

Abstract
Delivering apoptosis inducing peptides to cells is an emerging area in cancer and molecular therapeutics. Here, we have identified an alternative mechanism of action for the proapoptotic chimeric peptide D-NuBCP-9-r8. Integral to D-NuBCP-9-r8 is the Nur-77-derived D-isoform sequence fsrslhsll that targets Bcl-2, and the cell-penetrating peptide (CPP) octaarginine (r8) that is required for intracellular delivery. We find that the N-terminal phenylalanine of fsrslhsll acts in synergy with the cell-penetrating moiety to enhance peptide uptake at low nontoxic levels and cause rapid membrane blebbing and cell necrosis at higher (IC50) concentrations. These effects were not observed when a single phenylalanine–alanine mutation was introduced at the N-terminus of D-NuBCP-9-r8. Using primary samples from chronic lymphocytic leukemia (CLL) patients and cancer cell lines, we show that NuBCP-9-r8 induced toxicity, via membrane disruption, is independent of Bcl-2 expression. Overall, this study demonstrates a new mechanism of action for this peptide and cautions its use as a highly specific entity for targeting Bcl-2. For delivery of therapeutic peptides the work emphasizes that key amino acids in cargo, located several residues away from the cell-penetrating sequence, can significantly influence their cellular uptake and mode of action.
Introduction
Important regulators of apoptotic mechanisms are the Bcl-2 family of proteins including antiapoptotic variants Bcl-2 and Bcl-XL and proapoptotic Bax and Bak.1 The correct balance of activities, interaction, and expression of these proteins is critical for cell survival and central to the induction of apoptosis. Cancer cells are noted for having dysregulated apoptosis and this is often due to increased expression and thus activity of Bcl-2.1–5 The increase in Bcl-2 expression confers a survival advantage especially when the cells are challenged with chemotherapy during anticancer treatments. Thus targeting Bcl-2 is an active area of research in cancer, including lymphomas and leukemias, and a number of anti-Bcl-2 drugs are now in clinical trials.1,5,6
Most of these drugs are small-molecule entities that freely diffuse through the plasma membrane to interact with Bcl-2.1 Targeted peptides are also promising therapeutic entities but a major hurdle to their efficacy lies in the fact that they are generally membrane impermeable and additional components or vectors are required to allow them access to the cell interior.7 These include cell-penetrating peptides (CPP) or protein transduction domains that are typically <30 residues in length. Hundreds of different CPP sequences have now been described but all have a universal capacity to breach biological membranes and enter cells, either alone or associated with cargo.8,9 The best characterized are those with sequences enriched in cationic residues lysine and arginine; notable examples include the HIV-TAT peptide and synthetic oligoarginines R6-20. The mechanism by which CPPs enter cells is still largely unresolved despite intense studies over the past two decades.10,11 There is strong evidence for both uptake through endocytic pathways and directly across the plasma membrane; this is especially the case where the cargo is small, for example a fluorophore or short peptide.
Using CPPs to deliver apoptotic peptides is an active area of research and recently a short peptide sequence, FSRSLHSLL, as the D isomer fsrslhsll was found to be cytotoxic when delivered to cells by the CPP octaarginine, whilst a mutated control peptide with an alanine inserted in place of the N-terminal phenylalanine and the C-terminal leucine asrslhsla-r8 chimera was nontoxic.12 The targeting sequence is derived from Nur77, a member of the orphan receptor superfamily that interacts with Bcl-2 to tether it and its affector and effector proteins as antiapoptotic complexes. The CPP-linked Nur77-derived peptide, now commercially available as the Bcl-2 converter peptide D-NuBCP-9-r8, was shown to interact with Bcl-2 to expose a BH3 domain that ultimately activates and releases proapoptotic factors such as Bax.
Here, we initially investigated the capacity of this peptide to affect the viability of leukemia cell lines and primary tissue from patients with chronic lymphocytic leukemia (CLL). Using a combination of live cell confocal microscopy and viability assays we find that the presence of the N-terminal phenylalanine of the Nur77-derived sequence significantly enhances the ability of the peptide to interact with the plasma membrane, to gain access to cells and also mediate rapid cell death through a mechanism that is independent of Bcl-2 expression. These studies were confirmed in adherent cells highlighting the strong membrane interacting synergy between phenylalanine and octaarginine even when they are located 10 residues away from each other. These studies have important implications for the use of CPP as vectors for therapeutic peptides but also for the applicability of D-NuBCP-9-r8 as a Bcl-2 targeting entity.
Results
Viability of KG1a cells following incubation with r8-, asr-a-r8, and fsr-l-r8
Incubation of FSRSLHSLL- or fsrslhsll, attached to the CPP octaarginine-induced cytotoxicity in Bcl-2-transfected Jurkat cells, over parental cells, but a control L and D sequence ASRSLHSLA attached to the same CPP was relatively nontoxic.12 We initially investigated the viability of acute myeloid leukemia KG1a cells following 24-hour exposure to increasing concentrations of r8 alone or of r8 conjugated to fsrslhsll (fsr-l-r8) or asrslhsla (asr-a-r8, Table 1). These sequences, including the presence of the aminohexanoic acid bridge were as previously described12 with the exception that here the N-terminus was not acetylated and the C-terminal was further extended with GC to allow subsequent attachment of a fluorescent probe. The results (Figure 1a) show that r8 or asr-a-r8-treated cells are viable up to 20 µmol/l but fsr-l-r8 was toxic to KG1a cells (IC50 12 µmol/l); this is comparable to values observed in the previous study.12
Table 1. Name and sequence of peptides used in this study.
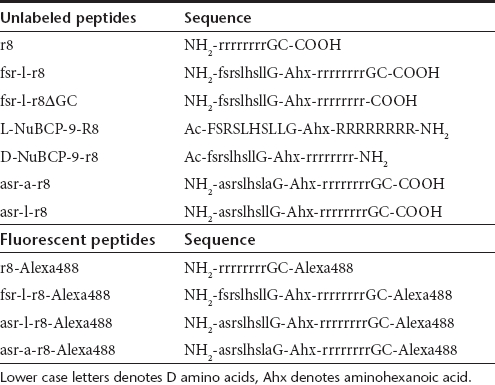
Figure 1.

Cell uptake and cytotoxicity of Bcl-2 targeted peptides in KG1a leukemia cells. (a) Viability of cells following 24-hour incubation with 0–20 µmol/l unlabeled r8, asr-a-r8, and fsr-l-r8, results represent means ± SD for three separate experiments performed in triplicate. (b) Viability of cells incubated with peptides for 1 hour in either complete medium (CM) or serum-free medium (SF) and then for 23 hours in the absence of peptides. Results represent means ± SD for two separate experiments performed in triplicate. (c) Morphological analysis of cells incubated in the absence (control) or presence of 20 µmol/l r8-, asr-a-r8, or fsr-l-r8 for 24 hours. (d) Cells were incubated at 37 °C with 2 or 10 µmol/l r8-, asr-a-r8-, or fsr-l-r8-Alexa488 for 1 hour before washing and analysis by confocal microscopy. Results shown represent maximum projection images of Alexa488 fluorescence, arrows show peptide enrichment in the nucleolus. (e) Media-containing 2 µmol/l r8-, asr-a-r8-, or fsr-l-r8-Alexa488 was added to cells before analyzing peptide uptake by time-lapse microscopy at 37 °C for 10 minutes. Bar = 10 µm. Available as Supplementary Videos S1–S3.
Incubation of cells for only 10 minutes with either of two proliferating cell nuclear antigen-binding domain peptides fused to the CPP HIV-TAT was sufficient to inhibit cell cycle progression of mouse myoblasts by 30% for up to 8 hours after peptide incubation.13 To investigate whether the toxicity of the fsr-l-r8 peptide was dependent on incubation time, peptide incubations were performed at 37 °C for 1 hour before removal of the extracellular peptide and further incubating the KG1a cells in peptide-free complete medium for a further 23 hours. The viability of the cells was compared with those incubated in the continued presence of the peptide for 24 hours. Incubation with the peptide for just 1 hour was sufficient to give similar IC50 values to those observed when the peptide was left on the cells for 24 hours (Figure 1b).
We also performed the initial 1-hour peptide incubation in medium lacking serum; CPPs such as R8 have been shown to bind strongly to serum proteins thus reducing their effective extracellular peptide concentration for cellular interaction and penetration.14 Cells were incubated with fsr-l-r8 for 1 hour in serum-free medium, washed of peptide and further incubated for 23 hours in complete medium. The IC50 for fsr-l-r8 decreased from 12 to 2 µmol/l in these experiments thus highlighting the effect of protein binding to peptide activity (Figure 1b). Comparative viability assays were also performed between peptide fsr-l-r8 and an identical sequence, lacking the terminal GC that we use for linking the fluorophore (fsr-l-r8ΔGC, Table 1) and the same cytotoxicity profiles were observed demonstrating no effect of addition of these two residues (Supplementary Figure S1). The cellular morphology of the cells after this 24-hour incubation was then assessed and cells incubated with fsr-l-r8 showed extensive morphological changes or were completely destroyed (Figure 1c).
Uptake and subcellular distribution of r8-, asr-a-r8, and fsr-l-r8-Alexa488 in KG1a cells
We investigated the subcellular distribution of Alexa488-labeled r8, asr-a-r8 and fsr-l-r8 following incubations with these cells. These were initially determined following 1-hour incubation at 2 µmol/l extracellular peptide concentration with KG1a cells at 37 °C. Both r8- and asr-a-r8-Alexa488 were endocytosed to label intracellular vesicles (Figure 1d). Unexpectedly, the subcellular distribution of fsr-l-r8 was very different showing diffuse cytoplasmic labeling and in most cases the nucleolus was also labeled. Cells incubated with 10 µmol/l Alexa488-labeled r8, asr-a-r8 or fsr-l-r8 displayed diffuse but strong cytoplasmic labelling.
We previously monitored the immediate cellular uptake of R8-Alexa488 in leukemia cells using time-lapse microscopy;15 these experiments were performed in the continued presence of the peptide thus excluding the requirement for the washing steps. The uptake of these peptides was similarly continuously monitored from the time of peptide addition for a 10-minute period. In cells incubated with either 2 µmol/l r8-Alexa488 or asr-a-r8-Alexa488 (Figure 1e, Supplementary Videos S1 and S2) the intracellular fluorescence was low throughout the incubation period and the peptides were predominantly located on the plasma membrane. Peptide fsr-l-r8-Alexa488 very rapidly localized to the periphery of cells and after 5 minutes most of the cells were diffusely labeled with peptide and the intensity increased further to the end of the incubation period (Figure 1e and Supplementary Video S3). When identical 2 µmol/l peptide experiments were performed at 4 °C, thus inhibiting endocytosis, all three peptides labeled the cytoplasm and nucleolus and this was more obvious when the peptide concentration was increased to 10 µmol/l (Supplementary Figure S2a). Quantification of uptake at 4 °C showed comparable fluorescence for all peptides (Supplementary Figure S2b).
Membrane activity of fsr-l-r8 is dependent on the N-terminal phenylalanine of the Nur77-derived sequence
Despite there being only two residue differences between the Bcl-2 targeting peptide and the control peptide they show major differences in their capacities to gain access to cells. To further investigate the link between peptide sequence and affect, asr-l-r8 was synthesized and evaluated; this differs from fsr-l-r8 by a single amino acid at the N-terminus which has been changed from f in fsr-l-r8 to a in asr-l-r8 (Table 1). This peptide did not reduce cell viability and had a cellular uptake profile that was the same as the previous control variant asr-a-r8 (Figure 2a,b). This demonstrated the significant influence of the N-terminal phenylalanine on the cellular dynamics of fsr-l-r8. We quantified the uptake of these peptides at 37 °C after 1-hour incubation at 2 µmol/l extracellular peptide concentration. Peptides asr-l-r8 and asr-a-r8-Alexa488 gave similar fluorescence values (Figure 2c) but fluorescence values of cells incubated with fsr-l-r8 were much higher thus confirming the earlier microscopy analysis.
Figure 2.

Contribution of the N-terminal phenylalanine residue to cellular uptake and rapid cytotoxicity of fsr-l-r8. (a) Viability of KG1a cells following incubations with 0–20 µmol/l unlabeled asr-a-r8, asr-l-r8, or fsr-l-r8 for 24 hours. Results represent means ± SD for three separate experiments performed in triplicate. (b) Cells were incubated at 37 °C with 2 or 10 µmol/l asr-a-r8-, asr-l-r8-, or fsr-l-r8-Alexa488 for 1 hour before washing and analysis by confocal microscopy. Results shown represent maximum projection images of Alexa488 fluorescence. (c) Cells were incubated at 37 °C with 2 µmol/l asr-a-r8-, asr-l-r8-, or fsr-l-r8-Alexa488 for 1 hour before processing for flow cytometry and quantification of peptide uptake. Results represent the geometric means ± SD from three separate experiments performed in duplicate, **P < 0.01. (d) Unlabeled asr-a-r8 or fsr-l-r8 and propidium iodide (PI) were added to KG1a cells (nested in microwells) that were then immediately imaged by confocal microscopy at 37 °C for 1 hour. Results represent single-section images of morphology and PI fluorescence. Bar = 10 µm. Full-time lapse series available as Supplementary Videos S4 and S5.
Manufacture of microwells for time lapse imaging of leukemia cells with fsr-l-r8
The use of time-lapse microscopy allowed us to analyze the immediate effects of apoptotic peptide-CPP chimeras on morphology and membrane integrity of adherent cells.16 Peptides in imaging medium-containing serum, and propidium iodide (PI) as a marker of plasma membrane integrity were placed on cells that were then sequentially imaged to allow real-time analysis of cell morphology and, via PI leakage, plasma membrane integrity. This is normally difficult with nonadherent cells settled on similar plane surfaces as they move from the field of view or focus following addition of solutions. Using laser ablation, we therefore designed and manufactured custom microwells to retain the cells in one position. A scanning electron micrograph of a manufactured microwell array is shown (Supplementary Figure S3). This technique allows for the design of wells of multiple shapes and volumes and for KG1a cells, having a mean diameter of 10.1 µm, wells of 15 µm diameter were manufactured. This allowed us to settle the cells into wells before addition of the peptides. For experiments shown in (Figure 2d) cells were incubated with 10 µmol/l asr-a-r8 or fsr-l-r8 and cellular morphology and PI fluorescence was monitored for 1 hour. Those incubated with asr-a-r8 retained their normal morphology and were PI negative after 1 hour (Figure 2d and Supplementary Video S4) whereas cells incubated with fsr-l-r8 showed initial (<10 minutes) evidence of microblebbing and then extensive blebbing terminating in swelling and PI intake (Supplementary Video S5). There was a degree of variation regarding the time at which these effects were observed with some cells leaking PI within 20 minutes and others taking longer to reveal themselves as PI positive. Identical results were obtained with fsr-l-r8ΔGC (data not shown). This confirms that this peptide causes rapid membrane blebbing and eventual loss of plasma membrane integrity.
Uptake, subcellular distribution, and cytotoxicity of fsr-l-r8 in primary cells
We then investigated the degree of cytotoxicity of this peptide relative to the control (asr-a-r8) peptide in six fresh primary samples from patients with CLL. Human peripheral blood mononuclear cells were isolated from blood and then incubated for 24 hours with 0–20 µmol/l peptide. There was no loss of viability in cells treated with the control peptide and a similar dose response profile was observed for all patient samples treated with fsr-l-r8, giving a IC50 range of 6–10 µmol/l (Figure 3a). Immunolabeling following sodium dodecyl sulfate-polyacrylamide gel electrophoresis confirmed Bcl-2 expression in all six samples and the ratio of Bcl-2 intensity against internal α-tubulin control revealed a range for between 0.6 and 2.3 (Figure 3b). There was no evidence of a correlation between Bcl-2 expression and fsr-l-r8-mediated cytotoxicity. As previously described for KG1a cells, peptide uptake was then investigated by monitoring CLL cells for 10 minutes immediately (from 30 seconds) after peptide addition. In cells incubated with 2 µmol/l asr-a-r8-Alexa488 no significant cellular fluorescence was observed within this timeframe (Figure 3c and Supplementary Video S6). In cells incubated with 2 µmol/l fsr-l-r8-Alexa488, cellular fluorescence was observed after only 2.5 minutes (Figure 3c and Supplementary Video S7) and the number of labeled cells increased throughout the 10-minute period, however, the proportion of cells displaying diffuse cytosolic labeling was much lower compared with KG1a cells under the same experimental setup.
Figure 3.

Viability and uptake of Bcl-2 targeting peptides in chronic lymphocytic leukemia (CLL) cells. (a) CLL cells from six individual patients were incubated with 0–20 µmol/l peptides for 24 hours, results are means ± SD for one experiment performed in triplicate. (b) Bcl-2 expression in samples analyzed in (a). Results are expressed as Bcl-2/α-tubulin ratios for each set of bands and samples were loaded in duplicate. (c) Complete media-containing 2 µmol/l asr-a-r8- or fsr-l-r8-Alexa488 was added to cells before analysis of peptide uptake by time-lapse microscopy at 37 °C for 10 minutes. Results represent single-section images of Alexa488 fluorescence. Bar = 10 µm. Full-time lapse series available as Supplementary Videos S6 and S7.
Effects of D-NuBCP-9-r8 is independent of Bcl-2 expression
Initial characterization of D-NuBCP-9-r8 was performed in Bcl-2- Jurkat cells that were found to be less sensitive to the peptide compared with cells transfected with Bcl-2.12 As we had shown that fsr-l-r8 causes PI leakage within 1 hour in KG1a cells we incubated Jurkat cells with 10 µmol/l of unlabeled asr-l-r8 or D-NuBCP-9-r8 which is now commercially available as the Bcl-2 converter peptide. Cells incubated with asr-l-r8 retained normal morphology throughout the experimental period and were PI negative (Figure 4a and Supplementary Video S8). For D-NuBCP-9-r8 there was differential sensitivity within the cell population with some cells seeming to be unaffected while others underwent rapid membrane disruption (Figure 4a and Supplementary Video S9). This resulted in the expulsion of cellular contents that became PI positive as they were ejected from the cell; often these cells were PI negative just before this event took place. Immunolabeling of cell lysates confirmed that the Jurkat cells used here were Bcl-2- in contrast to KG1a+ cells (Figure 4b).
Figure 4.

Membrane activity of Bcl-2 targeted peptides in Jurkat cells. (a) Time-lapse images of Jurkat cells incubated with propidium iodide (PI) and 10 µmol/l asr-l-r8 or D-NuBCP-9-r8 for 1 hour at 37 °C. Insert represent magnified image of five cells showing rapid cellular destruction. Results represent single-section images of morphology and PI fluorescence. Full-time lapse series available as Supplementary Videos S8 and S9. (b) Bcl-2 expression in KG1a and Jurkat cells. (c) Jurkat or (d) KG1a cells were incubated for 1 hour in the presence of unlabeled peptides and propidium iodide (PI) for 1 hour at 37 °C. Results show flow cytometry histograms (PI uptake and percentage PI positive) in untreated (control) and peptide-treated cells.
The effects of the peptides were quantified by flow cytometric analysis of PI uptake in Jurkat cells incubated with the peptides at 37 °C for 1 hour. D-NuBCP-9-r8 and fsr-l-r8 increased the number of dead cells to 20–30% of the cell culture within 1 hour while the viability of the cells treated with asr-l-r8 peptide was the same as untreated cells (Figure 4c). When the same experiments were performed in KG1a cells, the PI positive fraction was much larger, representing up to 75% of the analyzed cell population (Figure 4d).
Effects of fsr-l-r8 on adherent cell lines
To further investigate the cellular effects of these peptides we performed experiments in adherent epithelial HeLa cells that we have previously used to investigate CPP uptake.15,16 Comparative cellular uptake of Alexa488-conjugated peptides asr-a-r8, asr-l-r8, and fsr-l-r8 at 2 and 10 µmol/l extracellular concentration confirmed the results obtained in nonadherent cells as fsr-l-r8-Alexa488-treated cells had much stronger labeling and even at 2 µmol/l some peptide was located in the cytosol in addition to punctate structures (Figure 5a). Thus for this peptide the effects of the terminal phenylalanine on cell uptake were not limited to nonadherent cells.
Figure 5.

Cellular uptake and membrane activity of Bcl-2-targeted peptides in HeLa cells. (a) Cells were incubated at 37 °C for 1 hour with 2 or 10 µmol/l Alexa488-labeled asr-a-r8, asr-l-r8 or fsr-l-r8 before analysis by confocal microscopy. Results shown represent maximum projection images of Alexa488 fluorescence, Bar = 10 µm. (b) Time-lapse images of HeLa cells incubated with propidium iodide (PI) and 20 µmol/l asr-l-r8, L-NuBCP-9-R8, or D-NuBCP-9-r8 for 1 hour at 37 °C. Insert represent magnified image highlighting extensive blebbing before PI intake. Results represent single-section images of morphology and PI fluorescence. Bar = 50 µm. Full-time lapse series available as Supplementary Videos S10–S12.
We then performed similar time-lapse microscopy in HeLa cells incubated with 20 µmol/l of either asr-l-r8, D-NuBCP-9-r8, or the -isomer of this peptide (-NuBCP-9-R8) in serum containing medium-containing PI. Cells incubated with asr-l-r8 (Figure 5b and Supplementary Video S10) and r8 (data not shown) had normal morphologies and were PI negative after 1 hour. Cells incubated with D-NuBCP-9-r8, showed signs of blebbing after ~30 minutes and for the remainder of the incubation period some extensive membrane reorganization and swelling on distinct parts of the plasma membrane of cells was observed before leakage of PI (Figure 5b and Supplementary Video S11). Within the same population of cells, there was again a large variation in the response to the effects of the peptides with some remaining PI negative throughout the experiment. The effects of L-NuBCP-9-R8 on the cells (Supplementary Video S12) were less rapid than those observed with D-NuBCP-9-r8 and also fsr-l-r8 (data not shown), but extensive membrane reorganization was observed.
Discussion
CPP have in the last two decades received widespread attention as potential delivery vectors for macromolecular therapeutics.17 This includes a number of targeted peptides that by a variety of mechanisms are designed to influence apoptosis.18 We and others have highlighted that the plasma membrane of cells have distinct thresholds for excluding CPPs such as R8 and HIV-TAT from direct entry rather than through endocytosis.15,19,20 We initially extended on these studies here by analyzing the cellular dynamics and effects of the Nur77-derived sequence attached to r8. The specificity of the Nur77 targeting sequence was initially shown by simultaneously replacing respectively the N- and C-terminal phenylalanine and leucine residues for alanine.12
Confocal microscopy experiments highlighted large and unexpected differences in the extent of uptake and subcellular distribution of asr-a-r8- and fsr-l-r8-Alexa488 that was later pinpointed, via synthesis of another peptide differing in only one amino acid, to be a result of the NH2 phenylalanine residue. Addition of FF at the C-terminus of R9 (R9FFC) showed an enhanced capacity over other CPPs to deliver antisense morpholino oligomers to cells,21 but later studies suggested that the peptide also had some membrane disrupting effects.22 A recent study in HeLa and glioma cells found that addition of a penetration accelerating sequence FFLIPKG upstream of R8 increased uptake significantly compared to R8 alone.23 FFLIPKG-R8 was observed to diffusely label the cytoplasm within five minutes of addition to cells whereas R8 only labeled vesicles. This is similar to our comparative observations in adherent and nonadherent cell types incubated with relatively low concentrations of fsr-l-r8 and asr-a-r8. The presence of two phenylalanines at the NH2 terminus of histidine-containing amphipathic cationic peptides also significantly enhanced toxicity of these entities against mammalian cells and bacteria.24 A common feature of antimicrobial peptides and also CPPs such as penetratin is that they are either amphipathic throughout the sequence and helical, or that they contain a cationic sequence and a hydrophobic cluster that may be only two residues in length.9 The cationic residues are thought to locate the peptide, via electrostatic interactions, to the plasma membrane and hydrophobic residues allow for immersion into the hydrocarbon portion of the membrane.25 It is likely that the NH2 terminal phenylalanine, unlike alanine in the control peptide, was sufficient to promote the former and change the dynamics of the peptide with cells. By performing similar cell uptake experiments on ice we show that this effect is temperature-dependent, suggesting a requirement for a more fluidic membrane.
At higher concentrations (10 µmol/l) the plasma membrane was permeable to all peptides studied here but in the case of fsr-l-r8 there was also additional dramatic morphological damage. By continuous visualization of cells from the point of addition of the peptide, we observed extensive cell blebbing in HeLa cells and in KG1a cells. In the case of nonadherent cells, these observations were made possible by the generation of engineered grids to hold the cells in position. A proportion of these cells was also permeable to PI within 60 minutes suggesting strongly that here it is acting as a necrotic rather than an apoptotic factor. Currently we do not know why some cells in a single population are more sensitive than others.
Cells were equally sensitive to fsr-l-r8 when the peptide incubations were performed for 24 hours or 1 hour followed by washing and further incubation in peptide-free medium for 23 hours. Two cell cycle targeting peptides attached to the CPP Tat had bioactivity in mouse myoblasts for over 6 hours after only 10-minute incubation indicating that a biological response to CPP:peptide fusions is mediated within minutes and can last long after the peptides are washed away.13 The IC50 could also be reduced significantly by omitting serum from the incubation medium and our previous studies have shown extensive binding of R8 to serum proteins.14 In serum containing medium a large fraction of the peptide is sequestered by serum proteins thus the active concentration required for membrane penetration and/or perturbation is increased. Serum also contains proteases that can further reduce the potency of CPPs and cargo especially if they are of the -form. This may have caused the much delayed morphological effects in HeLa cells when they were incubated with L-NuBCP-9-R8 compared with D-NuBCP-9-r8. It agrees with the previous demonstration of diminished activity of D-NuBCP-9-r8 when fsrslhsll as cargo was substituted with FSRSLHSLL.12
Regarding a requirement for Bcl-2, three separate lines of evidence suggest that this protein does not play a role in the membrane blebbing caused by fsr-l-r8 and D-NuBCP-9-r8. Most importantly, we can clearly observe significant peptide-induced blebbing in Jurkat cells that are Bcl-2-. Secondly membrane damage and leakage in all cell lines occurs within 1 hour; a time scale that is faster than that typically observed for cellular apoptosis. Thirdly, in primary human cells from CLL patients, the cell death observed did not correlate with the level of expression of Bcl-2 found in these patients. Our data does not exclude that possibility that the presence of Bcl-2 could further increase the potency of peptide-induced cell death as KG1a cells, which are Bcl-2 positive, were more susceptible than Jurkat cells and the lack of correlation in primary cells could be due to a requirement for just low levels of Bcl-2 for this effect. However, our data cautions the use of D-NuBCP-9-r8 as a highly specific entity for targeting Bcl-2; the peptide is clearly membrane active thus giving it dual potency as a cytotoxic agent.
Overall the data questions the specificity of D-NuBCP-9-r8 and illustrates how a single amino acid residue in the cargo sequence can significantly alter the membrane activity of r8, highlighting, as previously described for another apoptotic peptide,16 that relatively small cargo can have large effects on the dynamics of CPPs with cells.
Materials and Methods
Alexa Fluor 488-C5-maleimide and all tissue culture reagents were from Invitrogen (Paisley, UK). Glass-bottomed culture dishes (35 mm) for microscopy were from MatTek (Ashland, MA). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide, PI and antibodies against Bcl-2 and α-tubulin were from Sigma (Gillingham, UK). Complete miniprotease inhibitor cocktail were from Roche (Mannheim, Germany). Horseradish peroxidase-conjugated anti-mouse antibody was from Pierce (Northumberland, UK). Menzel–Glazer cover slips (no. 0) were from Fisher Scientific (Loughborough, UK).
Peptide synthesis and conjugation. Sequences of the peptides used in this study are shown in Table 1; lower case letters denote D isoforms, Ahx denotes aminohexanoic acid. L-NuBCP-9-R8 was from Thermo Fisher Scientific (Ulm, Germany), D-NuBCP-9-r8 (Bcl-2 Converter) was from Merck KGaA (Darmstadt, Germany) and all other peptides were from Severn Biotech (Worcester, UK). Labeling of peptides with Alexa488 was performed through a C-terminal cysteine as previously described.15,20 All peptides were purified and characterized using reversed phase high-performance liquid chromatography on a C18 Luna 100 å 5-µm semipreparative column (Phenomenex, Macclesfield, UK) and peptide masses were confirmed using matrix-assisted laser-desorption ionization–time-of-flight spectrometry.
Cell lines and primary tissue. Human acute myeloid leukemia KG1a cells and acute T-cell lymphocytic Jurkat cells were cultured in a humidified 5% CO2 incubator at 37 °C and maintained at a confluency of 0.5–2 × 106 cells/ml in RPMI 1640 medium, supplemented with 10% (vol/vol) fetal bovine serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin. Human cervical carcinoma, HeLa cells were maintained as a subconfluent monolayer in D-MEM supplemented with 10% (vol/vol) fetal bovine serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin. Peripheral blood was collected from CLL patients with their informed consent in accordance with the Declaration of Helsinki and in keeping with the ethical approval obtained from South East Wales Research Ethics Committee (02/4806). Cells were isolated from freshly collected blood samples by density centrifugation and maintained for in RPMI 1640 medium, supplemented with 10% (vol/vol) fetal bovine serum, 100 IU/ml penicillin, and 100 µg/ml streptomycin.26
Cellular localization of Alexa488-labeled peptides in KG1a and HeLa cells. KG1a (5 × 105) cells were washed by centrifugation (800g for 2 minutes) in complete medium and equilibrated in this medium at 37 °C (or on ice) for 15 minutes. The medium was replaced with fresh temperature equilibrated (4 or 37 °C) medium-containing 2 or 10 µmol/l r8, asr-a-r8-, or fsr-l-r8-Alexa488 and incubated under tissue-culture conditions for 1 hour. Cells were then washed twice in serum-free RPMI 1640 medium (serum-free medium), once in imaging medium (serum-free medium without phenol red) and finally resuspended in 500 µl of imaging medium. A fraction of the cell suspension (100 µl) was transferred to the centre of glass-bottomed 35-mm culture dishes and the cells were allowed to settle for 30 seconds–1 minute. They were then analyzed on a Leica SP5 confocal laser-scanning microscope equipped with an Ar laser and a 63 × oil immersion objective. Cells were imaged through the z-axis to generate maximum projection profiles which were finally arranged using Adobe Photoshop. HeLa cells (1.8 × 105) were seeded into glass-bottomed, 35-mm culture dishes and allowed to adhere for 24 hours. The cells were then incubated in complete medium-containing 2 or 10 µmol/l Alexa488-labeled peptides for 1 hour at 37 °C before washing once in imaging medium and then analyzed by confocal microscopy as described above but using a 40 × oil immersion objective.
Time-lapse microscopy of KG1a and CLL cells incubated with Alexa488-labeled peptides. The experiments were performed as previously described.15 Briefly, KG1a (1 × 105) and CLL (2 × 105) cells were washed in complete medium, resuspended in imaging medium-containing 10% fetal bovine serum and transferred to 35-mm imaging dishes that were then placed on an imaging platform on the confocal microscope, equilibrated at 37 °C. Fluorescently labeled peptides were added to final concentrations of 2 or 10 µmol/l and the cells were allowed to settle for 30 seconds. Images were then acquired using the ×63 oil immersion objective every 30 seconds for 10 minutes using a laser exposure of 1.3 seconds to capture each frame. Images showing fluorescent profiles were selected at different time points and Supplementary Videos show Direct Interference Contrast bright field and fluorescence frames processed as side-by-side animations using NIH ImageJ software.
Time-lapse microscopy of unlabeled peptides in HeLa and Jurkat cells. HeLa cells (1.8 × 105) were seeded into glass-bottomed, 35-mm culture dishes and allowed to adhere for 24 hours. The cells were washed and replaced with 1.0 ml Dulbecco's modified Eagle's medium (no phenol red) containing 10% fetal bovine serum, 10 mmol/l Na-HEPES pH 7.4, 1.0 µg/ml PI, and peptides at 20 µmol/l. Jurkat cells (4 × 105 /ml) were washed 2× in phenol red-free RPMI medium-containing 10 mmol/l Na-HEPES pH 7.4, and then incubated in 1.0 ml of this medium-containing 1.0 µg/ml PI and peptides at 10 µmol/l. Cells were imaged every 30 seconds (laser exposure 1.5 seconds) for 1 hour at 37 °C by confocal microscopy capturing bright field and PI frames with a HeNe 543 laser.
Microwell manufacture and time-lapse microscopy in KG1a cells. The microwells were fabricated into glass cover slips using laser ablation. For this, Menzel–Glazer cover slips were first coated with a layer of liquid ink blackboard maker (Pentel easyflo) to provide a sacrificial layer before ablation.27 Microwells (15 µmol/l diameter, 18 µm pitch) in a 10 by 10 array were ablated individually into the cover slip using a 157 nm F2 eximer laser. Radiation of 157 nm was delivered from a coherent LPF220i laser source and coupled through an enclosed, N2-perfused, 3-m long beamline, to a 25× Schwarzschild projection lens. Beam shaping was achieved through a pair of 25 element spherical arrays manufactured from CaF2. Microwells were ablated with exposures of 200 shots per microwell and pulse energy of 24 mJ. Substrates were held on a precision vacuum chuck supported by X-Y-Z-ø, stage set with a resolution of 50 nm (MetaFAB, Cardiff, UK). Laser debris was removed from the surface by sonicating the surfaces for 20 minutes in each of ethanol, 1 mol/l HCl/H2O and water. Surfaces were further etched by treating in 7 mol/l KOH/H2O at 70 °C for 1 hour followed by sonication for 20 minutes in water. For experimentation, KG1a cells (1 × 105) were washed and resuspended in 600 µl imaging medium before being pipetted slowly on to the microwells and allowed to settle for 10 minutes under tissue-culture conditions. Imaging medium-containing 10% fetal bovine serum, Na-HEPES (pH 7.4), 1 µg/ml PI, and fsr-l-r8 or asr-a-r8 (final concentration 10 µmol/l) were added to the cells and these were imaged as described above.
Quantification of the cellular uptake of r8-, asr-a-r8-, asr-l-r8- and fsr-l-r8-Alexa488. KG1a cells (5 × 105) were washed once in complete medium and equilibrated at 4 or 37 °C for 15 minutes, before centrifugation and incubation for 1 hour with fresh temperature equilibrated medium (4 °C, 37 °C) containing 2 µmol/l Alexa488 peptides; incubations were performed at either 4 or 37 °C. Cells were washed once with ice-cold phosphate-buffered saline (PBS), incubated with 0.25 mg/ml trypsin/EDTA solution at 37 °C for 5 minutes and then placed as a suspension in 1.5-ml centrifuge tubes. The cells were washed once in ice-cold PBS, twice with PBS-containing 14 µg/ml heparin and finally resuspended in 200 µl ice-cold PBS. Trypsinization and heparin washes are required to remove surface bound peptides. Cellular fluorescence was then immediately quantified using the 488 nm excitation laser on a Becton Dickinson FACSCalibur analyzer. Live cells were gated on a forward and side scatter basis, and 10,000 viable cells were assayed.
Cell viability assays. KG1a (4 × 104 cells/well in a total volume of 200 µl) and CLL cells (3 × 105/well in a total volume of 200 µl) were seeded in 96-well plates and incubated with 0–20 µmol/l unlabeled peptides for 20 hours under tissue-culture conditions. Cell viability was then assessed using the MTT assays as previously described.20 For all experiments, viability is expressed as the percentage of viable cells relative to untreated controls. Cell viability was also measured using a Accuri C6 flow cytometer following incubation of the cells with 10 µmol/l peptide and 0.5 µg/ml PI at 37 °C for 1 hour. Changes in forward scatter and exclusion of PI were used to determine the death of cells.
Influence of incubation time, and serum on peptide cytotoxicity. KG1a cells were incubated with 0–20 µmol/l unlabeled peptides for 1 hour at 37 °C in complete medium or serum-free medium, washed and then further incubated in complete medium for 19 hours. MTT viability assays were then performed.
Bcl-2 expression in cell lines and primary samples. Cells were washed and harvested by centrifugation and an equal volume of cell pellet and ice-cold lysis buffer (50 mmol/l Tris–HCl, pH 8.0, 150 mmol/l NaCl, 0.02% sodium azide, 1% Triton X-100, 1 mmol/l DTT) containing protease inhibitor cocktail was added. These were mixed and incubated on ice for 10 minutes before centrifugation at 13,000g (4 °C) for 10 minutes. Supernatants corresponding to 15-µg protein were then mixed with SDS sample buffer and loaded on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. Following electrophoresis the proteins were transferred to nitrocellulose papers, probed with anti-Bcl-2 and α-tubulin antibodies, washed and incubated with horseradish peroxidase-conjugated antibodies. Subsequently bands were visualized using enhanced chemiluminescence. ImageJ was used to perform densitometric analysis and the Bcl-2 signal was divided by the tubulin signal to allow comparison between the different CLL samples. In order to facilitate quantitation, samples were run in duplicate.
Statistical methods. Statistical analysis was performed using one-way ANOVA followed by a Tukey–Kramer post hoc test.
SUPPLEMENTARY MATERIAL Figure S1. Viability of cells following 24-hour incubation with 0–20 µmol/l fsr-l-r8 or fsr-l-r8δGC. Figure S2. Cellular distribution and uptake of Alexa488-labeled peptides in KG1a cells at 4 °C. Figure S3. Scanning electron micrograph of an array of circular microwells Video S1. Uptake of 2 µmol/l Alexa488-labeled r8 in KG1a cells, supplement to Figure 1e. Video S2. Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in KG1a cells, supplement to Figure 1e. Video S3. Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in KG1a cells, supplement to Figure 1e. Video S4. KG1a cells incubated with 10 µmol/l asr-a-r8 while resting in microwells, supplement to Figure 2d. Video S5. KG1a cells incubated with 10 µmol/l fsr-l-r8 while resting in microwells, supplement to Figure 2d. Video S6. Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in CLL cells, supplement to Figure 3c. Video S7. Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in CLL cells, supplement to Figure 3c. Video S8. Jurkat cells incubated with 10 µmol/l asr-l-r8, PI added as a marker for membrane integrity, supplement to Figure 4a. Video S9. Jurkat cells incubated with 10 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 4a. Video S10. HeLa cells incubated with 10 µmol/l 20 µmol/l asr-l-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b. Video S11. HeLa cells incubated with 20 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b. Video S12. HeLa cells incubated with 20 µmol/l L-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
Acknowledgments
Funding for this work was obtained through BBSRC grant D013038 awarded to A.T.J. and EPSRC grant EP/P502381/1 awarded to D.B. Research in the Cardiff CLL Research group by P.B. and C.F. is funded by a leukemia and Lymphoma Research Specialist Programme. The authors declared no conflict of interest.
Supplementary Material
Viability of cells following 24-hour incubation with 0–20 µmol/l fsr-l-r8 or fsr-l-r8δGC.
Cellular distribution and uptake of Alexa488-labeled peptides in KG1a cells at 4 °C.
Scanning electron micrograph of an array of circular microwells.
Uptake of 2 µmol/l Alexa488-labeled r8 in KG1a cells, supplement to Figure 1e.
Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in KG1a cells, supplement to Figure 1e.
Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in KG1a cells, supplement to Figure 1e.
KG1a cells incubated with 10 µmol/l asr-a-r8 while resting in microwells, supplement to Figure 2d.
KG1a cells incubated with 10 µmol/l fsr-l-r8 while resting in microwells, supplement to Figure 2d.
Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in CLL cells, supplement to Figure 3c.
Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in CLL cells, supplement to Figure 3c.
Jurkat cells incubated with 10 µmol/l asr-l-r8, PI added as a marker for membrane integrity, supplement to Figure 4a.
Jurkat cells incubated with 10 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 4a.
HeLa cells incubated with 10 µmol/l 20 µmol/l asr-l-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
HeLa cells incubated with 20 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
HeLa cells incubated with 20 µmol/l L-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
REFERENCES
- Borden EC, Kluger H., and, Crowley J. Apoptosis: a clinical perspective. Nat Rev Drug Discov. 2008;7:959. doi: 10.1038/nrd2756. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov. 2008;7:1001–1012. doi: 10.1038/nrd2637. [DOI] [PubMed] [Google Scholar]
- Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9:501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KW., and, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C., and, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov . 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Jones AT. Gateways and tools for drug delivery: endocytic pathways and the cellular dynamics of cell penetrating peptides. Int J Pharm. 2008;354:34–38. doi: 10.1016/j.ijpharm.2007.10.046. [DOI] [PubMed] [Google Scholar]
- Foged C., and, Nielsen HM. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv. 2008;5:105–117. doi: 10.1517/17425247.5.1.105. [DOI] [PubMed] [Google Scholar]
- Henriques ST, Melo MN., and, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they. Biochem J. 2006;399:1–7. doi: 10.1042/BJ20061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerg C., and, Merkle HP. On the biomedical promise of cell penetrating peptides: limits versus prospects. J Pharm Sci. 2008;97:144–162. doi: 10.1002/jps.21117. [DOI] [PubMed] [Google Scholar]
- Fonseca SB, Pereira MP., and, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61:953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K.et al. (2008A short Nur77-derived peptide converts Bcl-2 from a protector to a killer Cancer Cell 14285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunnemann G, Martin RM, Haupt S, Patsch C, Edenhofer F., and, Cardoso MC. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J . 2006;20:1775–1784. doi: 10.1096/fj.05-5523com. [DOI] [PubMed] [Google Scholar]
- Kosuge M, Takeuchi T, Nakase I, Jones AT., and, Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug Chem. 2008;19:656–664. doi: 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- Watkins CL, Schmaljohann D, Futaki S., and, Jones AT. Low concentration thresholds of plasma membranes for rapid energy-independent translocation of a cell-penetrating peptide. Biochem J. 2009;420:179–189. doi: 10.1042/BJ20090042. [DOI] [PubMed] [Google Scholar]
- Watkins CL, Brennan P, Fegan C, Takayama K, Nakase I, Futaki S.et al. (2009Cellular uptake, distribution and cytotoxicity of the hydrophobic cell penetrating peptide sequence PFVYLI linked to the proapoptotic domain peptide PAD J Control Release 140237–244. [DOI] [PubMed] [Google Scholar]
- Heitz F, Morris MC., and, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart KM, Horton KL., and, Kelley SO. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org Biomol Chem. 2008;6:2242–2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R., and, Brock R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Fretz MM, Penning NA, Al-Taei S, Futaki S, Takeuchi T, Nakase I.et al. (2007Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells Biochem J 403335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton HM, Nelson MH, Hatlevig SA, Reddy MT., and, Iversen PL. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug Chem. 2004;15:290–299. doi: 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP.et al. (2006Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents J Control Release 116304–313. [DOI] [PubMed] [Google Scholar]
- Takayama K, Nakase I, Michiue H, Takeuchi T, Tomizawa K, Matsui H.et al. (2009Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas) J Control Release 138128–133. [DOI] [PubMed] [Google Scholar]
- Mason AJ, Moussaoui W, Abdelrahman T, Boukhari A, Bertani P, Marquette A.et al. (2009Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine-rich amphipathic cationic peptides J Biol Chem 284119–133. [DOI] [PubMed] [Google Scholar]
- Shental-Bechor D, Haliloglu T., and, Ben-Tal N. Interactions of cationic-hydrophobic peptides with lipid bilayers: a Monte Carlo simulation method. Biophys J. 2007;93:1858–1871. doi: 10.1529/biophysj.106.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper C, Ward R, Lin TT, Brennan P, Starczynski J, Musson M.et al. (2007Highly purified CD38+ and CD38- sub-clones derived from the same chronic lymphocytic leukemia patient have distinct gene expression signatures despite their monoclonal origin Leukemia 21687–696. [DOI] [PubMed] [Google Scholar]
- Shin DS, Lee JH, Suh J., and, Kim TH. Elimination of surface debris generated by KrF excimer laser ablation of polyimide. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing. 2006;416:205–210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Viability of cells following 24-hour incubation with 0–20 µmol/l fsr-l-r8 or fsr-l-r8δGC.
Cellular distribution and uptake of Alexa488-labeled peptides in KG1a cells at 4 °C.
Scanning electron micrograph of an array of circular microwells.
Uptake of 2 µmol/l Alexa488-labeled r8 in KG1a cells, supplement to Figure 1e.
Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in KG1a cells, supplement to Figure 1e.
Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in KG1a cells, supplement to Figure 1e.
KG1a cells incubated with 10 µmol/l asr-a-r8 while resting in microwells, supplement to Figure 2d.
KG1a cells incubated with 10 µmol/l fsr-l-r8 while resting in microwells, supplement to Figure 2d.
Uptake of 2 µmol/l Alexa488-labeled asr-a-r8 in CLL cells, supplement to Figure 3c.
Uptake of 2 µmol/l Alexa488-labeled fsr-l-r8 in CLL cells, supplement to Figure 3c.
Jurkat cells incubated with 10 µmol/l asr-l-r8, PI added as a marker for membrane integrity, supplement to Figure 4a.
Jurkat cells incubated with 10 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 4a.
HeLa cells incubated with 10 µmol/l 20 µmol/l asr-l-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
HeLa cells incubated with 20 µmol/l D-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.
HeLa cells incubated with 20 µmol/l L-NuBCP-9-r8, PI has been added as a marker for membrane integrity, supplement to Figure 5b.