

Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart muscle disease that is often familial, characterized by arrhythmias of right ventricular origin, due to transmural fatty or fibrofatty replacement of atrophic myocardium. ARVC is usually diagnosed in the clinical setting between 20 and 40 years of age. The disease is seldom recognised in infancy or under the age of 10, probably because the clinical expression of the disease is normally postponed to youth and adulthood. This review focuses its attention to the pediatric age, defined as the period of life raging from birth to 18 years. During this span of life, ARVC is not so rare as previously supposed and can be identified by applying the same diagnostic criteria proposed for the adult. Ventricular arrhythmias range from isolated ventricular arrhythmias to sustained ventricular tachycardia and fibrillation. Children and adolescents with ARVC must be carefully evaluated and followed-up especially when a family positive history is present, taking into account the high probability during this life-period that asymptomatic affected patients become symptomatic or that arrhythmias worsen during follow-up. The recent identification of the first defective gene opens new avenues for the early identification of affected subjects even when asymptomatic.
MeSH: Arrhythmogenic Right Ventricular Dysplasia/diagnosis, Arrhythmogenic Right Ventricular Dysplasia/therapy, Arrhythmogenic Right Ventricular Dysplasia/complications, Heart Ventricle/pathology, Magnetic Resonance Imaging, Syncope/etiology, Death, Sudden, Cardiac/etiology, Tachycardia, Ventricular/etiology
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart muscle disease of unknown etiology, often familial, characterized by arrhythmias of RV origin, due to transmural fatty or fibrofatty replacement of atrophic myocardium (Fig 1).1–4 In 1978 Frank et al5 described four patients with sustained ventricular tachycardia (VT) and diskinetic areas of the RV and the authors called this entity arrhythmogenic RV dysplasia, because they considered it as a result of a ‘maldevelopment’ of the RV myocardium.
Figure 1.
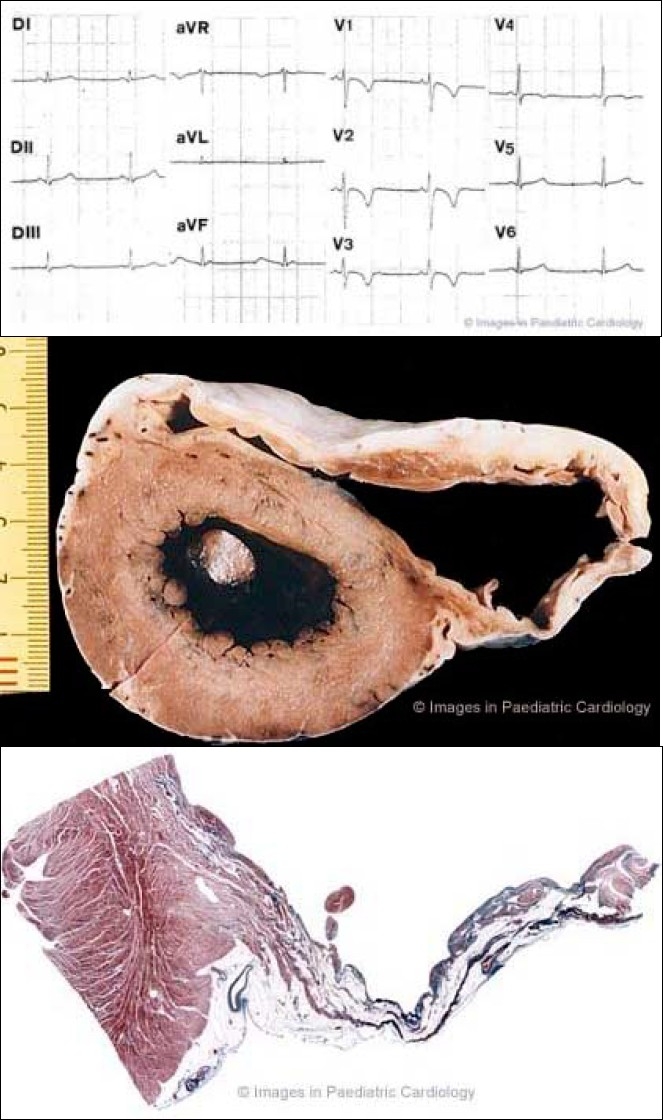
A 17 year old asymptomatic soccer player who died suddenly on effort 1a) 12-lead ECG at preparticipation screening for competitive sport activity: note typical inverted T-waves in the right precordial leads up to V4
1b) Cross section of the heart specimen showing diffuse involvement of the RV free wall with anterior and subtricuspid aneurysms
1c) Histology of the posterior aneurysm shows a transmural fibrofatty replacement of the atrophic myocardium accounting for a thin wall (Heidenhain Trichrome ×4)
Marcus et al6 first reported in 1982 the clinical profile of this disease; the typical patient described was a middle-aged male who presented with palpitations, tachycardia or syncope. Physical examination was normal, whereas the ECG showed incomplete or complete right bundle branch block and T wave inversion over V1 to V4. The clinical picture was characterised by sustained VT with left bundle branch block (LBBB) morphology and an enlarged RV with segmental wall motion abnormalities, particularly in the infundibulum. In the late 1980's Thiene et al discovered through autopsy studies that ARVC was a major cause of sudden death in the young1 and in competitive athletes.7 Subsequent clinical and histologic studies demonstrated that this disease was not congenital, namely a structural cardiac defect present at birth, but acquired and progressive in time, most often genetically determined.2 Thus ARVC has been recently added to dilated, hypertrophic and restrictive cardiomyopathies as a fourth heart muscle disease within the new classification of cardiomyopathies.8
A Task Force of the European Society of Cardiology has proposed the criteria for the clinical diagnosis of ARVC.9 Table 1 shows the major and minor criteria, based on the identification of structural abnormalities, fatty or fibrofatty replacement of RV myocardium, electrocardiographic changes, arrhythmias of RV origin, and familial disease. The diagnosis is fulfilled in the presence of two major criteria or one major plus two minor criteria or four minor from different groups. The sensitivity and specificity of these criteria are still unknown.
Table 1.
Diagnostic criteria (from McKenna et al9)

From the etiopathogenetic point of view, three basic mechanism have been postulated to explain progressive loss of RV myocardium with fibro-fatty replacement of the RV myocardium; i.e.
Apoptosis or programmed cell death
Injury and repair process in the setting of chronic myocarditis
First reports concerned sporadic cases of ARVC. In the paper by Marcus et al6 only one patient had a familial history of ARVC. This patient had been previously reported in 1974 by Waynberger et al12 who studied familial cases of VT with LBBB configuration. In the 1980s, other authors demonstrated a familial occurrence of the disease.13–16 In 1988 Nava et al17 conducted the first large epidemiological study on ARVC, by analyzing 72 members from 9 families, and advanced the hypothesis that the disease is transmitted by autosomal dominance inheritance with variable expression and penetrance. This genetic transmission was further confirmed by studies on twins.18,19 Six ARVC loci have been identified so far. Two loci have been mapped in close proximity of chromosome 14, and the others on chromosome 1, 2, 3 and 10.20–26 An autosomal recessive variant of ARVC that is associated with palmoplantar keratosis and woolly hair, (so-called ‘Naxos disease’) has been mapped on chromosome 17, and a mutation of plakoglobin, a protein implicated in cell to cell adhesion, has been found.26 More recently, we discovered the first mutation of the gene of the ARVC variant mapping to chromosome 1q42-43, which encodes for a defective cardiac ryanodin receptor.27 Familiar occurrence has been demonstrated in 50% of patients with ARVC by Nava et al.4,17
During its natural history, ARVC may manifest in the following clinical phases: concealed period, overt electrical disorder, RV failure, biventricular pump failure.4 The concealed phase is characterized by subtle RV changes, with or without minor ventricular arrhythmias. The pathologic process involves only one region of the so-called triangle of dysplasia: the subtricuspidal region, the apex and the outflow tract. In this phase patients are usually asymptomatic, present with or without non-complex ventricular arrhythmias, and nevertheless are at risk of sudden death. The overt clinical disorder is typical of symptomatic arrhythmic patients, who show overt RV functional and structural abnormalities, in the setting of an increased risk of sudden death. RV failure is caused by the progressive extension of RV disease, determining a global dysfunction with a relatively preserved left ventricular function. Biventricular pump failure is the final stage of ARVC, provoked by pronounced left ventricular involvement and often associated to atrial fibrillation and thromboembolic events.
ARVC in children and adolescents
ARVC is usually diagnosed in the clinical setting between 20 and 40 years of age.28 The disease has been less well studied in the pediatric age group. The prevalence of children affected by ARVC in non-selected clinical series usually comprises 5% to 30% of the series.6,29,30 ARVC may lead to sudden death even in pediatric age. In the study of Blomstrom-Lundqvist et al29 on long term follow-up of 15 patients with ARVC, one of the two patients who died suddenly was a child. Daliento et al31 reported two cases of sudden death in their series of 17 children with ARVC, followed for more than 7 years. Also in autoptic series the prevalence of children and adolescent is not trivial. In the original paper of Thiene at al1 on sudden death due to ARVC, children and adolescents comprised 50% of cases and sudden death was often the first sign of the disease. The youngest autoptic patient who died suddenly was described by Pawel et al32 in 1994. This patient was a 7-year-old boy of Italian descent, who also had histological involvement of the left ventricle.
ARVC in infants
Sporadic cases have been observed early in life, even in the embryological phase. Fontaine et al33 described a 27-week old fetus with RV aneurysm and arrhythmias observed in utero. Histology of his heart showed evidence of adipocytes interspersed with myocardial fibers. Neither fibrosis nor signs of inflammation were observed.
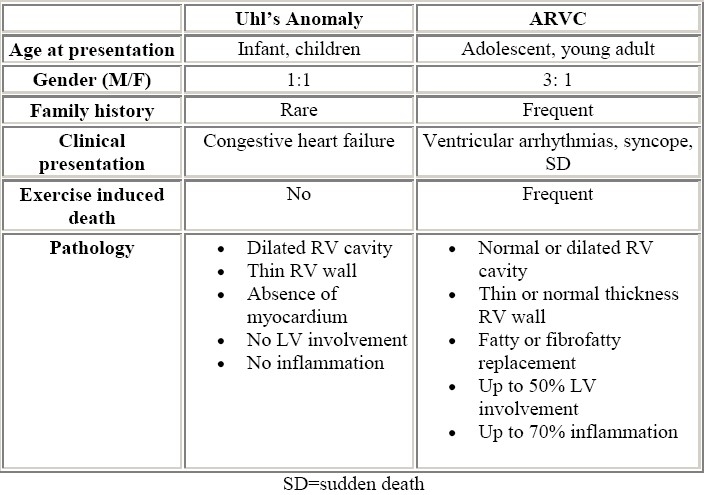
When dealing with right heart failure and death that occurs in few weeks or months old infants, ARVC must be differentiated from Uhl's disease (Table 2). The latter was described in 1952 by Uhl,34 who reported a case in which the wall of the RV was paper thin with “almost total absence of the myocardium of the right ventricle”. Although James et al35 suggested that Uhl's anomaly and ARVC share similar pathogenesis, Uhl's anomaly generally leads to congestive heart failure at an early age and death after few weeks or months without arrhythmias.36,37 The definitive differential diagnosis can be made only by autopsy.
Table 2.
Uhl's anomaly vs ARVC
The only ARVC infant presenting with heart failure was reported by Pinamonti et al,38 who studied a 5 month female, in whom unfortunately, autopsy was not performed.
In older cases, clinical presentation of ARVC is more typical. Chest x-ray is normal, ECG shows negative T wave up to V4, and invasive and non invasive imaging techniques demonstrate RV abnormalities. Antiarrhythmic treatment seems to protect from sudden death. Makanda et al39 reported a case of 16-month-old child admitted to hospital due to the occurrence of sustained VT that was pharmacologically cardioverted. Physical examination and chest X-ray were normal. An incomplete right bundle branch block was present at ECG. Two akinetic wall motion abnormalities were found at right angiogram. On follow-up this patient did well with antiarrhythmic therapy. Minor left ventricular abnormalities may also be associated with ARVC at a very young age. An 18-month-old female was described by Pinamonti et al,38 presenting with polymorphic non-sustained VT and negative T wave from V1 to V4 at ECG, and who did well in beta-blockers therapy.
Sometimes the disease is not recognized at first assessment, especially in the absence of arrhythmias. Matsuoka et al40 described a 4 year old boy evaluated prior to an operation for cerebral abscess, who at the age of 10 days had undergone a first hemodynamic study due to cyanosis during crying, that was considered at first normal. Chest X-ray showed an enlarged heart size; the ECG had a marked superior axis deviation of QRS and negative T wave up to V3. Invasive investigations demonstrated findings suggestive of ARVC, such as multiple diverticular outpouchings of RV wall, dilatation together with a reduction of ejection fraction of both ventricles. Ventricular arrhythmias had never been detected. Without antiarrhythmic protection, this patient died suddenly soon after the invasive examinations, but autopsy was not performed.
In general, the disease is seldom diagnosed below the age of 10 years, probably because the clinical expression of the disease is normally postponed to youth and adulthood.28
ARVC in children and adolescents (up to 18 years)
The distinctive phases of ARVC, i.e. concealed phase, overt electrical disorder, RV failure, biventricular pump failure can be well recognized in this span of life.
During the concealed phase, premature ventricular beats with LBBB pattern can be the only clinical manifestation of ARVC, also in asymptomatic children. Patterson et al41 reported a five-year-old girl with frequent ventricular extrasystoles, who had small apical saccular diverticulae at echocardiogram. Tomisawa et al42 studied two children, 5 and 12 years old respectively, admitted to hospital because of frequent extrasystoles with LBBB morphology. The ECG demonstrated notched T wave in V2 and V3 in the former and inverted T wave in V1-V4 in the latter. At angiography an aneurysm of RV outflow tract was found in both. Aneurysmectomy led to abolition of the premature ventricular complexes in the patient with the larger aneurysm. The other was successfully treated with beta-blockers. In the series of Nava et al30 of patients with concealed form of ARVC, four individuals were children (range 4 to 16 years). In this subgroup of patients, ventricular arrhythmias ranged from isolated ventricular beats (2 patients) to sustained VT (1 patient) or ventricular fibrillation (1 patient). All the patients showed a normal physical tolerance, a normal cardiac silhouette at X-ray (Fig. 2), negative precordial T wave in 75 % of cases, localized abnormalities of RV and fibro (3 cases) or fibro-fatty (1 case) replacement at endomyocardial biopsy. They all did well on antiarrhythmic drug therapy. Symptomatic children with overt clinical ARVC, ie RV structural abnormalities associated with ventricular arrhythmias, possibly leading to sudden death, are not as rare as previously reported. For many years ventricular fibrillation has been considered idiopathic when no recognizable major left-heart disease or coronary pathologic condition were present. In this setting RV was underestimated or ignored. Martini et al43 described six patients resuscitated from ventricular fibrillation and among the five patients with morphofunctional abnormalities of RV and typical ECG, two were children and showed minor left ventricular wall motion abnormalities.
Figure 2.
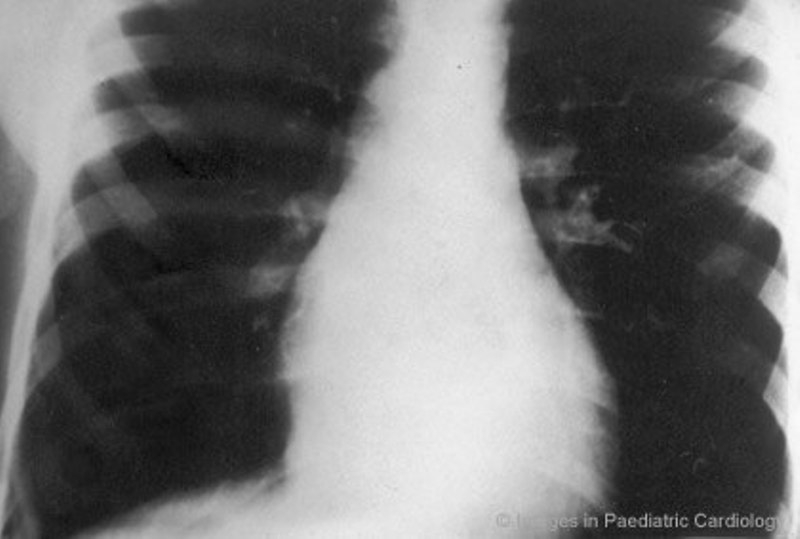
An 8-year-old boy with premature ventricular complex - posteroanterior chest x-ray shows a normal sized cardiac silhouette (modified from Nava et al30
Sustained VT with LBBB morphology was a common form of presentation in ARVC children in the 1980s. Dungan et al44 described three children presenting with VT at a very young age. Chest x-rays were normal, whereas RV was characterized by typical wall motion abnormalities at angiography. Sustained VT occurred spontaneously or during stress testing. All three patients remained asymptomatic with antiarrhythmic drug therapy during long-term follow-up. In the study of Reiter et al45 on clinical spectrum of VT with LBBB, seven patients had ARVC. Two patients were children and showed negative T wave on right precordial leads and typical RV abnormalities. VT was inducible in both patients. The younger, treated with quinidine died suddenly 19 months after evaluation. Of note, he presented a mild reduction of left ventricular ejection fraction, which probably could have represented a proarrhythmogenic factor.
Refractory VT has been sometimes treated with surgical procedure, especially in the 1980s. RV disconnection had been introduced for the treatment of drug refractory cases.46 This technique, that can be partial or total, is a surgical procedure that electrically isolates the arrhythmogenic RV from the rest of the heart. Despite the acceptable long-term results, this procedure has been seldom employed in adolescents because of hemodynamic alterations. Mc Lay et al47 described a 14 year old white girl who underwent RV total disconnection with good results during the subsequent follow-up. A surgical approach consisting of myocardial excision and cryocoagulation has been also used with contrasting results in the children. A 15-year-old boy died suddenly 5 months after the operation48 while another 14-year-old did well on follow-up after discharge.49
Global RV failure, although rare, may also appear in the pediatric age group. Kearney et al50 described an 11-year-old girl who was transplanted because of right heart failure with peripheral edema and hepatomegaly. Biventricular failure has also been reported in children. Smith et al51 studied the hearts of two sisters, aged 17 and 14, with no history of heart disease. The elder sister died suddenly, while the younger sister developed congestive heart failure and underwent cardiac transplantation.
Diagnostic criteria and follow-up
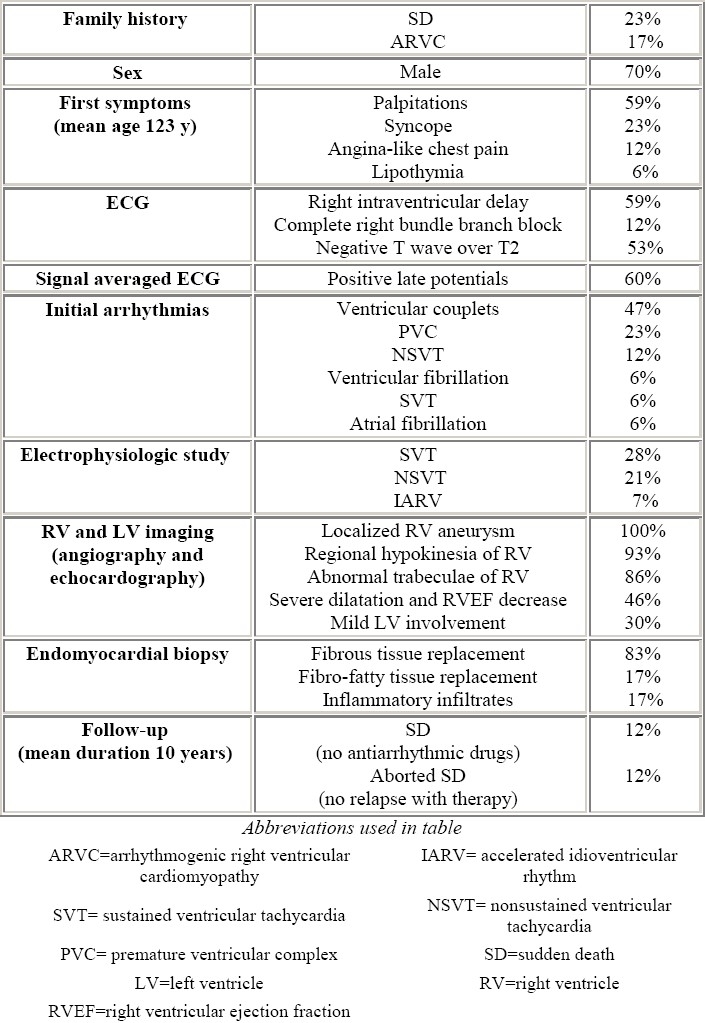
A clinical study on ARVC pediatric patients was conducted by Daliento et al in 1995.31 They examined 17 patients (Table 3) with a mean age at onset of symptoms of 12 years and a mean age at diagnosis of 15 years. As far as the presence of major and minor diagnostic criteria, family occurrence of ARVC was 41%. The symptoms were varied, with palpitations and syncope being the most frequent (58% and 23% respectively). Syncope was frequently related to sustained ventricular arrhythmias.
Table 3.
Main findings in 17 children and adolescents with ARVC (modified from Daliento et al54)
Negative T waves beyond V2 were present in half of the patients. Although localized prolongation of the QRS complex in right precordial leads was uncommon, two patients showed complete right bundle branch block. The incidence of epsilon wave that is conventionally defined as a distinct wave of small amplitude that occupies the QT segment in the right precordial leads, was not reported in that series. In pediatric age the frequency of epsilon wave is unknown, in contrast with an incidence of about 25-30% of adults (Fig. 3). Whether epsilon wave is uncommon in children or is simply underdiagnosed is uncertain. Late potentials were found in more than half of the patients at 40 Hz filter (Fig. 4).
Figure 3.
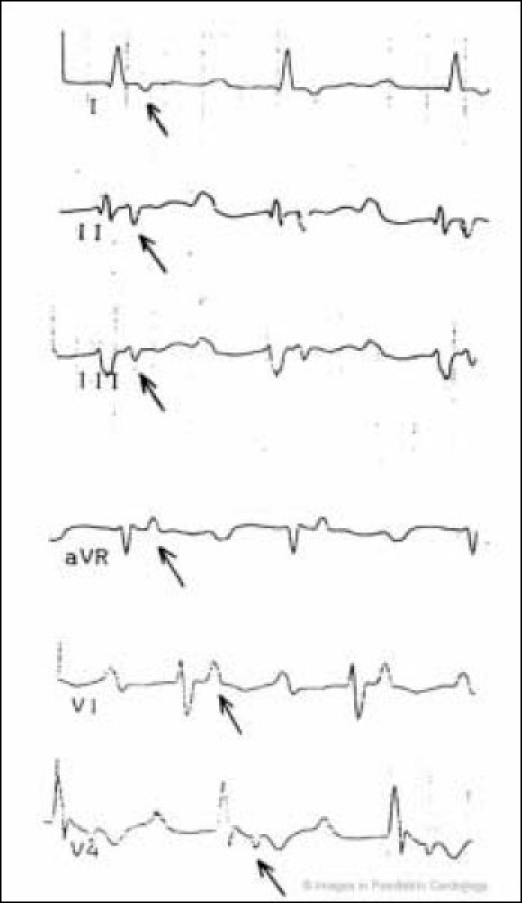
A 48-year old man with a severe form of ARVC. Epsilon wave (arrow) simulates atrial tachycardia in leads I, II, III, aVR, V1 and V4. The epsilon wave is different from P axis. Right atrial enlargement, increased PR segment, low voltage of QRS, negative T-wave from V1-V6 are also present (modified from Nava et al4)
Figure 4.
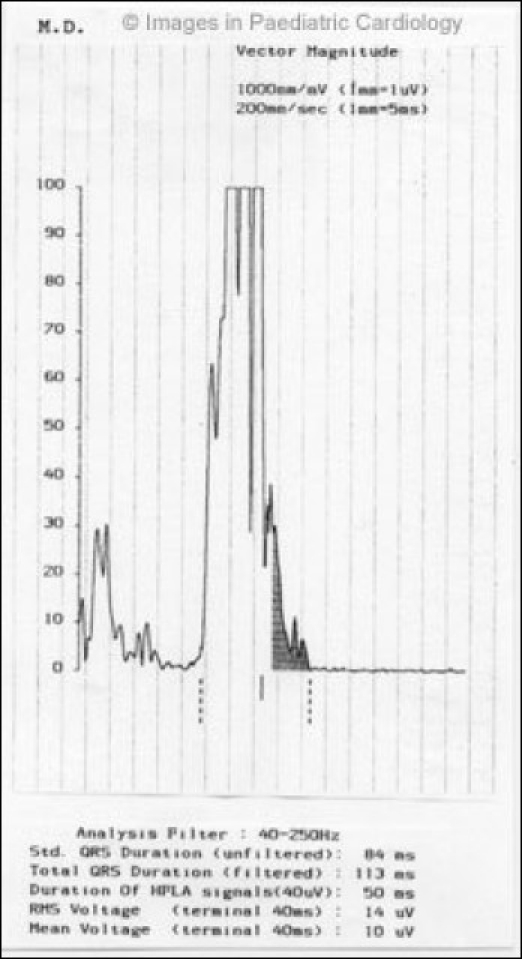
Presence of late potentials at 40 Hz filter in a 16 year-old child with non-sustained ventricular tachycardia
Initial spontaneous arrhythmias recorded on surface ECG or Holter were prevalently minor. During follow-up arrhythmic picture was mainly characterized by sustained and nonsustained VT (Fig. 5).
Figure 5.
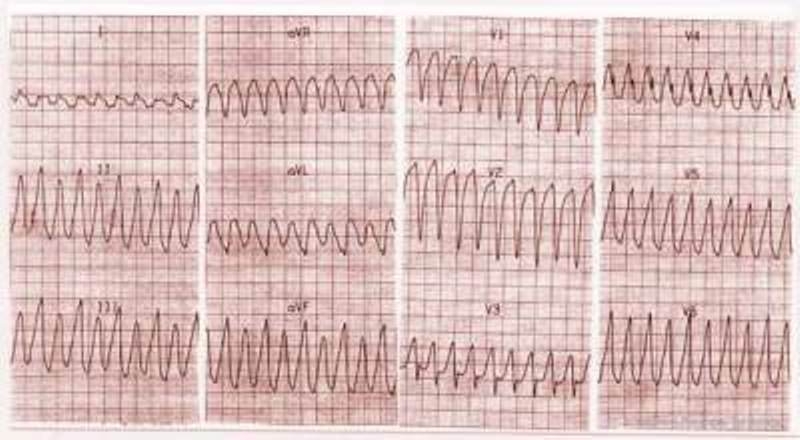
A 18-year-old boy with a concealed form of ARVC complained of palpitations. A monomorphic ventricular tachycardia with LBBB morphology was recorded. Echocardiogram showed only a diaphragmatic wall bulging and slight dilatation of RV outflow tract (modified from Nava et al4)
Localized subtricuspidal and/or anterior RV aneurysm was demonstrated in all by imaging techniques, together with a high incidence of regional apical and/or diaphragmatic hypokinesia as well as an abnormal trabecular pattern (Fig. 6–8). Severe dilatation and reduction of RV ejection fraction was found in nearly half of the patients. Mild left ventricular impairment was present in one third of the patients.
Figure 6.
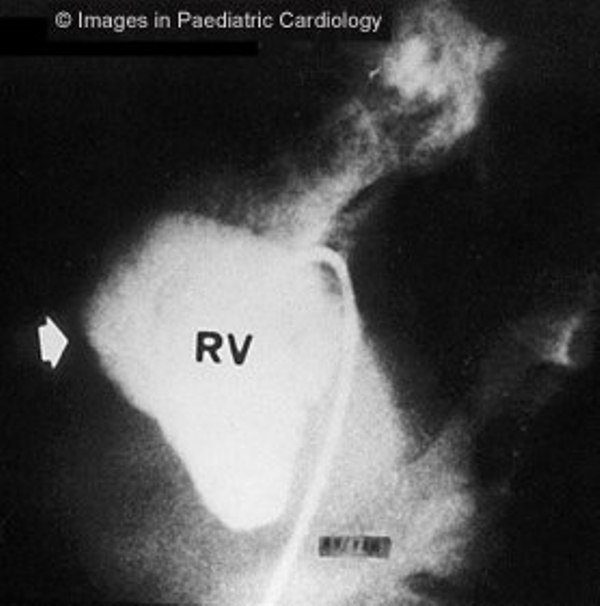
Angiography of the RV (left anterior oblique view) shows anterior bulging of infundibulum (arrow) in a 16 years old child with non-sustained VT (RV=right ventricle)
Figure 8.
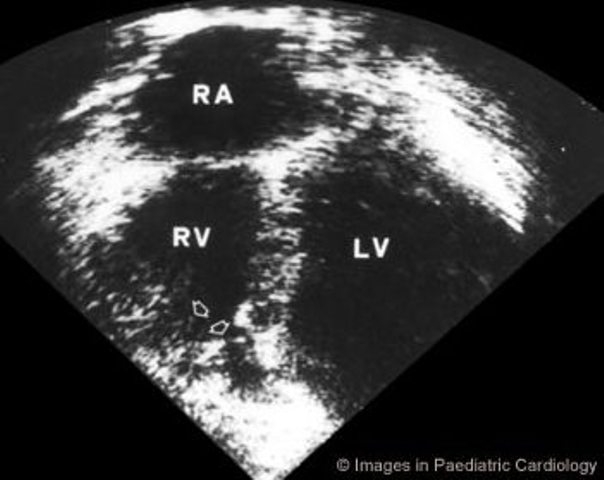
Apical four chamber echocardiogram showing RV dyskinesia of the apex (arrows) in a 13 year-old girl (LV=left ventricle, RA=right atrium, RV=right ventricle)
Figure 7.
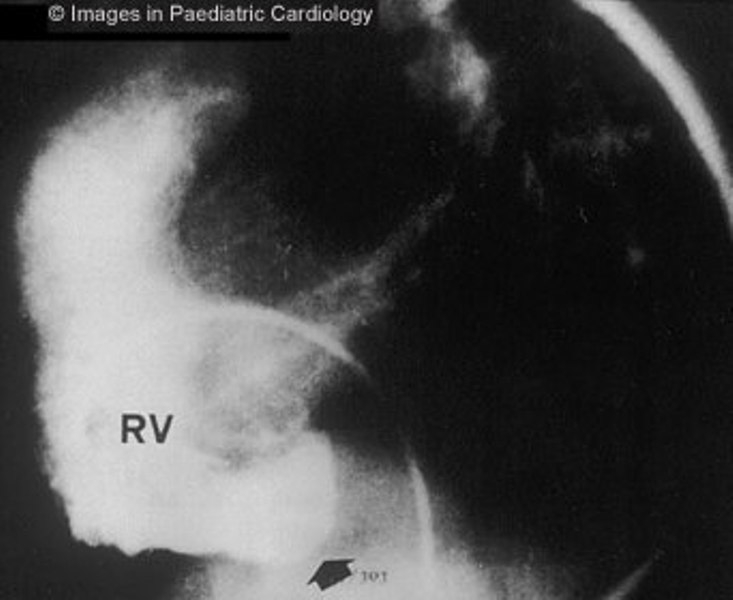
Angiography of the RV (lateral view) shows subtricuspidal bulging (arrow) in a 17 years old child with sustained VT (RV=right ventricle)
At endomyocardial biopsy, fibrous replacement (Fig. 9) was prevalent compared to fibro-fatty substitution. Inflammatory lymphocyte infiltrates and necrosis were rare and not associated to a peculiar histologic variant.
Figure 9.
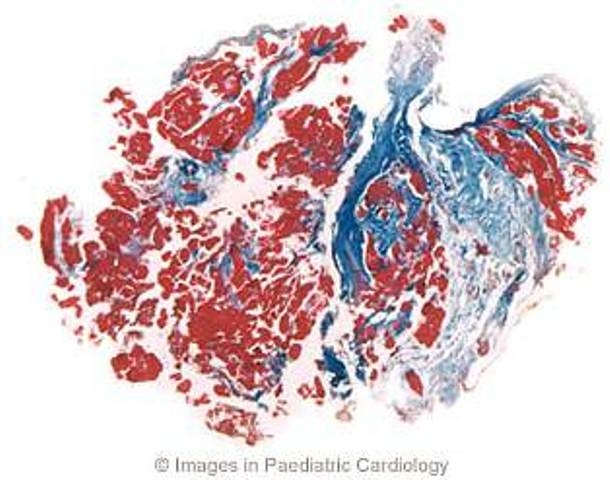
A 21 year old man with a family history of ARVC and spontaneous sustained VT with LBBB morphology. Endomyocardial biopsy, performed at 18 years of age, shows extensive replacement of the myocardium by fibrosis (Azan-Mallory stain, original magnification ×45) (modified from Menghetti et al52)
Magnetic resonance imaging was not performed in this study and its specificity is still unknown52 (Fig. 10). During a mean follow-up of seven years, two patients died suddenly, without antiarrhythmic drug protection. The two patients with aborted sudden death as first symptom had no relapses of sustained ventricular arrhythmias on beta-blockers and disopyramide plus beta-blockers, respectively. Sustained VT occurred in three patients; two patients had no recurrences of VT on disopyramide plus beta-blockers and sotalol, respectively. In the third subject, the combination of amiodarone plus beta-blocker therapy significantly reduced the number of attacks of VT. The other 10 patients, treated empirically with beta-blockers and/or class I and III antiarrhythmic drugs had no episode of sustained VT during the follow-up.
Figure 10.
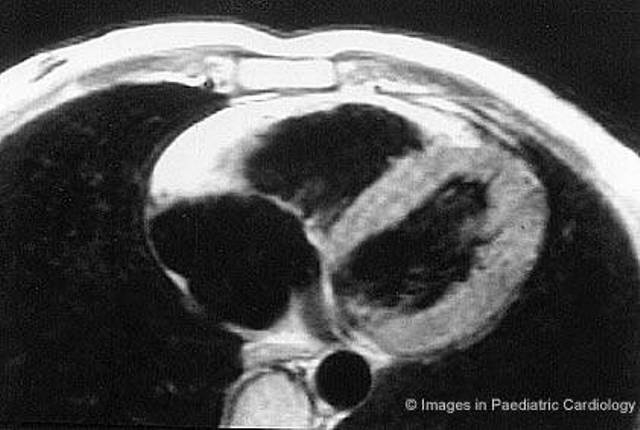
Same patient of figure 9. Short axis nuclear magnetic resonance shows a diffuse bright signal from the RV wall, suggestive for extensive myocardial fatty replacement (modified from Menghetti et al52)
Daliento et al31 demonstrated that the diagnostic criteria were valid also for the children and adolescents and that the risk of sudden death or ventricular fibrillation was higher in this span of life, especially when not protected by antiarrhythmic drug therapy. In this series, the pediatric patients were managed medically. Nowadays, an implantable defibrillator is the treatment of choice in patients resuscitated from cardiac arrest or judged to be at high risk of sudden death.53,54 Catheter ablation can sometime treat effectively ventricular tachyarrhythmias in selected patients. However the long-term efficacy of catheter ablation is controversial. This technique has a high acute success rate (60-90%), but VT recurrences are common and may lead to sudden arrhythmic death.
Peculiar aspects
ARVC in twins
The relationships between genetic and environmental factors have studied using twins. Identical twins may show different clinical course and prognosis17 or the same clinical findings and follow-up.18 Environmental factors such as history of myocarditis or exposure to toxin could play a role in the clinical manifestation of the disease, also in the setting of an identical genetic inheritance.
Myocarditis
An association between ARVC and an acute myocardial inflammation has been reported also in children. Whether myocarditis is a primary event or a complication of ARVC remains unclear. In some children the first manifestation of the disease is characterized by chest pain, ST segment elevation at ECG and an increase of in myocardial enzymes, in the setting of normal coronary arteries and typical morphofunctional abnormalities of the RV, together with inflammatory cells at endomyocardial biopsy. The onset of ventricular electrical instability may emerge in the setting of an inflammatory disorder as reported by Blomstrom-Lundqvist,29 or may be delayed. An interval of six years after an acute myocardial inflammation has been reported in a 15 year-old boy with ARVC by Daliento et al55 (Fig. 11). Myocarditis may be a precipitating factor in ARVC. Sabel et al56 described a 14-year-old boy with ARVC, who had a myocarditis, and died suddenly after a follow-up of 1.5 years, during mild exercise, although non-invasive tests were indicative of a low arrhythmic risk.
Figure 11.
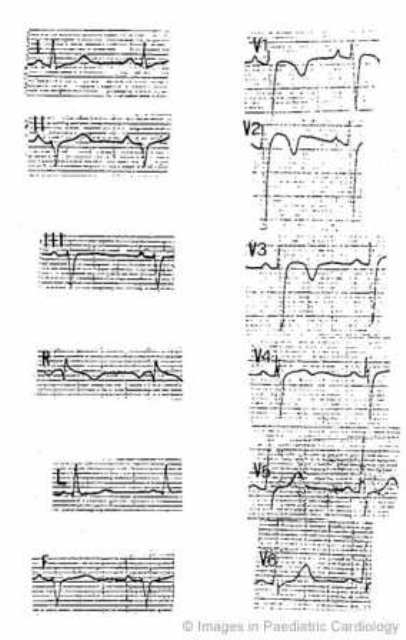
A 15-year-old boy who was admitted to the hospital for chest pain. Persistent ST segment elevation was present at ECG together with an increase in myocardial enzymes. Angiography revealed normal coronary arteries and morphofunctional abnormalities of the RV. He was resuscitated from ventricular fibrillation six years after investigation
In conclusion, ARVC is not as rare as previously thought in the pediatric population. It can be identified by applying the same diagnostic criteria that have been proposed for adults. The paediatric subgroup must be carefully evaluated and followed-up especially when a family positive history is present, taking into account the high probability that during this life-span, asymptomatic affected patients become symptomatic or that arrhythmias worsen during the follow-up. The recent identification of the first defective gene opens new avenues for the early identification of affected subjects even when asymptomatic.
Acknowledgments
Supported by MURST and Telethon, Rome and Fondazione Cassa di Risparmio, Padova, Italy
References
- 1.Thiene G, Nava A, Corrado D, Rossi L, Penneli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 2.Basso C, Thiene G, Corrado G, Angelini A, Nava A, Valente M. Arrhytmogenic right ventricular cardiomyopathy: dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 3.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestations of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Multicenter Study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 4.Nava A, Rossi L, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Amsterdam: Elsewier; 1997. [Google Scholar]
- 5.Frank F, Fontaine G, Vedel J, Mialet G, Sol C, Guiraudon G, Grosgogeat Y. Electrocardiologie de quatre cas de dysplasie ventriculaire droite arythmogène. Arch Mal Coer. 1978;71:963–972. [PubMed] [Google Scholar]
- 6.Marcus F, Fontaine G, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 7.Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden cardiac death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588–596. doi: 10.1016/0002-9343(90)90176-e. [DOI] [PubMed] [Google Scholar]
- 8.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connel J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 9.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomström-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardyomyopathy. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiene G, Basso C, Danieli GA, Rampazzo A, Corrado D, Nava A. Arrhythmogenic right ventricular cardiomyopathy: a still underecognized clinical entity. Trends Cardiovasc Med. 1997;7:84–90. doi: 10.1016/S1050-1738(97)00011-X. [DOI] [PubMed] [Google Scholar]
- 11.Valente M, Calabrese F, Thiene G, Angelini A, Basso C, Nava A, Rossi L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–484. [PMC free article] [PubMed] [Google Scholar]
- 12.Waynberger M, Courtadon M, Peltier JM, Ducloux G, Jallut H, Slama R. Tachycardies ventriculaires familiales. A propos de 7 observations. Nouv Presse méd. 1974;3:1857–1860. [PubMed] [Google Scholar]
- 13.Ruder MA, Winston SA, Davis JC, Abott JA, Eldar M, Scheinman MM. Arrhythmogenic right ventricular dysplasia in a family. Am J Cardiol. 1985;56:799–800. doi: 10.1016/0002-9149(85)91144-0. [DOI] [PubMed] [Google Scholar]
- 14.Rakovec P, Rossi L, Fontaine G, Sasel B, Markez J, Voncina D. Familial arrhythmogenic right ventricular disease. Am J Cardiol. 1986;58:377–378. doi: 10.1016/0002-9149(86)90089-5. [DOI] [PubMed] [Google Scholar]
- 15.Nava A, Scognamiglio R, Thiene G, Canciani B, Daliento L, Buja GF, Stritoni P, Fasoli G, Dalla Volta S. A polymorphic form of familial arrhythmogenic right ventricular dysplasia. Am J Cardiol. 1987;59:1405–1409. doi: 10.1016/0002-9149(87)90929-5. [DOI] [PubMed] [Google Scholar]
- 16.Laurent M, Descaves C, Biron Y, Deplace C, Almange C, Daubert JC. Familial form of arrhythmogenic right ventricular dysplasia. Am Heart J. 1987;113:827–829. doi: 10.1016/0002-8703(87)90728-9. [DOI] [PubMed] [Google Scholar]
- 17.Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja GF, Martini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia. A study involving nine families. J Am Coll Cardiol. 1988;12:1222–1228. doi: 10.1016/0735-1097(88)92603-4. [DOI] [PubMed] [Google Scholar]
- 18.Buja GF, Nava A, Daliento L, Scognamiglio R, Miorelli M, Canciani B, Alampi G, Thiene G. Right ventricular cardiomyopathy in identical and nonidentical young twins. Am Heart J. 1993;126:1187–1193. doi: 10.1016/0002-8703(93)90673-w. [DOI] [PubMed] [Google Scholar]
- 19.Hiraoka E, Masanobu K, Sakamoto S, Miki T, Ohga N, Suzuki S, Mizutani K, Kintaka T, Matsuo T. Identical twins with arrhythmogenic right ventricular dysplasia. Am J Cardiol. 1995;76:1099–1100. doi: 10.1016/s0002-9149(99)80312-9. [DOI] [PubMed] [Google Scholar]
- 20.Rampazzo A, Nava A, Danieli GA, Buja GF, Daliento L, Fasoli G, Scognamiglio R, Corrado D, Thiene G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum Molec Genet. 1994;3:959–962. doi: 10.1093/hmg/3.6.959. [DOI] [PubMed] [Google Scholar]
- 21.Severini GM, Kraijnovic M, Pinamonti B, Sinagra GF, Fioretti P, Brunazzi MC, Falaschi A, Camerini F, Giacca M, Mestroni L. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics. 1996;31:193–200. doi: 10.1006/geno.1996.0031. [DOI] [PubMed] [Google Scholar]
- 22.Rampazzo A, Nava A, Erne P, Eberhard M, Vian E, Slomp P, Tiso N, Thiene G, Danieli GA. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Molec Genet. 1995;4:2151–2154. doi: 10.1093/hmg/4.11.2151. [DOI] [PubMed] [Google Scholar]
- 23.Rampazzo A, Nava A, Miorin M, Fonderico P, Popa B, Tiso N, Livolsi B, Zimbello R, Thiene G, Danieli GA. ARVD 4, a new locus for ARVC, maps to chromosome 2 long arm. Genomics. 1997;45:259–263. doi: 10.1006/geno.1997.4927. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad F, Li D, Karibe A, Gonzalez O, Tapscott T, Hill R, Weilbaecher D, Blackie P, Furey M, Gardner M, Bachinsky LL, Roberts R. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation. 1998;98:2791–2795. doi: 10.1161/01.cir.98.25.2791. [DOI] [PubMed] [Google Scholar]
- 25.Li D, Ahmad f, Gardener MJ, Weilbaecher D, Hill R, Karibe A, Gonzalez O, Tapscott T, Sharratt GP, Bachinsky LL, Roberts R. The locus of a novel gene responsible for arrhythmogenic right ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet. 2000;66:148–156. doi: 10.1086/302713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney J, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Gen. 2001 doi: 10.1093/hmg/10.3.189. (in press) [DOI] [PubMed] [Google Scholar]
- 27.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 28.Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, Daliento L, Buja G, Corrado D, Danieli GA, Thiene G. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–2233. doi: 10.1016/s0735-1097(00)00997-9. [DOI] [PubMed] [Google Scholar]
- 29.Blomstrom-Lundqvist C, Sabel KG, Olsson SB. A long term follow-up of 15 patients with arrhythmogenic right ventricular dysplasia. Br Heart J. 1987;58:477–488. doi: 10.1136/hrt.58.5.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nava A, Thiene G, Canciani B, Martini B, Daliento L, Buja GF, Fasoli G. Clinical profile of concealed form of arrhythmogenic right ventricular cardiomyopathy presenting with apparently idiopathic ventricular arrhythmias. Int J Cardiol. 1992;35:195–206. doi: 10.1016/0167-5273(92)90177-5. [DOI] [PubMed] [Google Scholar]
- 31.Daliento L, Turrini P, Nava A, Rizzoli G, Angelini A, Buja GF, Scognamiglio R, Thiene G. Arrhythmogenic right ventricular cardiomyopathy in young versus adult patients: similarities and differences. J Am Coll Cardiol. 1995;25:655–664. doi: 10.1016/0735-1097(94)00433-Q. [DOI] [PubMed] [Google Scholar]
- 32.Pawel BR, de Chadarévian JP, Wolk JH, Donner RM, Vogel L, Braverman P. Sudden death in childhood due to right ventricular dysplasia: report of two cases. Ped Pathol. 1994;14:987–995. doi: 10.3109/15513819409037695. [DOI] [PubMed] [Google Scholar]
- 33.Fontaine G, Fontaliran F, Frank R. Arrhythmogenic right ventricular cardiomyopathies. Clinical forms and main differential diagnoses. Circulation. 1998;97:1532–1535. doi: 10.1161/01.cir.97.16.1532. [DOI] [PubMed] [Google Scholar]
- 34.Uhl HSM. A previously undescribed congenital malformation of the heart: almost total absence of the myocardium of the right ventricle. Bulletin of the John Hopkins Hospital. 1952;92:197–209. [PubMed] [Google Scholar]
- 35.James TN, Nichols MM, Sapire DW, DiPatre PL, Lopez SM. Complete heart block and fatal ventricular failure in an infant. Circulation. 1996;93:1588–1600. doi: 10.1161/01.cir.93.8.1588. [DOI] [PubMed] [Google Scholar]
- 36.Gerlis LM, Schmidt-Ott SC, Ho SY, Anderson RH. Dysplastic conditions of the right ventricular myocardium: Uhl's anomaly v arrhythmogenic right ventricular dysplasia. Br Heart J. 1993;69:142–150. doi: 10.1136/hrt.69.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcus FI, Fontaine G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: a review. PACE. 1995;18:1298–1314. doi: 10.1111/j.1540-8159.1995.tb06971.x. [DOI] [PubMed] [Google Scholar]
- 38.Pinamonti B, Sinagra G, Salvi A, Di Lenarda A, Morgera T, Silvestri F, Bussani R, Camerini F. Left ventricular involvement in right ventricular dysplasia. Am Heart J. 1992;123:711–724. doi: 10.1016/0002-8703(92)90511-s. [DOI] [PubMed] [Google Scholar]
- 39.Makanda A, Trémouroux-Wattiez M, Stijns-Cailteux M, de Jonghe D, Moretto M, Vliers A. Dysplasie arythmogène du ventricule droit chez un enfant de 16 mois. Arch Mal Cœur. 1989;82:811–814. [PubMed] [Google Scholar]
- 40.Matsuoka Y, Kawaguchi K, Okishima T, Hayakawa K. An infant with suspected right ventricular dysplasia presenting unique ventriculograms. Clin Cardiol. 1988;11:55–58. doi: 10.1002/clc.4960110119. [DOI] [PubMed] [Google Scholar]
- 41.Patterson MWH, De Souza E. Two-dimensional echocardiographic diagnosis of arrhythmogenic right ventricular dysplasia presenting as frequent ventricular extrasystoles in a child. Pediatr Cardiol. 1988;9:41–43. doi: 10.1007/BF02279883. [DOI] [PubMed] [Google Scholar]
- 42.Tomisawa M, Onouchi Z, Goto M, Nakata K, Mizukawa K, Kusunoki T. Right ventricular aneurysm with ventricular premature beats. Br Heart J. 1974;36:1182–1185. doi: 10.1136/hrt.36.12.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, Daliento L, Dalla Volta S. Ventricular fibrillation without apparent heart disease: description of six cases. Am Heart J. 1989;118:1203–1209. doi: 10.1016/0002-8703(89)90011-2. [DOI] [PubMed] [Google Scholar]
- 44.Dungan NT, Garson A, Jr, Gillette PC. Arrhythmogenic right ventricular dysplasia: a cause of ventricular tachycardia in children with apparently normal hearts. Am Heart J. 1981;102:745–750. doi: 10.1016/0002-8703(81)90101-0. [DOI] [PubMed] [Google Scholar]
- 45.Reiter MJ, Smith WM, Gallagher JJ. Clinical spectrum of ventricular tachycardia with left bundle branch morphology. Am J Cardiol. 1983;51:113–121. doi: 10.1016/s0002-9149(83)80021-6. [DOI] [PubMed] [Google Scholar]
- 46.Guiraudon GM, Klein GJ, Gulaumshein SS, Painvin GA, Del Campo C, Gonzales JC, Ko PT. Total disconnection of right ventricular free wall: surgical treatment of right ventricular tachycardia associated with right ventricular dysplasia. Circulation. 1983;67:463–470. doi: 10.1161/01.cir.67.2.463. [DOI] [PubMed] [Google Scholar]
- 47.Mc Lay JS, Norris A, Campbell RW, Kerr F. Arrhythmogenic right ventricular dysplasia: an uncommon cause of ventricular tachycardia in young and old. Heart. 1993;69:158–160. doi: 10.1136/hrt.69.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Misaki T, Watanabe G, Iwa T, Tsubota M, Ohtake H, Yamamoto K, Watanabe Y. Surgical treatment of arrhythmogenic right ventricular dysplasia: long-term outcome. Ann Thorac Surg. 1994;58:1380–1385. doi: 10.1016/0003-4975(94)91918-6. [DOI] [PubMed] [Google Scholar]
- 49.Imakita M, Yutani C, Ishibashi-Ueda H, Isobe F, Kamiya T. A patient with ventricular tachycardia showing remarkable fatty infiltration and lymphocytic myocarditis in the right ventricular wall. Am J Cardiovasc Pathol. 1990;3:337–340. [PubMed] [Google Scholar]
- 50.Kearney DL, Towbin JA, Bricker JT, Radovancevic B, Frazier OH. Case 5 Familial right ventricular dysplasia (cardiomyopathy) Ped Pathol & Labor Med. 1995;15:181–189. doi: 10.3109/15513819509026952. [DOI] [PubMed] [Google Scholar]
- 51.Smith M, Kickuk MR, Ratliff NB. Clinical and pathologic study of two siblings with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 1999;8:273–278. doi: 10.1016/s1054-8807(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 52.Menghetti L, Basso C, Nava A, Angelini A, Thiene G. Spin-echo magnetic resonance for tissue characterisation in arrhythmogenic right ventricular cardiomyopaty. Heart. 1996;76:467–470. doi: 10.1136/hrt.76.6.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirsch LR, Weinstock DJ, Magid MS, Levin AR, Gold JP. Treatment of presumed arrhythmogenic right ventricular dysplasia in an adolescent. Chest. 1993;104:298–300. doi: 10.1378/chest.104.1.298. [DOI] [PubMed] [Google Scholar]
- 54.Link MS, Wang PJ, Haugh CJ, Homoud MK, Foote CB, Costeas XB, Estes MA., III Arrhythmogenic right ventricular dysplasia: clinical results with implantable cardioverter defibrillators. J Inter Card Electropophysiol. 1997;1:41–8. doi: 10.1023/a:1009714718034. [DOI] [PubMed] [Google Scholar]
- 55.Daliento L, Turrini P, Baratella MC, Nava A. The disease in children and adolescents. In: Nava A, Rossi L, Thiene G, editors. In Arrhymogenic Right Ventricular Cardiomyopathy/Dysplasia. Amsterdam: Elsevier; 1997. pp. 61–68. [Google Scholar]
- 56.Sabel KG, Blomström-Lundqvist C, Olsson SB, Eneström S. Arrhythmogenic right ventricular dysplasia in brother and sister: is it related to myocarditis? Pediatr Cardiol. 1990;11:113–116. doi: 10.1007/BF02239576. [DOI] [PubMed] [Google Scholar]