.org, last accessed Aug 2011) which exposes the BEDTools file manipulation, feature parsing and overlap detection functions. In terms of speed and memory efficiency, pybedtools therefore compares favorably with Galaxy's (Giardine et al., 2005) bx-python, Kent source (Kent et al., 2002) and the original BEDTools software (Supplementary Fig. 1). Formats with different coordinate systems (e.g. BED vs GFF) are handled with uniform, well-defined semantics described in the documentation. Additional features and example scripts illustrating the library's functionality are in the documentation at http://packages.python.org/pybedtools.
3 APPLICATION
The pybedtools package employs a syntax that is intuitive to Python programmers. For example, given an annotation file of genes, hg19.gff, and a file containing relevant genetic variation, snps.bed, one can identify genes that contain SNPs with the following:
![]()

At this point, one can easily examine the genes that overlap SNPs:
![]()
or filter the results with simple boolean functions:
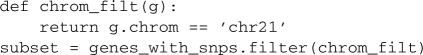
The underlying BEDTools commands send their results to ‘standard output’. To assist in managing intermediate files, pybedtools automatically saves these results as temporary files that are deleted when Python exits. Results can be explicitly saved with the saveas() method:
![]()
Given a FASTA file of the genome, hg19.fa, sequences for this subset of genes can be retrieved and saved with:
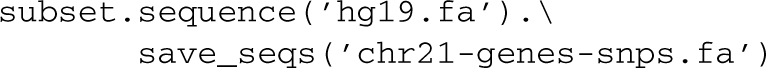
One of the more powerful extensions provided by the pybedtools interface is the ability to mix file operations with feature operations in a way that makes otherwise difficult tasks very accessible with minimal code. For example, the following identifies the closest gene (within 5 kb) to each intergenic SNP:

This example illustrates several powerful features of pybedtools that confer additional functionality and greatly simplify analyses as compared with the BEDTools command line utilities (see Supplementary Material for an analogous experiment with BEDTools). For example, set subtraction between BedTools is used to extract features that are unique to one file (snps - genes). Similarly, one may also use the addition operator to identify features in the first file that overlap features in multiple datasets (e.g. snps + novel_snps + genes). Moreover, there is essentially no limit to the number of files that can be compared with the + and − operators.
Arguments sent to BedTool objects are passed to BEDTools programs. The argument d=True tells the BEDTools closestBed program to append the distance (in base pairs) between each SNP and the closest gene to the end of each line, equivalent to the -d argument typically given on the command line.
Additionally, the argument stream=True indicates that the resulting BedTool object will stream results as a Python iterable of Interval objects instead of saving the results to a temporary file. This saves disk space and reduces file operations when performing many operations on large files.
Also note the indexing of the Interval object gene via [-1]. This retrieves the last item on the line, which, because of the d=True argument, represents the distance in base pairs between each SNP and gene. All elements of a line can be accessed from an Interval object by their integer index, and core attributes by their name.
Finally, although nearby represents results that are a composite of GFF and BED features (i.e. genes and snps), the operation that produced nearby was driven by the gene GFF file. Therefore gene.name is seamlessly extracted from the GFF ‘attributes’ field.
Pybedtools also allows one to integrate sequence alignments in the widely used SAM/BAM format into their analyses. The following example illustrates how one would use pybedtools to identify sequence alignments that overlap coding exons.

Alternatively, this analysis could be reduced to the following statement:
![]()
Some BEDTools programs require files containing chromosome sizes. Pybedtools handles these automatically with the genome keyword argument to methods that wrap such programs. For example, the following command creates a bedGraph file of read coverage for the hg19 assembly:
![]()
4 CONCLUSION
The pybedtools package provides a convenient and flexible interface to both the BEDTools command-line tools and efficient functions in the BEDTools C++ libraries. Pybedtools simplifies complicated analyses by extending the functionality in BEDTools and by providing, to our knowledge, the first Python library offering a common interface for manipulating datasets in diverse formats. Other new functionality includes: set operations on multiple datasets using a simple, intuitive syntax, the ability to filter features and select specific columns or attributes, a unified interface to common attributes (e.g. chromosome, start, end, name and strand) from many file formats, and a documented command history. Pybedtools provides researchers with a simple and efficient interface for exploring complex genomics datasets in widely used formats.
Funding: Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- 1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danacek P., et al. The Variant Call Format and VCFTools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium et al. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;10:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W., et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1106. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A., Hall I. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L. The case for cloud computing in genome informatics. Genome Biol. 2010;11:207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.