

Abstract
Noonan syndrome is a common genetic disorder characterized by facial anomalies, congenital heart defect, short stature, webbed neck, chest deformities and undescended testes. The phenotypic expression of Noonan syndrome is extremely variable, with some affected subjects showing only minor features of the syndrome. Cardiac malformations are also heterogeneous. Pulmonary stenosis, with or without dysplastic pulmonary valve and hypertrophic cardiomyopathy, are the “classic” cardiac defects reported in Noonan syndrome. However, atrial septal defect, atrioventricular septal defect, left-sided obstructive lesions, tetralogy of Fallot and patent ductus arteriosus have also been described. Autosomal dominant inheritance has been documented in some families, although many cases appear to be sporadic. The diagnosis of Noonan syndrome is at present purely clinical, because a “diagnostic” test is not available. Indeed, although a gene for Noonan syndrome has been recently mapped by linkage analysis to chromosome 12q, the gene or genes of the syndrome have not been yet cloned.
MeSH: Noonan syndrome, clinical genetics, heart defects, congenital, genetic counselling
Introduction
Noonan syndrome is one of the most common genetic diseases associated with congenital heart defect, being second for frequency only to Down syndrome. The overall incidence of Noonan syndrome is believed to be between 1/1000 and 1/2000 livebirths.1 The syndrome was first recognized as a clinical entity in the sixties by Noonan and Ehmke, when they described several patients with pulmonary stenosis associated with characteristic facial anomalies, short stature, webbed neck, chest deformity and undescended testes.2,3 Pulmonary stenosis with or without dysplastic pulmonary valve and hypertrophic cardiomyopathy are the “classic” cardiac defects reported in Noonan syndrome, but atrial septal defect, atrioventricular septal defect, left-sided obstructive lesions, tetralogy of Fallot and patent ductus arteriosus have also been described.4–17
There is a wide range of variability in the phenotypical expression of Noonan syndrome, with some affected subjects having minimal features.18,19 Noonan syndrome affects both males and females, and the karyotype is normal. Autosomal dominant inheritance with variable expressivity has been documented in many families.19–21 A gene for Noonan syndrome has been mapped by linkage analysis in several families to the long arm of chromosome 12.22 Nevertheless, the gene has not yet been cloned therefore the diagnosis of Noonan syndrome is purely clinical with no “diagnostic” test available.
Clinical features
The main phenotypical features of Noonan syndrome and their occurrence in our series of 190 children are listed in Table 1.
Table 1.
Phenotypical features in 190 patients with Noonan syndrome
Facial anomalies
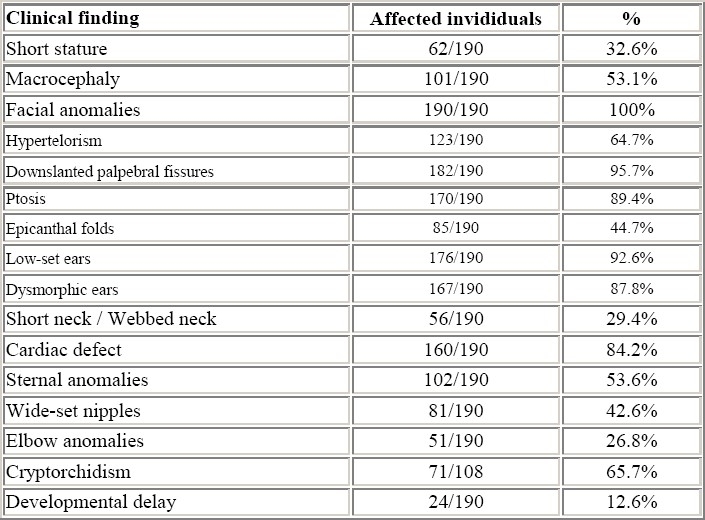
Facial features characteristic of Noonan syndrome are hypertelorism, epicanthic folds, antimongoloid slant of the palpebral fissures, ptosis, flat nasal bridge, micrognathia and low-set rotated ears with a thick helix (fig. 1a, b).
Figure 1.
Frontal (a) and lateral (b) appearance of a patient with Noonan syndrome, showing hypertelorism, flat nasal bridge, palpebral ptosis, thick lips, and low-set rotated ears with thick helix
Nevertheless, the phenotypical expression is highly variable and may change with age (fig. 2a, b, c).18,19,21,23,
Figure 2a.
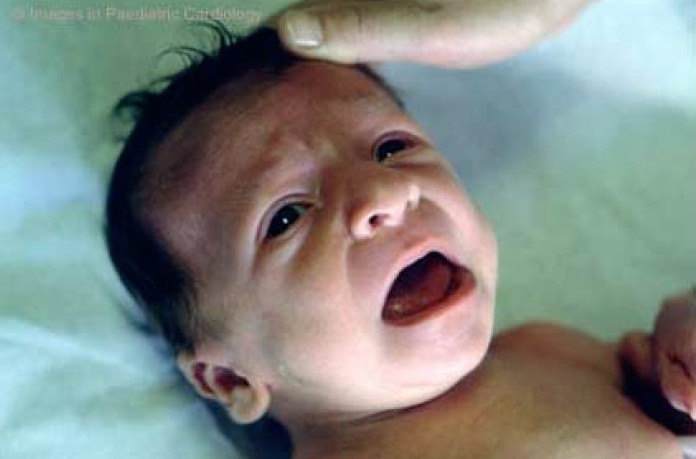
Facial appearance of a male patient with Noonan syndrome at different ages: At 4 months
Figure 2b.
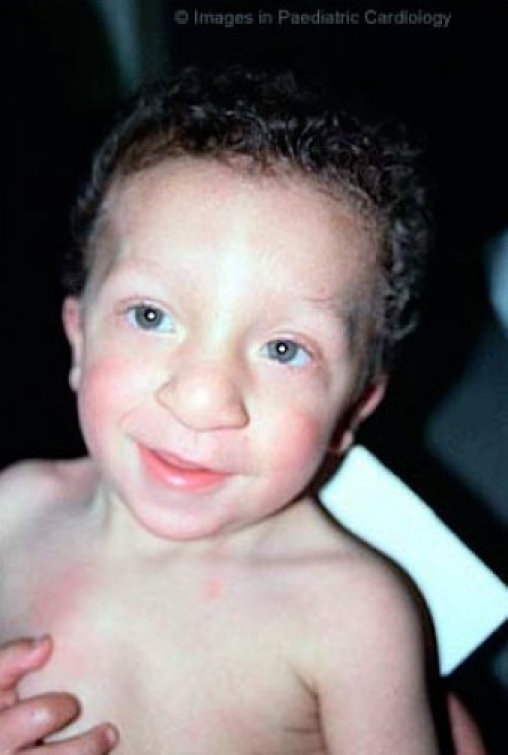
Facial appearance of a male patient with Noonan syndrome at different ages: At 3 years
Figure 2c.
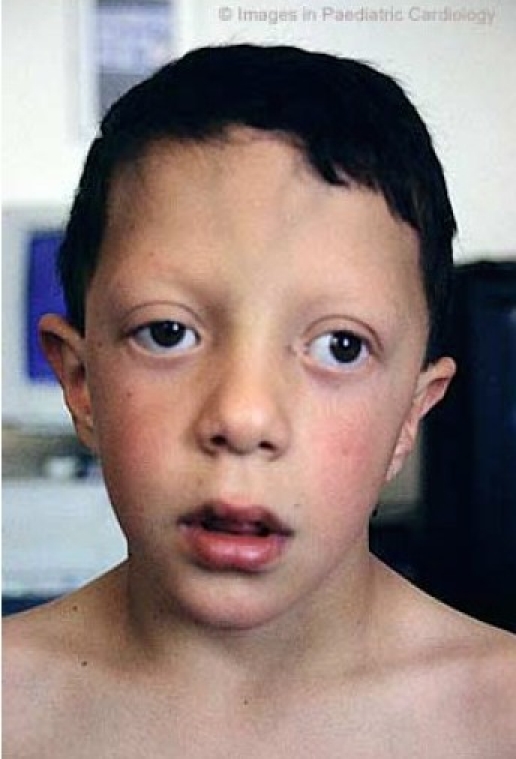
Facial appearance of a male patient with Noonan syndrome at different ages: At 9 years
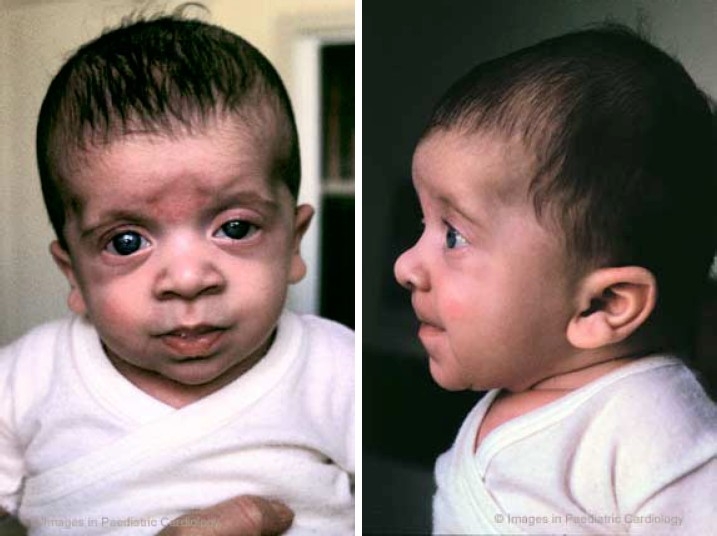
Facial anomalies can often be difficult to recognise in the newborn (fig. 3). In fact, the mean age at diagnosis from the literature is nine years, and the delay in recognising the distinctive characteristics probably reflects the evolving phenotype.1,23 In particular, ocular and nasal bridge anomalies become less evident with increasing age.
Figure 3.
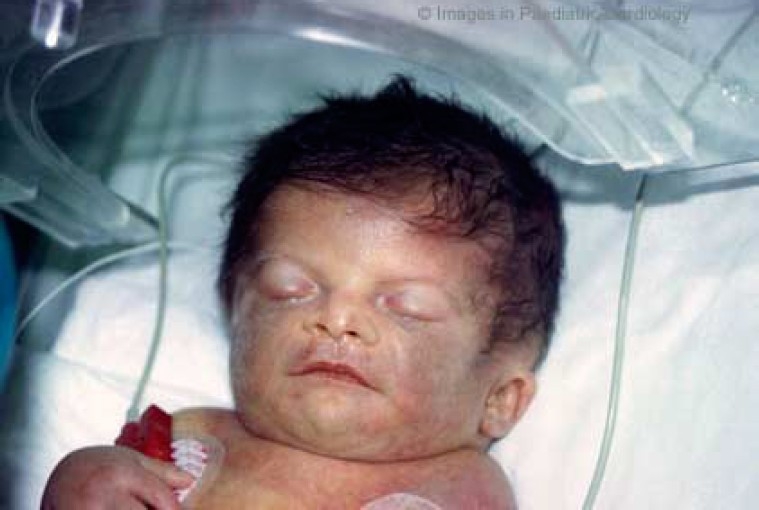
Facial appearance of a newborn with Noonan syndrome
It should also be noted that minor facial changes of Noonan syndrome are often found in one of the parents of affected children.18,21 In genetic counseling of families with a child with Noonan syndrome, in which one of the parents has mild features of the syndrome, a review of childhood photographs of the mother or the father may be a useful aid, particularly if resemblance to the affected offspring is shown.
Congenital heart defect
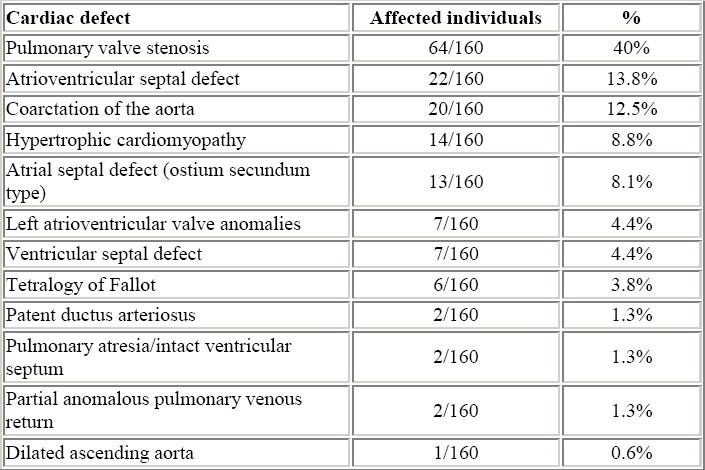
The prevalence of congenital heart defect in Noonan syndrome varies from 50 to 80% in the reported series from the literature.4–9 The percentage of occurrence of the different types of cardiac defect in our experience is shown in Table 2. Pulmonary stenosis and hypertrophic cardiomyopathy are generally the most common congenital heart defects found in Noonan syndrome.
Table 2.
Cardiac defects found in 160 individuals with Noonan syndrome
Pulmonary stenosis is often associated with a thickened and dysplastic valve. It is usually difficult to obtain a satisfactory result using the transcatheter balloon dilatation of such dysplastic valves, so surgical intervention is more likely to be needed.
Hypertrophic cardiomyopathy involves predominantly the ventricular septum as asymmetric septal hypertrophy, but may also affect the ventricular free walls. Left ventricular outflow tract obstruction may occasionally be produced.
Atrial septal defect, patent ductus arteriosus and tetralogy of Fallot are also found in Noonan syndrome. In our experience, left-sided obstructive lesions and atrioventricular septal defect are also frequently diagnosed. Left-sided anatomic obstruction can occur at the valvular or supravalvular level,9 in the subaortic position as a result of left atrioventricular valve abnormalities,11–12, or as coarctation of the aortic.13–15. Interestingly, the subgroup of patients with Noonan syndrome and aortic coarctation demonstrates male preponderance, and physical manifestations overlapping with those of Turner syndrome, so that the involvement of putative lymphogenic genes located on sex chromosomes has been suggested in these patients.24 The atrioventricular septal defect of subjects with Noonan syndrome is generally “partial” and frequently associated (25% of the cases in our series) with subaortic stenosis.16 The structural abnormalities causing subaortic stenosis include accessory fibrous tissue and/or anomalous insertion of the left atrioventricular valve and anomalous papillary muscle of the left ventricle. Interestingly, the anomalies of the left atrioventricular valve leaflets and of the subvalvular apparatus in patients with atrioventricular septal defect are similar to those reported in patients with hypertrophic cardiomyopathy.25,26
The pathogenesis of cardiac defects in Noonan syndrome has been attributed to a defect of cardiac jelly and extracellular matrix, in some cases,27 and the same mechanism is probably involved in the pathogenesis of the atrioventricular septal defect.28 Additionally, the development of leaflets and papillary muscles of the left atrioventricular valve is related to the morphogenesis of the left ventricular outflow tract and the atrioventricular and interventricular septations.29–32 Thereafter, in some patients with Noonan syndrome, the developmental mechanism of the left ventricular myocardium and the left atrioventricular valve may be altered.
Rare cardiac defects occurring in Noonan syndrome are pulmonary atresia with intact ventricular septum, which should be considered in the spectrum of pulmonary valve stenosis with dysplasia of the leaflets,33,34 and dilated ascending aorta.35
Neck anomalies
An obvious and evident webbing of the neck (fig. 4a, b), is generally present in only one fourth of the cases, while an apparent neck shortening with redundant skin on the neck and low posterior hairline are more commonly noted.
Figure 4a.
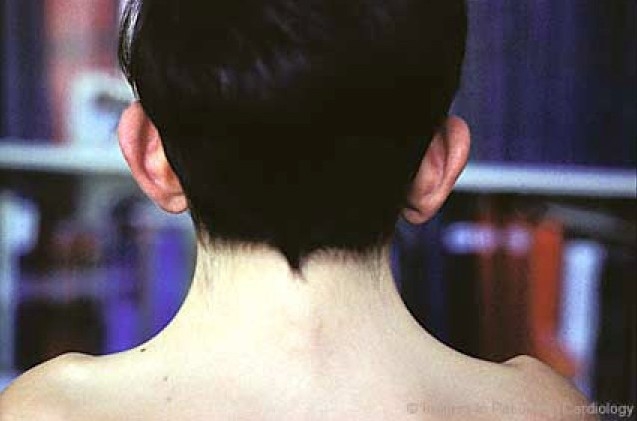
Webbed neck (a) and low posterior hairline (b) in a child with Noonan syndrome: Webbed neck
Figure 4b.
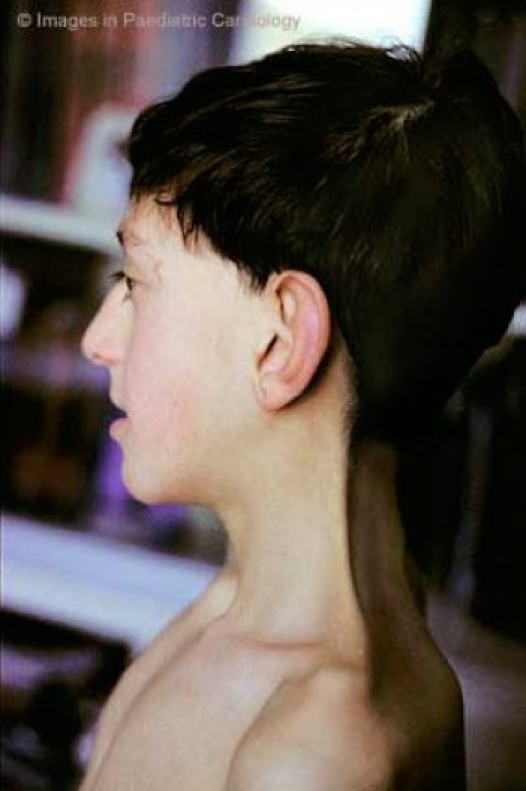
Webbed neck (a) and low posterior hairline (b) in a child with Noonan syndrome: Low posterior hairline
Skeletal anomalies
Thoracic deformities include the classic sternal changes of Noonan syndrome, often associated with shield chest (Figure 5). Kyphosis and thoracic scoliosis are frequently found, although medical or surgical treatment are rarely required. Bilateral elbow deformity is a frequent upper limb abnormality.
Figure 5.
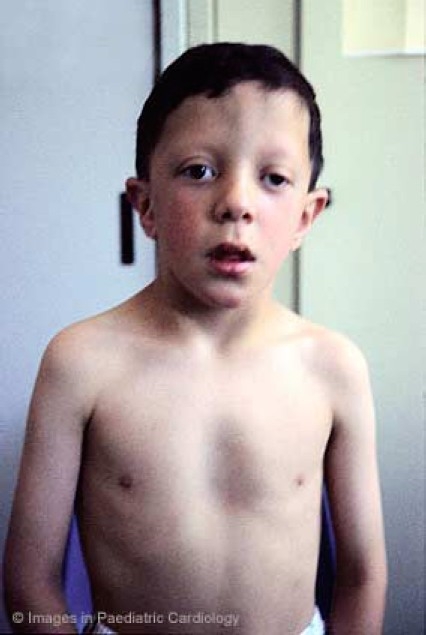
Characteristic thoracic and sternal deformities of Noonan syndrome
Genital anomalies
Up to three quarters of boys with Noonan syndrome may have undescended testes.23 Cryptorchidism adversely affects male fertility. Azoospermia or oligozoospermia may occasionally be found following orchidopexy for bilateral undescended testes.36 It remains to be seen whether earlier surgery would improve the outcome. In the presence of normally descended testes, men with Noonan syndrome are likely to be fertile. Pubertal development is delayed is both sexes, and fertility does not appear to be affected in females.
Developmental anomalies
The majority of children with Noonan syndrome (65% to 85% according to different series) have normal mental capacities and attend regular schools.1,21,23,37 Early delay in motor milestones could probably be explained by the presence of hypotonia and hyperextensibility in the younger child.
A recent study, investigating patterns of cognitive functioning in school-aged children, showed that Noonan syndrome is not associated with substantial deficits at the level of intellectual functioning.37 However, the correlation between phenotypic and cognitive expression revealed that a severe Noonan syndrome phenotype, i.e. more severe cardiac defect, more evident facial and skeletal anomalies, was associated with a specific pattern of deficits and capacities in cognitive functioning.37 Generally, patients with Noonan syndrome are at risk for speech delay,38,39 which could be related to hearing loss or recurrent otitis media that frequent occurs in this syndrome. Tests for hearing are recommended in children with Noonan syndrome, as are tests for vision, because of the association of strabismus and refractive errors in this syndrome.23
Growth
Growth retardation is an important feature of Noonan syndrome, and short stature is found in 50-70% of cases in previously reported series.21,23,40 In our experience (Table 1), the percentage of subjects with height below the 3rd percentile is lower. We believe that in older series, patients with a more complete expression of the syndrome may have been enrolled, while the more recent reports include also patients with milder phenotype Noonan syndrome such that the impact of growth abnormalities may be less evident.
The pattern of growth is similar in males and females, and weight-height ratio is normal. A delayed pubertal growth spurt has been documented in both sexes. Bone age is also usually delayed. The mean adult height reaches, in general, the lowest level of the normal percentiles.
Non-cyanotic heart defects are not found to have any major impact on overall growth patterns.
Coagulation factor deficiencies
Easy bruising after minor trauma and abnormal bleeding are frequently reported in patients with Noonan syndrome, and a variety of coagulation factor deficiencies have been detected.41 The most common abnormality is a partial factor XI deficiency, but VIII and XII factor anomalies have also been described. Combined coagulation factor deficiencies may also occur. The involvement of several factors, and the possible presence of combined abnormalities implies that anomalies in regulatory factors of the coagulation system, which is under chromosome genetic control may be the cause.
Familial transmission
Approximately half of the patients are sporadic cases in the families and may represent new mutations. In the remaining 50% of cases, Noonan syndrome is familial, with most of the pedigrees being consistent with autosomal dominant inheritance.2,3,15,19,22,42 A mild or subtle phenotype must be searched for in parents of affected children.
A predominance of maternal transmission is noted in familial cases. This has been thought to be due to infertility in affected males, which may be related to cryptorchidism.36
Molecular studies
The result of a linkage analysis of a three-generation family with eight affected individuals and 20 two-generation families performed by Jamieson et al.22 localised a gene for Noonan syndrome to chromosomal region 12q22qter. Nevertheless, the gene is at present not cloned, so that a laboratory test to confirm a clinical diagnosis of Noonan syndrome is not yet available. In searching the gene for Noonan syndrome, it would be interesting to look at candidate genes that might explain cardiac defects, particularly valvular abnormalities and cardiac hypertrophy.
Prenatal diagnosis
Prenatal diagnosis by molecular testing is not possible. Nevertheless ultrasonographic findings can suggest the diagnosis in utero. Reported sonographic signs include cystic hygroma, hydrothorax, polyhydramnios, and congenital heart defect.43 However, Noonan syndrome has an evolving phenotype, since early nuchal region findings can regress in later pregnancy and detectability of characteristic cardiac defects of Noonan syndrome can be low, since pulmonary stenosis can be difficult to diagnose in utero and asymmetric septal hypertrophy is a progressive anomaly that becomes more evident postnatally. The variability of the phenotype must also be taken in consideration, since patients with mild phenotypic expression have only subtle sonographic manifestations, which can be overlooked. Conversely, it has been noted that second-trimester manifestations may predict a severe postnatal course.
Conclusions
Noonan syndrome is a genetic condition with characteristic phenotypic anomalies, but a wide range of clinical expression. Cardiac malformations are also heterogeneous. At present, it is not known whether clinical variability reflects variable expression of the same genetic defect or, on the contrary, genetic heterogeneity may be involved by causing similar phenotypes due to different genetic entities.
It is important to point out that clinical aspects and associated malformations of Noonan syndrome can be early identified and favourably treated. Patients with Noonan syndrome can have good physical health and a normal grade of intelligence, with a positive integration in social life.
Although a gene for Noonan syndrome has been mapped by linkage analysis to chromosome 12q, the gene or genes of the syndrome have not been yet identified. The association of Noonan syndrome with peculiar types of cardiac defects could be used to address the search for the gene of Noonan syndrome to specific cardiac candidate genes. It is hopful that the rapidly evolving progresses in genetic mapping and identification could lead also to the understanding of the pathogenetic mechanism of this peculiar and fascinating syndrome.
References
- 1.Allanson JE. Noonan syndrome. J Med Genet. 1987;24:9–13. doi: 10.1136/jmg.24.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noonan JA, Ehmke DA. Associated non cardiac malformations in children with congenital heart disease. J Pediatr. 1963;63:468–470. [Google Scholar]
- 3.Noonan JA. Hypertelorism with Turner phemotype. A new syndrome with associated congenital heart disease. Am J Dis Child. 1968;116:373–380. doi: 10.1001/archpedi.1968.02100020377005. [DOI] [PubMed] [Google Scholar]
- 4.Nora JJ, Torres FG, Sinha AK, McNamara DG. Characteristic cardiovascular anomalies of XO Turner syndrome, XX and XY phenotype, and XO/XX Turner mosai. Am J Cardiol. 1970;25:639–641. doi: 10.1016/0002-9149(70)90612-0. [DOI] [PubMed] [Google Scholar]
- 5.Nora JJ, Lortscher RH, Spangler RD. Echocardiographic studies of left ventricular disease in Ullrich-Noonan syndrome. Am J Dis Child. 1975;129:1417–1420. doi: 10.1001/archpedi.1975.02120490031010. [DOI] [PubMed] [Google Scholar]
- 6.Siggers DC, Polani PE. Congenital heart disease in male and female subjects with somatic features of Turner's syndrome and normal sex chromosomes (Ullrich's and related syndromes) Br Heart J. 1972;34:41–46. doi: 10.1136/hrt.34.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phornphutkul C, Rosenthal A, Nadas AS. Cardiomyopathy in Noonan syndrome. Br Heart J. 1973;35:99–102. doi: 10.1136/hrt.35.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van der Hauwaert LG, Fryns JP, Dumoulin M, Logghe N. Cardiovascular malformations in Turner's and Noonan's syndrome. Br Heart J. 1978;40:500–509. doi: 10.1136/hrt.40.5.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burch M, Sharland M, Shinebourne E, Smith G, Patton M, McKenna W. Cardiologic abnormalities in Noonan syndrome: phenotypic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment in 119 patients. J Am Coll Cardiol. 1993;22:1189–1192. doi: 10.1016/0735-1097(93)90436-5. [DOI] [PubMed] [Google Scholar]
- 10.Digilio MC, Marino B, Giannotti A, Dallapiccola B. Exclusion of 22q11 deletion in Noonan syndrome with tetralogy of Fallot. Am J Med Genet. 1996;62:413–414. doi: 10.1002/ajmg.1320620404. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch HD, Gelband H, Garcia O, Gottlieb S, Tamer DM. Rapidly progressive obstructive cardiomyopathy in infants with Noonan's syndrome. Circulation. 1975;52:1161–1165. doi: 10.1161/01.cir.52.6.1161. [DOI] [PubMed] [Google Scholar]
- 12.Marino B, Gagliardi MG, Digilio MC, Polletta B, Grazioli S, Agostino D, Giannotti A, Dallapiccola B. Noonan syndrome: structural abnormalities of the mitral valve causing subaortic obstruction. Eur J Pediatr. 1995;154:949–952. doi: 10.1007/BF01958636. [DOI] [PubMed] [Google Scholar]
- 13.Marino B, Digilio MC, Giannotti A, Dallapiccola B. Noonan syndrome with left-sided cardiac obstructions. Am J Hum Genet. 1996;59(suppl):A98. doi: 10.1007/s004390050357. [DOI] [PubMed] [Google Scholar]
- 14.Digilio MC, Marino B, Giannotti A, Dallapiccola B. Noonan syndrome with cardiac left-sided obstructive lesions. Hum Genet. 1997;99:289. doi: 10.1007/s004390050357. [DOI] [PubMed] [Google Scholar]
- 15.Digilio MC, Marino B, Picchio F, Prandstraller D, Toscano A, Giannotti A, Dallapiccola B. Noonan syndrome and aortic coarctation. Am J Med Genet. 1998;80:160–162. [PubMed] [Google Scholar]
- 16.Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–706. doi: 10.1016/s0022-3476(99)70088-0. [DOI] [PubMed] [Google Scholar]
- 17.Danetz JS, Donofrio MT, Embrey RP. Multiple left-sided cardiac lesions in one of Noonan's original patients. Cardiol Young. 1999;9:610–612. doi: 10.1017/s1047951100005679. [DOI] [PubMed] [Google Scholar]
- 18.Allanson JE, Hall JG, Hughes HE, Preus M, Witt RD. Noonan syndrome: The changing phenotype. Am J Med Genet. 1985;21:507–514. doi: 10.1002/ajmg.1320210313. [DOI] [PubMed] [Google Scholar]
- 19.Sharland M, Morgan M, Smith G, Burch M, Patton MA. Genetic counselling in Noonan syndrome. Am J Med Genet. 1993;45:437–440. doi: 10.1002/ajmg.1320450407. [DOI] [PubMed] [Google Scholar]
- 20.Nora JJ, Nora AH, Sinha AK, Spangler RD, Lubs HA. The Ullrich Noonan syndrome (Turner phenotype) Am J Dis Child. 1974;127:48–55. doi: 10.1001/archpedi.1974.02110200050007. [DOI] [PubMed] [Google Scholar]
- 21.Mendez HMM, Opitz JM. Noonan syndrome: A review. Am J Med Genet. 1985;21:493–506. doi: 10.1002/ajmg.1320210312. [DOI] [PubMed] [Google Scholar]
- 22.Jamieson CR, van der Burgt I, Brady AF, van Reen M, Elsawi MM, Hol F, Jeffery S, Patton MA, Mariman E. Mapping a gene for Noonan syndrome to the long arm of chromosome 12. Nat Genet. 1994;8:357–360. doi: 10.1038/ng1294-357. [DOI] [PubMed] [Google Scholar]
- 23.Sharland M, Burch M, McKenna WM, Patton MA. A clinical study of Noonan syndrome. Arch Dis Child. 1992;67:178–183. doi: 10.1136/adc.67.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasegawa T, Ogata T, Hasegawa Y, Honda M, Nagai T, Fukushima Y, Nakahori Y, Matsuo N. Coarctation of the aorta and renal hypoplasia in a boy with Turner/Noonan surface anomalies and a 46,XY karyotype: A clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata. Hum Genet. 1996;97:564–567. doi: 10.1007/BF02281861. [DOI] [PubMed] [Google Scholar]
- 25.Klues HG, Roberts WC, Maron BJ. Anomalous insertion of papillary muscle directly into anterior mitral leaflet in hypertrophic cardiomyopathy. Significance in producing left ventricular outflow obstruction. Circulation. 1991;84:1188–1197. doi: 10.1161/01.cir.84.3.1188. [DOI] [PubMed] [Google Scholar]
- 26.Klues HG, Maron BJ, Dollar AL, Roberts WC. Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation. 1992;85:1651–1660. doi: 10.1161/01.cir.85.5.1651. [DOI] [PubMed] [Google Scholar]
- 27.Amman G, Sherman FS. Myocardial dysgenesis with persistent sinusoids in a neonate with Noonan's phenotype. Pediatr Pathol. 1992;12:83–92. doi: 10.3109/15513819209023283. [DOI] [PubMed] [Google Scholar]
- 28.Clark EB. Mechanisms in the pathogenesis of congenital heart defects. In: Pierpont ME, Moller JM, editors. The genetics of cardiovascular disease. Boston: Martinus-Nijoff; 1986. pp. 3–11. [Google Scholar]
- 29.De La Cruz MV, Giménez-Ribotta M, Saravalli O, Cayré R. The contribution of the inferior endocardial cushion of the atrioventricular canal to cardiac septation and to the development of the atrioventricular valves: study in the chick embryo. Am J Anat. 1983;166:63–72. doi: 10.1002/aja.1001660105. [DOI] [PubMed] [Google Scholar]
- 30.Garcia Pelàez I, Dìaz Gòngora G, Artega Martinez M. Contribution of the superior atrioventricular cushion to the left ventricular infundibulum. Experimental study on the chick embryo. Acta Anat. 1984;118:224–230. [PubMed] [Google Scholar]
- 31.Oosthoek PW, Wenink ACG, Wisse LJ, Gittenberger-de Groot AC. Development of the papillary muscles of the mitral valve: morphogenetic background of parachute-like asymmetric mitral valves and other mitral valve anomalies. J Thorac Cardiovasc Surg. 1998;116:36–46. doi: 10.1016/S0022-5223(98)70240-5. [DOI] [PubMed] [Google Scholar]
- 32.Webb S, Brown NA, Anderson RH. Formations of the atrioventricular septal structures in the normal mouse. Circ Res. 1998;82:645–656. doi: 10.1161/01.res.82.6.645. [DOI] [PubMed] [Google Scholar]
- 33.Klinge T, Laursen HB. Familial pulmonary stenosis with underdeveloped or normal right ventricle. Br Heart J. 1975;37:60–64. doi: 10.1136/hrt.37.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dennis NR, Warren J. Risks to offspring of patients with some common congenital heart defects. J Med Genet. 1981;18:8–16. doi: 10.1136/jmg.18.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin AE, Garver KL, Allanson J. Aortic-root dilatation in Noonan's syndrome. New Engl J Med. 1987;317:1668–1669. doi: 10.1056/NEJM198712243172616. [DOI] [PubMed] [Google Scholar]
- 36.Elsawi MM, Pryor JP, Klufio G, Barnes C, Patton MA. Genital tract function in men with Noonan syndrome. J Med Genet. 1994;31:468–470. doi: 10.1136/jmg.31.6.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van der Burgt I, Thoonen G, Roosenboom N, Assman-Hulsmans C, Gabreels F, Otten B, Brunner HG. Patterns of cognitive functioning in school-aged children with Noonan syndrome associated with variability in phenotypic expression. J Pediatr. 1999;135:707–713. doi: 10.1016/s0022-3476(99)70089-2. [DOI] [PubMed] [Google Scholar]
- 38.Wilson M, Dyson A. Noonan syndrome: Speech and language characteristics. J Comm Disorders. 1982;15:347–352. doi: 10.1016/0021-9924(82)90002-8. [DOI] [PubMed] [Google Scholar]
- 39.Cornish KM. Verbal-performance discrepancies in a family with Noonan syndrome. Am J Med Genet. 1996;66:235–236. doi: 10.1002/(SICI)1096-8628(19961211)66:2<235::AID-AJMG21>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 40.Ranke MB, Heidemann P, Knupfer C, Enders H, Schmalz AA, Bierich JR. Noonan syndrome: growth and clinical manifestations in 144 cases. Eur J Pediatr. 1988;148:220–227. doi: 10.1007/BF00441408. [DOI] [PubMed] [Google Scholar]
- 41.Sharland M, Patton MA, Talbot S, Chitolie A, Bevan DH. Coagulation-factor deficiencies and abnormal bleeding in Noonan's syndrome. Lancet. 1992;339:19–21. doi: 10.1016/0140-6736(92)90141-o. [DOI] [PubMed] [Google Scholar]
- 42.Collins E, Turner G. The Noonan syndrome - a review of the clinical and genetic features of 27 cases. J Pediatr. 1973;83:941–950. doi: 10.1016/s0022-3476(73)80527-x. [DOI] [PubMed] [Google Scholar]
- 43.Achiron R, Heggesh J, Grisaru D, Goldman B, Lipitz S, Yagel S, Frydman M. Noonan syndrome: a cryptic condition in early gestation. Am J Med Genet. 2000;92:159–165. doi: 10.1002/(sici)1096-8628(20000529)92:3<159::aid-ajmg1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]