

Abstract
Many persistent pain states (pain lasting for hours, days, or longer) are poorly treated because of the limitations of existing therapies. Analgesics such as nonsteroidal anti-inflammatory drugs and opioids often provide incomplete pain relief and prolonged use results in the development of severe side effects. Identification of the key mediators of various types of pain could improve such therapies. Here, we tested the hypothesis that hitherto unrecognized cytokines and chemokines might act as mediators in inflammatory pain. We used ultraviolet B (UVB) irradiation to induce persistent, abnormal sensitivity to pain in humans and rats. The expression of more than 90 different inflammatory mediators was measured in treated skin at the peak of UVB-induced hypersensitivity with custom-made polymerase chain reaction arrays. There was a significant positive correlation in the overall expression profiles between the two species. The expression of several genes [interleukin-1β (IL-1β), IL-6, and cyclooxygenase-2 (COX-2)], previously shown to contribute to pain hypersensitivity, was significantly increased after UVB exposure, and there was dysregulation of several chemokines (CCL2, CCL3, CCL4, CCL7, CCL11, CXCL1, CXCL2, CXCL4, CXCL7, and CXCL8). Among the genes measured, CXCL5 was induced to the greatest extent by UVB treatment in human skin; when injected into the skin of rats, CXCL5 recapitulated the mechanical hypersensitivity caused by UVB irradiation. This hypersensitivity was associated with the infiltration of neutrophils and macrophages into the dermis, and neutralizing the effects of CXCL5 attenuated the abnormal pain-like behavior. Our findings demonstrate that the chemokine CXCL5 is a peripheral mediator of UVB-induced inflammatory pain, likely in humans as well as rats.
INTRODUCTION
During inflammation, molecules such as hydrogen ions, purines, lipids (for example, prostanoids), and immune-related agents (cytokines and chemokines) can sensitize nociceptors (pain-sensitive cells), typically by the phosphorylation of various intracellular kinases, and evoke hyperalgesia or increased sensitivity to noxious stimuli (1). Resident or infiltrating inflammatory cells such as macrophages, neutrophils, lymphocytes, and mast cells can release a wide range of cytokines and chemokines, such as interleukin-1β (IL-1β), tumor necrosis factor–α (TNFα), and nerve growth factor (NGF) (2, 3), which induce and maintain pain-related hypersensitivity. In the skin, keratinocytes release cytokines after injury and during inflammation (4, 5). Cytokines and chemokines are important in recruiting different types of immune cells and can themselves directly influence transduction mechanisms in nociceptors (6–8).
Despite this understanding, there are still few drugs for inflammatory pain that have made the transition from bench to bedside. One issue that has been blamed for this poor conversion rate is the over-dependency of the drug discovery process on animal models, in which factors such as species, strain, and sex influence the ability of models to predict analgesia in human patients (9). Experiments on healthy human volunteers in which a defined injurious stimulus is followed by assessment of sensory processing can help to bridge this gap (10). In human studies, nociceptors can be activated directly by agents applied to skin, including adenosine triphosphate (ATP) (11), capsaicin or mustard oil (12), thermal burn (13), freeze injury (14), or histamine or cowhage (15).
Also, inflammation from a UVB burn produces a robust dose-dependent hypersensitivity to thermal and mechanical stimulation in man and rodents, which peaks between 24 and 48 hours after irradiation (16–19). UVB-induced hypersensitivity is confined to the irradiated area in both species and does not depend on N-methyl-D-aspartate (NMDA) receptor recruitment (20). After a UVB burn, rat primary afferent nociceptors do not develop spontaneous activity, but heat-insensitive C fibers show an enhanced response to suprathreshold mechanical stimuli, and heat-sensitive C fibers develop an increased response to noxious heat. These findings suggest that this extra-responsive pain state is induced by mediators acting peripherally (16, 17, 20). UVB irradiation increases the expression of inflammatory mediators such as IL-1β, IL-6, and IL-10 in vivo (21, 22). Additionally, irradiation of cells in vitro, such as mast cells, which are abundant in the skin, can up-regulate the mRNA for the major inflammatory chemokine CXCL8 (23). We used UVB irradiation as a stimulus to compare mediator expression in human and rat skin in a systematic fashion and to identify peripheral mediators of hyperalgesia. With custom-made polymerase chain reaction (PCR) array cards, we measured transcripts from more than 90 different putative inflammatory mediators at the peak of pain-related hypersensitivity in human and rat UVB-irradiated skin.
RESULTS
UVB irradiation causes sensory changes in human and rat skin
To compare the expression of cytokines and chemokines during UVB-induced inflammation in human and rat skin, we chose a level of irradiation known to result in increased blood flow as well as in mechanical and thermal pain-related hypersensitivity (16, 17). We selected a time point of 40 hours after irradiation because sensory changes are known to peak between 1 and 2 days. In human forearm skin, three minimal erythema doses (MEDs) of UVB applied to an area of 10 mm2 caused a significant increase in blood flow to the irradiated area [230.1 ± 29.2 arbitrary units (AUs)] compared to a nonirradiated control skin site (14.4 ± 3.2 AU) (Fig. 1A). In rats, the plantar surface of the hindlimb was irradiated with UVB (1000 mJ/cm2), also producing a significant increase in blood flow (169.7 ± 25.8 AU) when compared to the contralateral limb’s nonirradiated skin surface (39.3 ± 4.4 AU) (Fig. 1B). A clear mechanical and thermal hyperalgesia was observed in both human and rat skin irradiated with UVB. The mean 50% pain threshold to mechanical stimulation was found to be 17.6 ± 5.8 g in control skin from human volunteers. This was significantly reduced to 1.4 ± 0.3 g (Fig. 1C) within the irradiated area. Mean heat pain thresholds were also significantly reduced to 40.1 ± 0.5°C from 47.2 ± 0.7°C in nonirradiated skin (Fig. 1E). In rats, UVB irradiation caused a significant drop in mechanical withdrawal thresholds from 11.2 ± 1.5 g in the contralateral nonirradiated skin to 4.1 ± 0.5 g in irradiated skin (Fig. 1D). Thermal withdrawal latencies were also significantly reduced from 14 ± 0.5 s on the control side to 7.4 ± 0.6 s in the irradiated site (Fig. 1F). Despite the use of general anesthetics in the rats, which may have delayed the inflammatory process (24), the cardinal signs of inflammation were present, and these results demonstrate similarity in mechanical and thermal hypersensitivity between the human and rat when equivalent doses of UVB are used (16, 17).
Fig. 1.

UVB-induced sensory changes in human and rat skin 40 hours after UVB irradiation. (A and B) Three MEDs and UVB (1000 m J/cm2) to human and rat skin, respectively, caused a significant increase in blood flow to the irradiated area when compared to control skin sites. (C to F) In the same area, UVB irradiation caused a significant reduction in thresholds to mechanical (C and D) and thermal (E and F) stimulation in human and rat skin, respectively (paired t test or Wilcoxon signed-rank test (depending on data distribution). n = 10 (A, C, and E). n = 8 (B, D, and F).**P < 0.01; ***P < 0.001. All data are presented as the means ± SEM. AU, arbitrary unit.
UVB irradiation regulates inflammatory mediators
We designed custom-made Taqman array cards to determine the expression of a large number of the inflammatory mediators (mainly chemokines and cytokines) in UVB-treated rat and human skin 40 hours after irradiation. Wherever possible, rat and human orthologs of the mediators were included to enable direct comparison between species. All data from rat and human cards are shown in tables S1 and S2, respectively. Data are displayed as a fold change (FC) in the relative expression of the transcript in UVB, setting the value from control skin as 1.0. Table 1 shows the top 20 up-regulated transcripts, ranked by FC, in rat and human, most of which were statistically significant when compared to control. A number of transcripts with a known role in inflammatory hyperalgesia were up-regulated in both species. For example, IL-1β and the enzyme cyclooxygenase-2 (COX-2) were both significantly up-regulated {rat: 5.0 (1.3 to 18.7) [FC (±1 SD range)], 4.6 (2.0 to 10.6), human: 10.3 (5.9 to 17.8), 5.3 (3.0 to 9.5), respectively} in both species. A number of other mediators exhibited even larger increases; many of these were chemokines (7 of the top 10 in Table 1). Of particular interest was the chemokine CXCL5, the top up-regulated transcript in both species [rat: 51.3 (20.5 to 128.2), human: 82.5 (45.4 to 150.0)]. This chemokine has not previously been implicated in nociceptive processing but is elevated in certain chronic pain syndromes (25). Using the more readily available rat tissue, we confirmed both the up-regulation and significance levels for a number of the top up-regulated mediators (fig. S1) using conventional quantitative PCR (qPCR). The results with conventional qPCR were highly consistent with those obtained with Taqman array PCR, hence validating our array data.
Table 1.
Increase in expression of the top 20 inflammatory mediators in UVB-treated skin. Transcript levels of more than 90 inflammatory mediators were measured in irradiated and nonirradiated skin at the peak of sensory changes with custom-made Taqman array cards in both the human and rat model. The level of significance was determined by t tests comparing irradiated versus control skin for each species. Because of multiple testing, P values were adjusted with the FDR. FC = UVB/Control. Data are expressed as mean FC ± SD (n = 8). ND, not detected; CXCL, chemokine (C-X-C motif) ligand; CCL, chemokine (C-C motif) ligand; IL, interleukin; G-CSF, granulocyte colony-stimulating factor; KGF, keratinocyte growth factor; COX-2, cyclooxygenase-2.
| Gene name | FC in rat | FC in human |
|---|---|---|
| CXCL5 | 51.3 (20.5 to 128.2)*† | 82.5 (45.4 to 150.0)* |
| iNOS | 34.3 (3.1 to 385.1)‡ | −1.1 (−2.2 to 1.8) |
| IL−24 | 32.7 (8.3 to 128.8)*† | 63.7 (44.5 to 91.3)* |
| CXCL2 | 24.6 (3.1 to 198.4)§† | 12.0 (8.0 to 18.0)* |
| CCL4 | 15.4 (6.6 to 35.8)* | 2.5 (1.4 to 4.5)‡ |
| IL-6 | 14.8 (4.1 to 53.9)§† | 54.7 (30.3 to 99.0)* |
| CCL2 | 14.6 (5.3 to 40.6)* | 5.1 (3.8 to 7.0)* |
| CCL7 | 14.2 (6.2 to 32.6)*† | 13.8 (4.2 to 44.8)* |
| CXCL7 | 14.0 (2.8 to 70.5)§ | 4.0 (1.8 to 8.6) |
| CCL11 | 11.6 (5.9 to 22.9)*† | 4.2 (1.1 to 16.6) |
| IL-10 | 10.7 (5.4 to 21.2)* | 8.0 (4.1 to 15.8)* |
| IL-3 | 9.0 (3.4 to 23.3)§ | ND |
| G-CSF | 7.1 (−1.2 to 62.2) | 25.0 (10.7 to 58.5)* |
| IL-19 | 6.2 (3.0 to 12.6)§ | ND |
| CCL3 | 6.0 (2.9 to 18.9)§ | 16.6 (10.7 to 25.7)* |
| CXCL4 | 6.0 (3.9 to 9.2)* | 2.1 (−1.1 to 5.0) |
| KGF | 5.8 (3.2 to 10.5)* | 4.3 (2.9 to 6.5)* |
| CXCL1 | 5.4 (1.9 to 15.5)§ | 18.9 (12.6 to 28.4)* |
| IL-1 β | 5.0 (1.3 to 18.7)‡ | 10.3 (5.9 to 17.8)* |
| COX-2 | 4.6 (2.0 to 10.6)§ | 5.3 (3.0 to 9.5)* |
P < 0.001.
Validated with individual qPCR.
P < 0.05.
P < 0.01. Mean FC (±1 SD range).
UVB-induced gene changes are correlated in human and rat
Seventy-three of the 92 different target transcripts detected were represented on Taqman array cards for the two species. Using an average of their FC in a log2 format, we plotted UVB-induced gene changes from the human and rat against each other. Calculation of the Pearson’s correlation coefficient (r = 0.435) determined that there was an overall positive correlation between the two sets of data (Fig. 2). In addition, this relationship was significant (P = 0.00012), suggesting that the inflammatory process induced by UVB irradiation and required for the development of thermal and mechanical hypersensitivity was similar in both species. The concordance between the two models is further emphasized by comparison of the FC values from the mediators that showed the top transcriptional changes (Table 1). Nevertheless, there were some genes (Fig. 2, triangles) that showed differential regulation in human and rat (CCL20, IL-1α, IL-20, and iNOS). When the distribution of the difference between the human and rat FC value for each gene was plotted, these four genes were situated 2 SDs away from the norm and therefore could be potential outliers. Without these four genes, there is positive shift in the Pearson’s correlation coefficient, to r = 0.69. Human volunteers consisted of both males and females, whereas only male rats were used for the PCR array. Because females have a higher incidence of some inflammatory diseases, we compared inflammatory mediator expression between genders and found a strong positive and significant correlation (r = 0.88, P < 0.001) (fig. S2), justifying our direct comparison of the human data with the rat data.
Fig. 2.
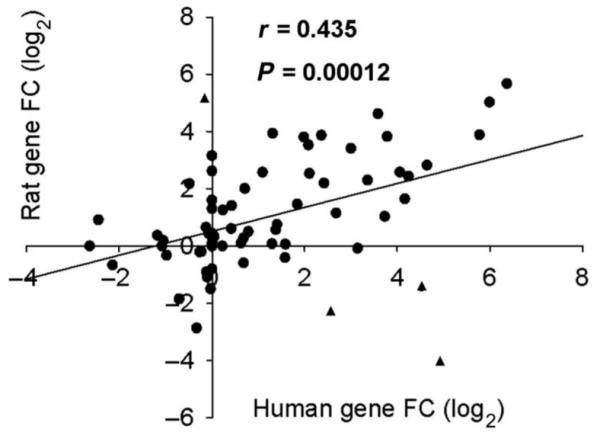
Correlation of UVB-induced chemokine and cytokine expression in the human and rat. Human and rat gene expression after UVB irradiation was plotted against each other. The Pearson’s correlation coefficient, r = 0.435, showed that there is a positive relationship between the two data sets. This relationship is also significant with P < 0.001. Potential outliers (▲, see Results). n = 73.
The chemokine CXCL5 causes mechanical pain-related hypersensitivity and induces infiltration of neutrophils and macrophages
CXCL5 expression was up-regulated at the peak of UVB-induced hypersensitivity in both species 40 hours after UVB treatment. In addition, the nonsteroidal anti-inflammatory drug (NSAID) piroxicam significantly attenuated the increased CXCL5 expression in the rat UVB model (fig. S3). We know that UVB-induced hypersensitivity is susceptible to NSAID treatment (16, 26), and therefore, we next investigated the possible algogenic, or pain-producing, role of this chemokine. Intraplantar injection of CXCL5 caused a dose-dependent reduction in mechanical pain thresholds at 0.5 (5.1 ± 1.2 g) and 6 (5.7 ± 0.9 g) hours after treatment when compared to vehicle control (10.4 ± 1.4 g and 11.9 ± 1.2 g, respectively) (Fig. 3A). Mechanical thresholds returned to baseline levels by 24 hours. In the same animals, there was no significant alteration in thermal withdrawal latencies at any dose (Fig. 3B) compared to vehicle-treated animals.
Fig. 3.

The chemokine CXCL5 causes mechanical pain-related hypersensitivity and induces an infiltration of neutrophils and macrophages. (A) When compared to control rats, intraplantar injection of CXCL5 (3 μg) produced a significant reduction in paw withdrawal thresholds to mechanical stimulation at 0.5 and 6 hours after treatment. No significant changes were measured in mechanical thresholds at doses of 0.1 and 1 μg. (B) In the same animals, no change was observed in withdrawal latencies to thermal stimulation at any dose [**P < 0.001; *P < 0.05, two-way repeated-measures analysis of variance (ANOVA); vehicle, 0.1 μg, 1 μg (n = 11), and 3 μg (n = 6)].(C and D) Plantar skin preparations stained with H&E showed a marked inflammatory cell infiltrate within the dermis 6 hours after intraplantar injection of CXCL5 (3 μg). A representative section at low magnification is shown in (C) (scale bar, 200 μm). In (D), a high-magnification section is shown (scale bar, 25 μm) and infiltrating cells, both monocytes (red arrows) and PMNs (blue arrows), are indicated. (E and F) A low level of macrophages (as shown by IBA1 staining) is seen in the dermis of control animals (E), which markedly increases at 6 hours after CXCL5 (3 μg) injections (F) (scale bar, 200 μm). (I) The increase in IBA1 expression after CXCL5 injections was confirmed with qPCR compared to mRNA expression in vehicle-treated skin. (G and H) Immunopositive CD3 cells (a marker of lymphocytes) were detected at low levels in control skin (G) and did not change after CXCL5 treatment (H). (J) There was no difference in the mRNA levels of CD3 in treated versus control animals. (K and L) The mRNA of the neutrophil marker G-CSFR (K) and the cognate receptor for CXCL5, CXCR2 (L), were significantly up-regulated in the chemokine-treated skin compared to control [*P < 0.05; **P < 0.01, Mann-Whitney rank sum test or t test (depending on data distribution); n = 3 to 5]. All data are presented as means ± SEM.
CXCL5, like all chemokines, can cause chemoattraction of certain immune cells, in particular neutrophils (27). Therefore, we investigated whether its ability to induce pain-related mechanical hypersensitivity was associated with cellular infiltration into the skin. A hematoxylin-eosin (H&E) stain revealed the presence of both monocytes and polymorphonuclear (PMN) cells within the dermis 6 hours after injection of the chemokine (Fig. 3, C and D).
Using an antibody against IBA1, a marker of cells from the monocyte lineage and hence macrophages (28), we found intense staining in rat skin 6 hours after CXCL5 treatment compared to vehicle (Fig. 3, E and F). With qPCR, we found that the increase in IBA1 staining was accompanied by a significant increase in the relative expression of IBA1 mRNA in CXCL5-treated skin (2.7 ± 0.5) compared to control (1.0 ± 0.1) (Fig. 3I). Low levels of CD3, a pan T cell marker, were detected in rat skin after vehicle and CXCL5 treatment (Fig. 3, G and H), and no difference was measured using qPCR (Fig. 3J). To measure the levels of neutrophils using qPCR, we used primers against the granulocyte colony-stimulating factor receptor (G-CSFR). This receptor is primarily found on myeloblasts and mature neutrophils (29) and therefore, in skin, is a good marker of infiltrating PMN cells that have matured. In agreement with the H&E staining results, we found a significant increase in G-CSFR mRNA relative expression of 16.3 ± 2.9 compared to vehicle-treated skin (Fig. 3K). CXCL5 acts largely through the G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor CXCR2 (26). This receptor is found primarily on neutrophils, but can also be expressed on macrophages and mast cells (30). After CXCL5 treatment, we also saw a significant increase in CXCR2 mRNA (Fig. 3L), possibly from infiltrating cells attracted by CXCL5, suggesting that this chemokine is acting in a receptor-specific manner.
UVB-induced hypersensitivity and CXCL5 expression peak at 48 hours
UVB irradiation causes a prolonged inflammatory pain state with pain-related hypersensitivity in the rat lasting for 10 to 14 days (16). Thresholds to mechanical stimulation were significantly reduced 24 and 48 hours after irradiation when compared to naïve animals but had returned to normal by 10 days after irradiation (Fig. 4A). In the same animals, we saw a significant reduction in withdrawal latency to a heat stimulus 24 and 48 hours after UVB treatment when compared to naïve animals; withdrawal latency returned to normal by 10 days (Fig. 4B). We next measured the expression of CXCL5 in the irradiated skin taken from these animals. Compared to mRNA levels in naïve skin and in agreement with the Taqman array data (Table 1), CXCL5 was significantly up-regulated most prominently 48 hours after irradiation, with a relative mRNA expression of 339.6 ± 171.6 versus control (1.0 ± 0.3) (Fig. 4C). There was also more but nonsignificant expression at 24 hours (7.4 ± 2.4) versus control. We also measured protein levels when CXCL5 mRNA expression was at its peak. In nonirradiated skin, CXCL5 protein was not detected; however, 48 hours after UVB treatment, a significant amount was seen in the inflamed skin (Fig. 4D). At the peak of pain-related hypersensitivity and CXCL5 expression (48 hours), we also measured macrophage, neutrophil, and T cell makers. Compared to control, 48 hours after UVB treatment, skin showed a significant increase in IBA1 (2.8 ± 0.7) and G-CSFR (32.7 ± 11.1) relative mRNA expression, similar to that seen after CXCL5 treatment alone (Fig. 4, E and G). We also see a small increase in CXCR2 expression (1.46 ± 0.25) compared to control (1.0 ± 0.1). However, this was not significant. Again, no differences were measured in the mRNA expression of CD3 (Fig. 4F), suggesting a lack of T cell influence in UVB-induced inflammation. Because we used both genders in the human group and because the inflammatory response, particularly with regard to neutrophil attraction, shows sex differences (31), we repeated these experiments in female rats. As in male rats, females showed a significant hypersensitivity 48 hours after UVB treatment. This was accompanied by significant increases in CXCL5, IBA1, and G-CSFR relative mRNA expression, indicating a comparable inflammatory response in female rats (fig. S4, A to D).
Fig. 4.

Time course of CXCL5 expression and pain-related hypersensitivity in the rodent UVB model. (A and B) UVB irradiation caused a significant reduction in mechanical (A) and thermal thresholds (B) at 24 and 48 hours after treatment. Mechanical thresholds were also reduced 8 hours after UVB treatment. (C) CXCL5 mRNA expression measured at 0, 4, 8, 24, and 48 hours and 10 days after UVB (1000 m J/cm2). Compared to 0 hours, CXCL5 expression significantly increased at 48 hours (y axis has a logarithmic scale). No difference was measured at other time points (Kruskal-Wallis one-way ANOVA on ranks, n = 4 to 5).(D) Expression of CXCL5 protein was measured with a specific ELISA. Control skin samples contained nondetectable levels of CXCL5. However, 48 hours after UVB irradiation, CXCL5 was significantly increased (n = 4 per group, Mann-Whitney rank sum test, P = 0.029). (E to H) UVB irradiation increased the expression of IBA1 (E) and G-CSFR (G), but not CXCR2 (H) or CD3 (F) [*P < 0.05; **P < 0.01; ***P < 0.001, Mann-Whitney rank sum test or t test (depending on data distribution); n = 5 to 7]. All data are presented as means ± SEM.
Neutralization of CXCL5 attenuates UVB-induced mechanical hypersensitivity and decreases the number of infiltrating cells
We next tested whether and to what extent CXCL5 contributes to the mechanical hypersensitivity caused by UVB treatment. In irradiated rats, repeated doses of a neutralizing antibody against CXCL5 significantly attenuated the reduced mechanical withdrawal thresholds 30 hours after UVB irradiation (6.0 ± 0.8 g) when compared to the group receiving a control immunoglobulin G (IgG) antibody (3.6 ± 0.7 g) (Fig. 5A), whereas no difference between groups was observed for thermal hypersensitivity at any time point (Fig. 5B). The neutralization of CXCL5 in the irradiated paw did not alter withdrawal thresholds or change the expression of IBA1 and G-CSFR at the early 8-hour time point (Fig. 5, C and D). Thirty hours after UVB irradiation, however, the attenuation of UVB-induced mechanical hypersensitivity was associated with a reduction in infiltrating macrophages and neutrophils. IBA1 staining was reduced in irradiated rat skin treated with the CXCL5 antibody compared to those treated with the IgG antibody (Fig. 5, I and K). This reduction was significant when measured by qPCR (Fig. 5E). Again, very few cells were positively stained for CD3 (Fig. 5, J and L), and no difference was measured in CD3 mRNA expression (Fig. 5F). Significant reductions in G-CSFR relative mRNA expression and CXCR2 were also measured after local neutralization of CXCL5 (Fig. 5, G and H).
Fig. 5.

CXCL5 contribution to UVB-induced pain-related hypersensitivity. (A and B) Animals received either a functional blocking CXCL5 antibody (Ab, 10 μg) or a control nonimmune IgG (10 μg) immediately after UVB irradiation, and again at 10 and 20 hours [shown by arrows in (A) and (B)]. Antibody neutralization of CXCL5 significantly attenuated the reduction in mechanical thresholds 30 hours after UVB (1000 mJ/cm2) (A). However, neutralization of this chemokine did not have an effect on thermal thresholds after UVB irradiation (B) (two-way repeated-measures ANOVA, n = 11). (C and D) At 8 hours after UVB irradiation, there was no change in infiltrating cell markers between treated and control animals. IBA1 mRNA expression was similar between CXCL5 antibody and the control IgG groups (C). Also, G-CSFR mRNA expression showed no change between groups (D). (E to H) At 30 hours after UVB irradiation, a time point at which the CXCL5-neutralizing antibody is effective in decreasing hypersensitivity behaviors, there was a significant reduction in IBA1 (E), G-CSFR (G), and CXCR2 mRNA expression (H) in animals treated with CXCL5-neutralizing antibody. No difference was found in CD3 mRNA expression (F) (*P < 0.05, t test; n = 4 to 6) (I to L) IBA1 staining in irradiated skin was reduced in animals treated with the CXCL5 antibody (K) versus vehicle (I). Low levels of CD3 staining were similarly detected in both groups (J and L) (scale bar, 200 μm). All data are presented as means ± SEM.
CXCL5 triggers calcium responses and increases migration in macrophages in vitro
All chemokines bind to G protein–coupled receptors, which when activated cause changes in intracellular calcium levels (32). We found that CXCL5 caused a calcium response in cultured macrophages (Fig. 6, A to D). At a dose of 100 nM, CXCL5 activated 43.3 ± 5% of the macrophages, significantly more than vehicle (0%) (Fig. 6E). An example of this response is shown in Fig. 6F. CXCL5 (10 nM) did not have an effect on calcium concentrations (Fig. 6E). No response was seen in sensory neurons cultured from the dorsal root ganglia (DRG) (Fig. 6G), suggesting that CXCL5 does not directly activate this cell type.
Fig. 6.

Exposure of macrophages to CXCL5 produced an increase in intracellular Ca2+ concentration ([Ca2+]i). (A to D) Macrophages in which application of ATP (25 μM) produced a change in [Ca2+]i [as seen in (D)] were included in the analysis (thus excluding dead or nonfunctional cells). CXCL5 at a dose of 100 nM increased [Ca2+]i in a number of cells as indicated by the red arrows (C). (E) The percentage of cells responding to 100 nM CXCL5 was significantly larger than after vehicle treatment (E) (Kruskal-Wallis one-way ANOVA on ranks, n = 3 to 6 independent experiments with at least 100 cells per concentration, data are presented as mean% ± SEM). (F and G) Representative response traces of a single cell for macrophages (F) and DRG neurons (G).
CXCL5 promote chemotaxis of neutrophils (33, 34); however, its chemotactic effects on macrophages have not been examined. Using a Boyden chamber, in which cells cross a concentration gradient through a porous filter, we found that a significantly increased percentage of macrophages migrated toward wells containing 1 nM (1392.7 ± 133%) and 10 nM (1142.9 ± 239%) CXCL5 over those containing control solution (100%) (Fig. 7A). The cells in control and CXCL5 (10 nM)–treated wells are shown in Fig. 7, B and C. To assess whether CXCL5 is a chemotactic or a chemokinetic factor, we used a checkerboard analysis (35). When the concentration of the chemokine was equal in both upper and lower wells, we noted enhanced migration as a result of increased motility in a nondirectional manner, a process termed chemokinesis. However, when there was a concentration gradient between the upper and lower chambers, migration was significantly higher, indicating that CXCL5 is a true chemotactic factor for macrophages (Fig. 7D).
Fig. 7.

CXCL5 attracts cultured peritoneal macrophages. (A) When compared to control, 1 and 10 nM CXCL5 significantly increased the migration of macrophages (Kruskal-Wallis one-way ANOVA on ranks, n = 3 to 5).(B and C) Counterstained filters show cells in a control well (B) and a well containing CXCL5 (10 ng/ml) (C). (D) To test whether CXCL5 induces migration through a gradient (chemotaxis) or solely random nondirectional migration (chemokinesis), we used the checkerboard analysis. In a Boyden chamber, cells were suspended in increasing concentrations of CXCL5 and allowed to migrate toward increasing concentrations of CXCL5 in the lower wells. The gray boxes show the results obtained when the same concentration was used in both wells (no gradient). When there is an increasing gradient, migration is further enhanced, indicating that CXCL5 induces directional chemoattraction on macrophages (Kruskal-Wallis one-way ANOVA on ranks, n = 3 to 5). Significant migration across a gradient versus migration when the concentration is the same. *P < 0.05 compared to 0 versus 0 nM; +P < 0.05 compared to 1 versus 1 nM; #P < 0.05 compared to 10 versus 10 nM. All data are presented as the means ± SEM.
DISCUSSION
Quantified doses of UVB irradiation applied to small patches of skin induce a marked hyperalgesia with a parallel time course and magnitude in rats and humans (16–19). The mechanism of UVB-induced hyperalgesia is not definitively established, but a major component is the peripheral sensitization of nociceptive afferent terminals in the irradiated skin (20). The mediators driving this peripheral sensitization are themselves not defined, but anti-inflammatory drugs reduce the sensory sequelae of UVB irradiation (16, 21, 26).
First, we used PCR to characterize the expression of inflammatory mediators in rat and human. PCR arrays are sensitive enough to assess the expression of a large number of transcripts in small tissue samples, samples of a size that can be realistically obtainable in a clinical setting from a range of patient groups. Our arrays assessed the expression of a broad range of inflammatory mediators with a particular emphasis on cytokines and chemokines. At the peak of UVB-induced hypersensitivity (40 hours), in both humans and rats, a number of chemokines and cytokines were transcriptionally up-regulated. Angst et al. (21) used microdialysis to assay the protein levels of a more restricted number of putative mediators 24 hours after UVB irradiation in human skin. As in our study, they found elevated levels of IL-6, IL-8, IL-10, G-CSF, and CCL4, and no change in TNFα and NGF. In contrast to our results, however, they did not find a significant increase in CCL2 or IL-1β.
Our second aim was to increase the likelihood that our results could be translated to the clinic. Pain mechanisms have in the past largely been explored in animal models, some of which have an uncertain relationship to chronic pain states in patients, which has hampered clinical translation. Although there have been several efforts to introduce animal models with greater face validity [for example, chemotherapy agent–induced neuropathies in rodents (36, 37)], there are still uncertainties about translatability across species. Our data demonstrate that after UVB irradiation, humans and rodents show transcriptional up-regulation of similar genes. This significant correlation of gene expression between species was not necessarily expected because the immune response has been thought to be more species-specific than other mechanisms. We categorized the response of inflammatory mediators to UVB irradiation. One group showed little alteration by UVB, changing less than twofold and comprising about 50 to 60% of transcripts studied. A second group of about 15 factors was moderately up-regulated in both species and included several previously identified genes with known roles in pain modulation such as CCL2, CCL3, COX-2, and IL-1β. A small number of factors were markedly up-regulated in both species by UVB treatment, and these are CXCL5, CXCL2, IL-24, and IL-6. Four factors were regulated in a discordant manner between species.
The degree of correlation in gene expression suggested a similar underlying biological response, at least in terms of inflammatory mediators, in humans and rats at the peak of UVB-induced hypersensitivity. These other persistent pain states in humans may also share mechanisms with rodents, which would facilitate the development of treatments. One example is osteoarthritis (OA) in which most pain arises from peripheral tissues (38), driven by peripheral mediators. These mediators are unidentified, and although the animal models of OA show joint damage and sensory abnormalities, it is still notclear whether they do so by releasing the same mediators that drive OA pain in patients. The approach we describe here may help to identify the clinically relevant factors.
The highly up-regulated transcripts by UVB have not been previously described as potential pain mediators. This group consisted mainly of chemokines: CCL4, CCL7, CCL11, CXCL2, CXCL5, and CXCL7, as well as the cytokine IL-24. The chemokine family has more than 50 members, and our data support the notion that this family contains a relatively unexplored group of promising pain mediators. Two chemokines, CCL2 and CX3CL1, have been extensively studied in chronic pain, either due to their actions on glia cells (39–41) or in the case of CCL2 also via neuronal interactions (42, 43). Other chemokines act peripherally. For example, the individual intraplantar injection of several chemokines (CCL2, CCL22, CCL5, CXCL1, CXCL8, and CXCL12) induces pain-related hypersensitivity in normal rats (7, 44, 45). A number of these previously described pro-nociceptive chemokines were significantly up-regulated in our UVB model (CCL2, CXCL1, and CXCL8).
Our data on UVB-induced mechanical hypersensitivity points to a previously unrecognized role for the chemokine CXCL5. CXCL5, also known as epithelial-derived neutrophil-activating peptide-78 in humans and lipopolysaccharide-induced CXC chemokine (LIX) in rodents, is involved in the recruitment and activation of leukocytes during inflammatory conditions (27). For example, this chemokine is elevated in the knee joints of arthritic patients and contributes to neutrophil recruitment (46). CXCL5 is one of a subclass of CXC chemokines that carry a glutamate-leucine-arginine (ELR) motif and act mainly via the CXCR2 receptor, although it may also act through other receptors (47, 48). The CXCR2 receptor is present on neutrophils, monocytes, and endothelial cells and is important in neutrophil chemoattraction and monocyte arrest during inflammation (27). CXCL5 can be expressed by cells of the epidermis such as keratinocytes (49), as well as infiltrating monocytes (50), and it is plausible that both of these cell types contribute to the increased expression of CXCL5 after UVB irradiation. The non-selective COX inhibitor piroxicam, like all NSAIDs, reduces inflammation in general and consequently has analgesic properties. At the peak of pain-related hypersensitivity, 48 hours after UVB treatment, the expression of CXCL5 mRNA was at its highest and could be partially ameliorated by piroxicam treatment. In addition, we also detected a significant level of CXCL5 protein at this time point. When injected into the plantar surface of the rat paw, CXCL5 evoked a mechanical but not thermal pain-related hypersensitivity. This hypersensitivity was associated with an infiltration of macrophages and neutrophils into the dermis. CXCL5 is a chemotactic agent for neutrophils (33, 34). Our data show that CXCL5 potently attracts cultured peritoneal macrophages and elicits sizeable calcium responses in these cells.
Irradiating the skin with UVB causes damage to DNA and apoptosis of epidermal cells (51). In mice, the subsequent inflammation results in recruitment of neutrophils and macrophages into the dermis (52, 53). We found that blocking the action of CXCL5 in the UVB-treated rodent significantly reduced the levels of macrophages and neutrophils in the skin at the time of maximal mechanical hypersensitivity. CXCL5 is therefore likely to contribute to the sensory changes evoked by UVB irradiation through the recruitment of inflammatory cells and the subsequent release of pro-algesic mediators. Both neutrophils and macrophages have been implicated in the development of abnormal pain states (54–57). Reduction of neutrophil recruitment attenuates pain-related hypersensitivity in some laboratory models of inflammation, for instance, in those associated with carrageenan and zymosan treatment (58, 59). Other chemokines with the ability to attract neutrophils such as CXCL1, CXCL2, and CXCL8 (in humans) were also significantly up-regulated after UVB treatment. These molecules may also have contributed to the infiltration of PMN cells and the subsequent sensory changes measured after UVB irradiation in our experiments. Pain-related hypersensitivity arising from nerve ligation or transection in rodents is partially dependent on macrophage recruitment (54, 60). Other chemokines with the ability to attract macrophages such as CCL2, CCL3, and CCL7 were also up-regulated in UVB-treated skin, suggesting that this cell type may also contribute to UVB-induced hypersensitivity.
CXCL8 and CXCL1 are other members of the CXC chemokine family with ELR motifs that can induce pain-related hypersensitivity when injected peripherally (44, 61). Both of these agents can act through the CXCR2 receptor. Antagonism of this receptor can attenuate pain-related hypersensitivity in a number of experimental pain states in rodents. These include the collagen-induced arthritis model; inflammatory pain arising from carrageenan, complete Freund’s adjuvant (CFA), or zymosan injection; and neuropathic-like pain as a result of partial nerve injury (55, 62). Attenuation of the hypersensitivity in these models, by CXCR2 blockade, is often associated with a decreased neutrophil infiltration.
There is a significant increase in mechanical pain-related hypersensitivity 0.5 hour after CXCL5 application that is not associated with leukocyte infiltration. CXCL5 therefore may act either directly on nociceptive terminals or via other resident cells expressing CXCR2. We have not been able to demonstrate direct effects of CXCL5 on dissociated cultured DRG neurons using calcium imaging, although there are some data suggesting that CXCR2 receptors are expressed in DRG cells (63). CXCL5 is, however, known to amplify the inflammatory cascade in endothelial cells by activating nuclear factor κB(NF-κB) and increasing the expression of multiple proinflammatory mediators (64). CXCR2 receptors can also be expressed on mast cells and tissue-resident macrophages, and when activated with CXCL5, these cells can quickly release pro-algesic factors such as TNFα (30). The injection of CXCL1 and another ELR member, CXCL2 (both up-regulated in this study by UVB), does not evoke thermal or mechan cal pain-related hypersensitivity despite PMN cell recruitment (65), suggesting that the pro-algesic properties of CXCL5 may be mediated through its action on resident or infiltrating macrophages.
Our studies demonstrate the utility of experimental pain models in animals and humans to understand pathophysiological mechanisms likely to be relevant in man. They underline the importance of inflammatory mediators, in particular thechemokines, in the development of abnormal pain states, emphasizing their significance as targets for the development of new analgesics.
MATERIALS AND METHODS
Animals
Experiments were performed with male Wistar rats (~250 g, Harlan) in accordance to the UK Home Office regulations. Food and water were available ad libitum, and animals were housed under standard conditions with a 12-hour light/dark cycle.
Volunteers
A total of 10 healthy volunteers (6 male and 4 female) were used for the study. All volunteers had skin of either type II or III (66) as established via an initial screen. Individuals with known skin hypersensitivity and dermatological conditions such as eczema or dermatitis were excluded from all experiments. All participants were asked to refrain from analgesics and anti-inflammatory or anti-histamine medication for 4 hours before irradiation and until all testing was completed. Caffeine or nicotine was forbidden for 1 hour before psychophysical testing. This study was approved by the Ethics Committee of King’s College London, and informed consent was obtained in writing from all participants.
UVB irradiation
Rats were anesthetized with a combination of medetomidine (Domitor, 0.25 mg/kg) and ketamine (Vetalar, 60 mg/kg) given intraperitoneally. When completely anesthetized, rats were covered, exposing only the plantar surface of the hind paw or the depilated lower limb, and placed under TL01 fluorescent bulbs with a maximum wavelength of 311 nm. The irradiance output was measured with a photometer (IL1400A with SEL240/UVB-1/TD filter, ABLE Instruments and Controls) placed at the distance of the exposed skin. This reading was used to calculate the time needed to deliver UVB (1000 mJ/cm2). After the procedure and at least 45 min after anesthetization, animals were given a subcutaneous injection of atipamezole hydrochloride (1 mg/kg).
Volunteers received UVB irradiation at three times MED to an area of 10 mm2 volar forearm using the TL01 fluorescent bulbs. The time to reach this dose was calculated from an initial screening protocol in which 1 MED was defined as the time needed to produce uniform reddening of the area 24 hours after irradiation. This involved irradiating 6 × 1 cm2 patches of volar forearm skin with varying doses, beginning at 307.5 mJ/cm2 and increasing the dose by 1.25 mJ/cm2 every square.
Behavioral testing
Mechanical hyperalgesia was assessed with the up-down method (67)by measuring withdrawal responses to a range of calibrated Von Frey hairs applied to the plantar surface of the rat hind paw. Thermal hyperalgesia was assessed by applying a radiant heat source to the same area and measuring the latency to withdrawal (68). For all behavioral experiments, rats were acclimatized for 1 week and baseline readings were acquired. All behavior was performed with the experimenter blind to treatment.
Psychophysical testing in humans
Mechanical pain thresholds were assessed in human volunteers by applying a range of calibrated Von Frey hairs of incremental force to the UVB-treated volar forearm skin 40 hours after irradiation. The subject was asked to rate each stimulus as painful or not painful, and the up-down method was again applied to calculate their 50% withdrawal threshold. Heat pain threshold was derived as the arithmetic mean of three consecutive measurements with a thermal sensory analyzer (TSA 2001-II, MEDOC) thermode held against the skin (69). Thresholds were obtained with ramped stimuli (0.5°C/s), which terminated when the subject pressed a button to indicate first percept of pain. Temperature of the thermode ramped down to baseline temperature of 32°C (center of neutral range) at a rate of ~5°C/s and remained at 32°C during 10-s interstimulus interval. The contact area of the thermode was 10 mm2. A maximal cut-off temperature of 50°C was used whereby the TSA would automatically return to baseline temperature. Thermal testing was first demonstrated over an area that was not used for testing during the experiment.
Blood flow analysis
In both species, blood flow to irradiated and unirradiated sites was measured with a laser Doppler flow meter (Moor-LAB, Moor Instruments). Rats were anesthetized and a Doppler probe was placed on the skin surface of the depilated lower limb. In humans, a similar probe was placed on the skin surface of the volar forearm. In both cases, the average flux was calculated over a period of 15 s.
Tissue preparation and RNA extraction
Tissue samples, from irradiated and unirradiated skin, were taken with a biopsy punch (3.0 mm, Stiefel) and immediately frozen in liquid nitrogen. In rats, samples were taken from depilated hairy skin of the lower hindlimb. Three-millimeter punch biopsies were taken from control and irradiated skin in human volunteers. Skin samples were homogenized, and total RNA was obtained with a “hybrid” method of phenol extraction (Trizol, Invitrogen) and column purification (RNeasy, Qiagen). This helped to achieve the extraction of high-quality RNA without a significant drop in yield (all 260:280 A ratios were in the range of 1.94 to 2.13). All samples were deoxyribonuclease (DNase) (Qiagen)–treated to prevent genomic contamination. This was confirmed with a RNA 6000 Nano Chip (Agilent) to ensure sufficient RNA integrity. RNA was subsequently synthesized into complementary DNA (cDNA) with the SuperScript II reverse transcriptase kit (Invitrogen) and following the manufacturer’sprotocol.
Taqman array setup and quantitative real-time PCR
Taqman array cards were custom-made and designed with the Applied Biosystems Web site (70). Both human and rat 384-well cards were based on microfluidic technology and contained 4 sets of 96 different primer pairs, which included four housekeeping genes [glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 18S, β-actin, and β2-microglobulin]. Each cDNA sample was diluted with PCR-grade water and added in a 1:1 ratio to Taqman Universal master mix, producing a final concentration of 1 ng/μl. Samples were fed into the appropriate loading ports (1 μl for each well) and prepared according to the manufacturer’s guidelines. Cards were placed into a 7900HT Fast Real-Time PCR system (Applied Biosystems), and cDNA samples were subjected to 40 cycles of amplification. Expression of each transcript was measured with the ΔΔCt (cycling time) method (71) normalized to the geometric mean of the four housekeeping genes with the R package NormqPCR (https://r-forge.r-project.org/projects/qpcr/). Relative changes in transcript levels are presented as fold change (FC = UVB/Control). Where transcript numbers were undetermined for a given detector in less than 50% of samples, the average Ct value was calculated with the remaining data values. Where transcript numbers were undetermined in more than 50% of transcripts for a given sample type (that is, case or control), the sample was given a default Ct of 38. If this occurred in both, no FC value was calculated.
For validation of UVB-induced gene changes and relative changes in the expression of cell markers, individual qPCR was performed with the Corbett Rotor-Gene 6000. Samples were processed in duplicate and amplified with the Roche LightCycler Master Mix containing SYBR Green for the detection of real-time changes. The efficiency of all primers was in the range of 0.8 to 1.2. Transcript levels were measured again with the ΔΔCt method normalized against GAPDH. Relative mRNA expression is presented as the amount of transcript in the treatment group compared to control. Primer pairs were designed with the Primer Blast software (72), except for CD3 (73), andare shown in Table 2.
Table 2.
Primer sequences used in individual qPCR.
| Gene | Direction | Sequence (5′ > 3′) |
|---|---|---|
| GAPDH | Forward | ATGGGAAGCTGGTCATCAAC |
| Reverse | CCACAGTCTTCTGAGTGGCA | |
| IBA1 | Forward | TCCCCACCTAAGGCCACCAGC |
| Reverse | CGTCTCCTCGGAGCCACTGGA | |
| CD3 | Forward | TTGAAGAACGAGCAGCTGTATCA |
| Reverse | CGGCTGTACTGGGCATCAT | |
| CXCL2 | Forward | CACCTCCACACTGTGATAGAGATTGG |
| Reverse | ATAACAGTCGTCCCGCCCTTCT | |
| CXCL5 | Forward | GCATTTCTGCTGCTGTTCACACT |
| Reverse | GGTTAAGCAAACACAGCGTAGCT | |
| CCL7 | Forward | CTCCAAAGCCCTGAAGACAG |
| Reverse | GTTCCTACCCCTTAGGACCG | |
| CCL11 | Forward | CTGCTGCTTTACCATGACCA |
| Reverse | GACCCACTTTTTCTTGGGGT | |
| IL6 | Forward | TCTCTCCGCAAGAGACTTCC |
| Reverse | CCGGACTTGTGAAGTAGGGA | |
| IL24 | Forward | TCTGCAGAATGTCTCGGATG |
| Reverse | AGGAAGTTATCCGGATTGGC | |
| G-CSFR | Forward | GGAGGGCTGCGGGCAAATCA |
| Reverse | GGGACCCGTCAGGCAGGTGA | |
| CXCR2 | Forward | GGCCATCGTCCACGCCACAA |
| Reverse | AAGATGACCCGCATGGCCCG |
Detection of CXCL5 protein by enzyme-linked immunosorbent assay
We used the Rat CXCL5/LIX DuoSet ELISA (enzyme-linked immunosorbent assay) kit (detection range: 62.5 to 4000 pg/ml, DY543, R&D Systems) to detect the presence of CXCL5 protein in rat skin. Protein was extracted from depilated hairy skin by mechanical homogenization in NP-40 lysis buffer [20 mM tris (pH 8), 137 mM NaCl, 10% glycerol, 1% NP-40, 2 mM EDTA, 20 μM leupeptin, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail]. Recombinant rat CXCL5/LIX standards and protein lysate (5 mg/ml) were run in duplicate according to the manufacturer’s instructions. Samples were read at 450 nm with wavelength correction at 540 nm. A reading within the range of the standard curve was considered positive and quantified. Protein was extracted from the ipsilateral and contralateral hairy skin of male Wistar rats 48 hours after being irradiated with UVB light (1000 mJ/cm2).
Histology
Rats were transaortically perfused with cold physiological saline, and the plantar skin, to the depth of the hypodermis, was removed and placed in 4% paraformaldehyde overnight. The tissue was embedded in paraffin and 6-μm sections were cut. The presence of inflammatory cells was determined with the counterstain H&E. Other sections were incubated with primary antibodies against ionized calcium-binding adaptor molecule 1 (IBA1, Wako, 1:1000) and CD3 (DakoCytomation, 1:3000) to detect the presence of macrophages and T cells, respectively. Briefly, sections were dewaxed and hydrated with a serial alcohol dilution. Endogenous peroxidases were inhibited with 3% H2O2, and the mounted sections were washed in buffer. Once blocked, sections were incubated with primary antibody for 2 hours. After further wash steps, sections were incubated with a goat anti-rabbit biotinylated secondary antibody (Vector Laboratories, 1:300) for 30 min. Specific binding of the primary antibody was revealed with the Vectastain ABC kit (Vector Laboratories) with diaminobenzidine (DAB).
Drug treatment
Animals were randomized into treatment groups and received intraplantar injections of vehicle [phosphate-buffered saline (PBS), 0.1% bovine serum albumin (BSA)] and 0.1, 1, or 3 μg of recombinant rat CXCL5/LIX (R&D Systems), respectively, in a volume of 50 μl using a 0.3-ml insulin syringe (Terumo). Mechanical and thermal thresholds were measured in the treated skin at 0.5, 3, 6, and 24 hours after chemokine injection.
To evaluate the effects of a CXCL5 antibody on UVB irradiation, rats received a dose of UVB (1000 mJ/cm2) to the plantar surface of the left hind paw. Immediately after irradiation, an intraplantar injection of either a function blocking goat anti-rat CXCL5/LIX antibody (R&D Systems) or normal goat IgG (R&D Systems) was given at a dose of 10 μg in 50 μl. Further antibody treatment was given directly after assessment of mechanical thresholds. Behavior was carried out 8, 20, and 30 hours after UVB. The area of skin receiving treatment was taken for RNA extraction and histological assessment.
In vitro assays
Macrophage culture
Adult rats were killed with CO2, and 20 to 50 ml of sterile Hanks’ balanced salt solution (HBSS) was injected into the peritoneal cavity. After 5 min, the buffer was retrieved from the cavity and spun to obtain a cell pellet. Cells were resuspended in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and incubated at 37°C.
Calcium imaging
For calcium imaging, cultured macrophages were incubated for 60 to 90 min at 37°C with 2 μM Ca2+ indicator Fura2-AM (Invitrogen) in HBSS. Cells were transferred to a perfusion system attached to an inverted microscope (Nikon) equipped with a monochromator (Photon Technology). Recordings were carried out with a standardized protocol. Average Ca2+ concentration–based fluorescent ratios (340/380-nm excitation) were taken over the first 2 min of each run under continuous perfusion (3 ml/min) with HBSS buffer solution (containing 10 mM Hepes, pH 7.4) and defined as the baseline level. A single manual application of CXCL5 was followed by 3 min without continuous perfusion. After a 2-min washout period, 25 μM ATP (Sigma) or 20 mM KCl was used as a positive control to define viable macrophages or dorsal root ganglion (DRG) neurons, respectively. DRG neurons were cultured as previously described (74). HBSS perfusion buffer for recordings with DRG neurons contained 10 mM Hepes and 15 mM glucose. All experiments were conducted at room temperature. Ratiometric changes of the 340/380-nm wavelengths after manual application were normalized to the averaged baseline ratio. To be defined as a responder, cells needed a ratio change greater than 20% from baseline and a positive response to the application of ATP (macrophages) or KCl (DRG neurons).
Chemotaxis
To assess chemotactic ability, we used the Boyden chamber (Neuroprobe). Briefly, varying concentrations of CXCL5, diluted in serum-free DMEM, were placed into the lower wells. Macrophages were suspended in serum-free DMEM and placed into the relevant top wells (50,000 per well). A polycarbonate filter containing 5-μm pores was used to separate the wells. After 3 hours in the incubator at 37°C, the membrane was removed and cells from the top were wiped away. Cells that had migrated to the chemokine side were stained with RapiDiffII (Biostain Ready Reagents), membranes were cover-slipped, and cells were counted.
Statistical analysis
For Taqman array cards, statistical significance was calculated by running t tests in R (two-sided, Welch’s t test) on the ΔCt values. To control for multiple hypothesis testing, we adjusted the P values using the false discovery rate (FDR) correction as proposed by Benjamini and Hochberg (75). All other statistical analysis was carried out with the SigmaStat software. Where the data were not normally distributed and had unequal variance, nonparametric tests were used.
Supplementary Material
Acknowledgments
Funding: S.B.M. and D.L.H.B. are members of the Wellcome Trust–funded London Pain Consortium. D.L.H.B. is a Wellcome Trust Clinical Scientist. J.M.D. is funded by the Biotechnology and Biological Sciences Research Council and has a case studentship with AstraZeneca. M.C. is sponsored by the Chilean National Scholarship Program for Graduate Studies. J.R.P., K.J.P., and H.K. are students of the London Pain Consortium and funded by the Wellcome Trust. T.K.Y.K. is the recipient of the National Sciences and Engineering Research Council of Canada post-graduate scholarship, Queen Elizabeth II Centennial Scholarship (Ministry of Advanced Education, British Columbia), and the Overseas Research Student Award (Universities UK). This work is part of the Europain project and funded by the Innovative Medicines Initiative Joint Undertaking (IMI JU) grant no. 115007.
Footnotes
Competing interests: The authors declare that they have no competing interests.
www.sciencetranslationalmedicine.org/cgi/content/full/3/90/90ra60/DC1
Fig. S1. Validation of up-regulated transcripts (as measured by PCR array cards) with qPCR.
Fig. S2. Correlation of PCR array data between male and female volunteers.
Fig. S3. The increased expression of CXCL5 mRNA is attenuated in the UVB model by piroxicam treatment.
Fig. S4. UVB-induced changes in female rats.
Table S1. Rat PCR array data.
Table S2. Human PCR array data.
REFERENCES AND NOTES
- 1.Woolf CJ, Ma Q. Nociceptors—noxious stimulus detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Rittner HL, Machelska H, Stein C. Leukocytes in the regulation of pain and analgesia. J. Leukoc. Biol. 2005;78:1215–1222. doi: 10.1189/jlb.0405223. [DOI] [PubMed] [Google Scholar]
- 3.Verri WA, Jr., Cunha TM, Parada CA, Poole S, Cunha FQ, Ferreira SH. Hypernociceptive role of cytokines and chemokines: Targets for analgesic drug development? Pharmacol. Ther. 2006;112:116–138. doi: 10.1016/j.pharmthera.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Albanesi C, De Pità O, Girolomoni G. Resident skin cells in psoriasis: A special Look at the pathogenetic functions of keratinocytes. Clin. Dermatol. 2007;25:581–588. doi: 10.1016/j.clindermatol.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Pastore S, Mascia F, Girolomoni G. The contribution of keratinocytes to the pathogenesis of atopic dermatitis. Eur. J. Dermatol. 2006;16:125–131. [PubMed] [Google Scholar]
- 6.Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1β sensors. J. Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mogil JS. Animal models of pain: Progress and challenges. Nat. Rev. Neurosci. 2009;10:283–294. doi: 10.1038/nrn2606. [DOI] [PubMed] [Google Scholar]
- 10.Schmelz M. Translating nociceptive processing into human pain models. Exp. Brain Res. 2009;196:173–178. doi: 10.1007/s00221-009-1809-2. [DOI] [PubMed] [Google Scholar]
- 11.Coutts AA, Jorizzo JL, Eady RA, Greaves MW, Burnstock G. Adenosine triphosphate-evoked vascular changes in human skin: Mechanism of action. Eur. J. Pharmacol. 1981;76:391–401. doi: 10.1016/0014-2999(81)90110-2. [DOI] [PubMed] [Google Scholar]
- 12.Koltzenburg M, Lundberg LE, Torebjörk HE. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219. doi: 10.1016/0304-3959(92)90262-A. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen JL, Kehlet H. Hyperalgesia in a human model of acute inflammatory pain: A methodological study. Pain. 1998;74:139–151. doi: 10.1016/s0304-3959(97)00160-7. [DOI] [PubMed] [Google Scholar]
- 14.Kilo S, Schmelz M, Koltzenburg M, Handwerker HO. Different patterns of hyperalgesia induced by experimental inflammation in human skin. Brain. 1994;117(Pt 2):385–396. doi: 10.1093/brain/117.2.385. [DOI] [PubMed] [Google Scholar]
- 15.Sikand P, Shimada SG, Green BG, LaMotte RH. Similar itch and nociceptive sensations evoked by punctate cutaneous application of capsaicin, histamine and cowhage. Pain. 2009;144:66–75. doi: 10.1016/j.pain.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop T, Hewson DW, Yip PK, Fahey MS, Dawbarn D, Young AR, McMahon SB. Characterisation of ultraviolet-B-induced inflammation as a model of hyperalgesia in the rat. Pain. 2007;131:70–82. doi: 10.1016/j.pain.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Bishop T, Ballard A, Holmes H, Young AR, McMahon SB. Ultraviolet-B induced inflammation of human skin: Characterisation and comparison with traditional models of hyperalgesia. Eur. J. Pain. 2009;13:524–532. doi: 10.1016/j.ejpain.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Harrison GI, Young AR, McMahon SB. Ultraviolet radiation-induced inflammation as a model for cutaneous hyperalgesia. J. Invest. Dermatol. 2004;122:183–189. doi: 10.1046/j.0022-202X.2003.22119.x. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann RT, Schmelz M. Time course of UVA- and UVB-induced inflammation and hyperalgesia in human skin. Eur. J. Pain. 1999;3:131–139. doi: 10.1053/eujp.1998.0106. [DOI] [PubMed] [Google Scholar]
- 20.Bishop T, Marchand F, Young AR, Lewin GR, McMahon SB. Ultraviolet-B-induced mechanical hyperalgesia: A role for peripheral sensitisation. Pain. 2010;150:141–152. doi: 10.1016/j.pain.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Angst MS, Clark JD, Carvalho B, Tingle M, Schmelz M, Yeomans DC. Cytokine profile in human skin in response to experimental inflammation, noxious stimulation, and administration of a COX-inhibitor: A microdialysis study. Pain. 2008;139:15–27. doi: 10.1016/j.pain.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 22.Saadé NE, Nasr IW, Massaad CA, Safieh-Garabedian B, Jabbur SJ, Kanaan SA. Modulation of ultraviolet-induced hyperalgesia and cytokine upregulation by interleukins 10 and 13. Br. J. Pharmacol. 2000;131:1317–1324. doi: 10.1038/sj.bjp.0703699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endoh I, Di Girolano N, Hampartzoumian T, Cameron B, Geczy CL, Tedla N. Ultraviolet B irradiation selectively increases the production of interleukin-8 in human cord blood-derived mast cells. Clin. Exp. Immunol. 2007;148:161–167. doi: 10.1111/j.1365-2249.2007.03332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuentes JM, Talamini MA, Fulton WB, Hanly EJ, Aurora AR, De Maio A. General anesthesia delays the inflammatory response and increases survival for mice with endotoxic shock. Clin. Vaccine Immunol. 2006;13:281–288. doi: 10.1128/CVI.13.2.281-288.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochreiter WW, Nadler RB, Koch AE, Campbell PL, Ludwig M, Weidner W, Schaeffer AJ. Evaluation of the cytokines interleukin 8 and epithelial neutrophil activating peptide 78 as indicators of inflammation in prostatic secretions. Urology. 2000;56:1025–1029. doi: 10.1016/s0090-4295(00)00844-x. [DOI] [PubMed] [Google Scholar]
- 26.Sycha T, Gustorff B, Lehr S, Tanew A, Eichler HG, Schmetterer L. A simple pain model for the evaluation of analgesic effects of NSAIDs in healthy subjects. Br. J. Clin. Pharmacol. 2003;56:165–172. doi: 10.1046/j.0306-5251.2003.01869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 28.Calvo M, Zhu N, Tsantoulas C, Ma Z, Grist J, Loeb JA, Bennett DL. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J. Neurosci. 2010;30:5437–5450. doi: 10.1523/JNEUROSCI.5169-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78:2791–2808. [PubMed] [Google Scholar]
- 30.Vieira SM, Lemos HP, Grespan R, Napimoga MH, Dal-Secco D, Freitas A, Cunha TM, Verri WA, Jr., Souza-Junior DA, Jamur MC, Fernandes KS, Oliver C, Silva JS, Teixeira MM, Cunha FQ. A crucial role for TNF-α in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br. J. Pharmacol. 2009;158:779–789. doi: 10.1111/j.1476-5381.2009.00367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Coupade C, Gear RW, Dazin PF, Sroussi HY, Green PG, Levine JD. β2-Adrenergic receptor regulation of human neutrophil function is sexually dimorphic. Br. J. Pharmacol. 2004;143:1033–1041. doi: 10.1038/sj.bjp.0705972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werry TD, Wilkinson GF, Willars GB. Mechanisms of cross-talk between G-protein-coupled receptors resulting in enhanced release of intracellular Ca2+ Biochem. J. 2003;374:281–296. doi: 10.1042/BJ20030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, López-Otín C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walz A, Burgener R, Car B, Baggiolini M, Kunkel SL, Strieter RM. Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J. Exp. Med. 1991;174:1355–1362. doi: 10.1084/jem.174.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinet Y, Martinet N, Vignaud JM, Plenat F. Blood monocyte chemotaxis. J. Immunol. Methods. 1994;174:209–214. doi: 10.1016/0022-1759(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 36.Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 37.Tanner KD, Reichling DB, Levine JD. Nociceptor hyper-responsiveness during vincristine-induced painful peripheral neuropathy in the rat. J. Neurosci. 1998;18:6480–6491. doi: 10.1523/JNEUROSCI.18-16-06480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawker G, Wright J, Coyte P, Paul J, Dittus R, Croxford R, Katz B, Bombardier C, Heck D, Freund D. Health-related quality of life after knee replacement. J. Bone Joint Surg. Am. 1998;80:163–173. doi: 10.2106/00004623-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, Miller RJ. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J. Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunha FQ, Lorenzetti BB, Poole S, Ferreira SH. Interleukin-8 as a mediator of sympathetic pain. Br. J. Pharmacol. 1991;104:765–767. doi: 10.1111/j.1476-5381.1991.tb12502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin X, Wan Y, Wang X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J. Neurosci. Res. 2005;82:51–62. doi: 10.1002/jnr.20612. [DOI] [PubMed] [Google Scholar]
- 46.Grespan R, Fukada SY, Lemos HP, Vieira SM, Napimoga MH, Teixeira MM, Fraser AR, Liew FY, McInnes IB, Cunha FQ. CXCR2-specific chemokines mediate leukotriene B4–dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis Rheum. 2008;58:2030–2040. doi: 10.1002/art.23597. [DOI] [PubMed] [Google Scholar]
- 47.Lüttichau HR. The cytomegalovirus UL146 gene product vCXCL1 targets both CXCR1 and CXCR2 as an agonist. J. Biol. Chem. 2010;285:9137–9146. doi: 10.1074/jbc.M109.002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith E, McGettrick HM, Stone MA, Shaw JS, Middleton J, Nash GB, Buckley CD, Ed Rainger G. Duffy antigen receptor for chemokines and CXCL5 are essential for the recruitment of neutrophils in a multicellular model of rheumatoid arthritis synovium. Arthritis Rheum. 2008;58:1968–1973. doi: 10.1002/art.23545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005;174:3695–3702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 50.Walz A, Schmutz P, Mueller C, Schnyder-Candrian S. Regulationand function of the CXC chemokine ENA-78 in monocytes and its role in disease. J. Leukoc. Biol. 1997;62:604–611. doi: 10.1002/jlb.62.5.604. [DOI] [PubMed] [Google Scholar]
- 51.Matsumura Y, Ananthaswamy HN. Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol. 2004;195:298–308. doi: 10.1016/j.taap.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Paz ML, Ferrari A, Weill FS, Leoni J, Maglio DH. Time-course evaluation and treatment of skin inflammatory immune response after ultraviolet B irradiation. Cytokine. 2008;44:70–77. doi: 10.1016/j.cyto.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Toichi E, Lu KQ, Swick AR, McCormick TS, Cooper KD. Skin-infiltrating monocytes/ macrophages migrate to draining lymph nodes and produce IL-10 after contact sensitizer exposure to UV-irradiated skin. J. Invest. Dermatol. 2008;128:2705–2715. doi: 10.1038/jid.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barclay J, Clark AK, Ganju P, Gentry C, Patel S, Wotherspoon G, Buxton F, Song C, Ullah J, Winter J, Fox A, Bevan S, Malcangio M. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225–234. doi: 10.1016/j.pain.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 55.Cunha TM, Barsante MM, Guerrero AT, Verri WA, Jr., Ferreira SH, Coelho FM, Bertini R, Di Giacinto C, Allegretti M, Cunha FQ, Teixeira MM. Treatment with DF 2162, a non-competitive allosteric inhibitor of CXCR1/2, diminishes neutrophil influx and inflammatory hypernociception in mice. Br. J. Pharmacol. 2008;154:460–470. doi: 10.1038/bjp.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cunha TM, Verri WA, Jr., Schivo IR, Napimoga MH, Parada CA, Poole S, Teixeira MM, Ferreira SH, Cunha FQ. Crucial role of neutrophils in the development of mechanical inflammatory hypernociception. J. Leukoc. Biol. 2008;83:824–832. doi: 10.1189/jlb.0907654. [DOI] [PubMed] [Google Scholar]
- 57.Levine JD, Gooding J, Donatoni P, Borden L, Goetzl EJ. The role of the polymorphonuclear leukocyte in hyperalgesia. J. Neurosci. 1985;5:3025–3029. doi: 10.1523/JNEUROSCI.05-11-03025.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ting E, Guerrero AT, Cunha TM, Verri WA, Jr., Taylor SM, Woodruff TM, Cunha FQ, Ferreira SH. Role of complement C5a in mechanical inflammatory hypernociception: Potential use of C5a receptor antagonists to control inflammatory pain. Br. J. Pharmacol. 2008;153:1043–1053. doi: 10.1038/sj.bjp.0707640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, Serhan CN, Ji RR. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010;16:592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Myers RR, Heckman HM, Rodriguez M. Reduced hyperalgesia in nerve-injured WLD mice: Relationship to nerve fiber phagocytosis, axonal degeneration, and regeneration in normal mice. Exp. Neurol. 1996;141:94–101. doi: 10.1006/exnr.1996.0142. [DOI] [PubMed] [Google Scholar]
- 61.Lorenzetti BB, Veiga FH, Canetti CA, Poole S, Cunha FQ, Ferreira SH. Cytokine-induced neutrophil chemoattractant 1 (CINC-1) mediates the sympathetic component of inflammatory mechanical hypersensitivitiy in rats. Eur. Cytokine Netw. 2002;13:456–461. [PubMed] [Google Scholar]
- 62.Manjavachi MN, Quintão NL, Campos MM, Deschamps IK, Yunes RA, Nunes RJ, Leal PC, Calixto JB. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. Eur. J. Pain. 2010;14:23–31. doi: 10.1016/j.ejpain.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 63.Wang JG, Strong JA, Xie W, Yang RH, Coyle DE, Wick DM, Dorsey ED, Zhang JM. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol. Pain. 2008;4:38. doi: 10.1186/1744-8069-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chandrasekar B, Melby PC, Sarau HM, Raveendran M, Perla RP, Marelli-Berg FM, Dulin NO, Singh IS. Chemokine-cytokine cross-talk. The ELR+ CXC chemokine LIX (CXCL5) amplifies a proinflammatory cytokine response via a phosphatidylinositol 3-kinase-NF-κB pathway. J. Biol. Chem. 2003;278:4675–4686. doi: 10.1074/jbc.M207006200. [DOI] [PubMed] [Google Scholar]
- 65.Rittner HL, Mousa SA, Labuz D, Beschmann K, Schäfer M, Stein C, Brack A. Selective local PMN recruitment by CXCL1 or CXCL2/3 injection does not cause inflammatory pain. J. Leukoc. Biol. 2006;79:1022–1032. doi: 10.1189/jlb.0805452. [DOI] [PubMed] [Google Scholar]
- 66.Astner S, Anderson RR. Skin phototypes 2003. J. Invest. Dermatol. 2004;122:xxx–xxxi. doi: 10.1046/j.1523-1747.2003.22251.x. [DOI] [PubMed] [Google Scholar]
- 67.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 68.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 69.Fruhstorfer H, Lindblom U, Schmidt WC. Method for quantitative estimation of thermal thresholds in patients. J. Neurol. Neurosurg. Psychiatry. 1976;39:1071–1075. doi: 10.1136/jnnp.39.11.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. http://www.appliedbiosystems.com.
- 71.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 72. http://www.ncbi.nlm.nih.gov/tools/primer-blast/
- 73.Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 2009;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong LF, Yip PK, Battaglia A, Grist J, Corcoran J, Maden M, Azzouz M, Kingsman SM, Kingsman AJ, Mazarakis ND, McMahon SB. Retinoic acid receptor β2 promotes functional regeneration of sensory axons in the spinal cord. Nat. Neurosci. 2006;9:243–250. doi: 10.1038/nn1622. [DOI] [PubMed] [Google Scholar]
- 75.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.