

Abstract
Marfan syndrome is an autosomal dominant heritable connective tissue disorder which involves primarily the skeletal, ocular and cardiovascular system. The incidence of MS is on average 1: 10000 with 25-30% of cases caused by sporadic mutations.
The leading cause of premature death in these patients is progressive dilatation and subsequent dissection of the ascending thoracic aorta resulting in cardiac tamponade, and left ventricular failure due to aortic regurgitation. Life expectancy is primarily determined by the severity of cardiovascular involvement, and has improved substantially over the last 20 years due to the advances in surgical and medical management.
The optimum management of Marfan patients includes a lifelong surveillance with particular emphasis placed on aortic behaviour. Preventive replacement of various portions of the aorta has been a major contribution for improved life expectancy in these patients. The different surgical and interventional treatment options currently available will be further outlined in this review.
MeSH: Marfan syndrome, Surgical techniques, Thoracic Surgery, Long term outcome, Cardiovascular complications
Introduction
In 1896 Antoine Marfan, a professor of paediatrics, presented for the first time a 5 year old girl with the phenotype of a connective tissue disorder. In 1931 the mode of inheritance with an autosomal dominant trait was discovered.
During the 20th century, additional components of this syndrome became apparent, particularly the cardiovascular involvement with dilatation of the aortic root and the high risk of subsequent aortic dissection which was first reported in 1943.1 Diagnosis mainly relied on clinical criteria, until recently when the underlying gene defect on chromosome 15q21 was discovered in 1990.2
Cardiothoracic surgery, has become an accepted and proven treatment for elective as well as emergency surgery for aortic aneurysm and dissection, and thereby can be considered as the major contributing factor for the increased life expectancy in patients with Marfan syndrome (MFS).
Pathophysiolgy and genetics
By immunhistochemical studies of Marfan patients it was shown, that most of the patients with MFS display an abnormal pattern of microfibrils in the extracellular matrix. Further studies revealed a 350 kD protein named Fibrillin which was characterised to be the main component of the extracellular microfibrils, to be responsible for these abnormalities.3 Subsequently the fibrillin gene could be mapped to the long arm of chromosome 15 and in situ hybridisation assigned the gene locus to 15q15-21.2 However, genetic testing proved to be less reliable than expected, with only 66% mutation detection rate of patients diagnosed with MFS by the Ghent nosology. The reason for these fair results are based on the fact that there are a variable number of mutations, which precludes broad spectrum genetic screening. Furthermore Fibrillin mutations have also been detected in Marfan related disorders.3
Diagnosis
In 1986 the Berlin nosology of heritable connective tissue disorder was published which classified certain organ manifestations into groups with major and minor criteria.4 Further refinements were included in the Gent criteria and recently revised by De Paepe et al in 1996.5 The criteria are subdivided into the following organ systems: skeletal, ocular, cardiovascular, pulmonary, skin, dura and family respectively genetic history. For a clinical diagnosis of MS, in the absence of a positive family history, a person suspected to have MS should at least display major criteria in two organ systems and involvement of a third organ system. When patients have a positive family history, major criteria in one and minor criteria in another organ system is required.5
Particular emphasis should be placed on the predominance of skeletal features in childhood, as this can lead to earlier diagnosis and more appropriate surveillance, of cardiovascular status. In younger patients with a family history of MFS who do no fulfil the diagnostic criteria as well as young patients who fail to meet criteria in one organ system are required to repeat evaluations on a regular basis until the age of 18.
Cardiovascular complications usually do not occur before the age of 16 years. A this point the aortic root dimensions approached the adult size so that one can refer to the recommendations for the adult population regarding aortic root dimensions which should be always measured in relation to body surface area.6
Infrequently, the infantile form of Marfan syndrome occurs, which is characterised by mitral valve regurgitation and aortic root dilatation which become apparent during the first three months of life. Mortality is substantial with 14% of affected children dying during the first year of life.7
Conservative and surgical considerations
Cardiovascular manifestations in MFS include mitral valve prolapse and regurgitation, left ventricular dilatation, pulmonary artery dilatation, and aortic valve regurgitation secondary to aortic root dilatation which can be regarded the most common cause of morbidity through the increased risk of dissection and rupture when the aortic root diameter exceeds 5.5 cm. Obviously the outcome of prophylactic aortic root surgery is superior over emergency surgery in the case of aortic dissection.
Dissection involving the ascending aorta is an absolute indication for operation with the intention to replace the sinus of Valsalva and the ascending aorta and eventually, if affected, the aortic arch (figures 1–3). Treatment options regarding the aortic valve are outlined below.
Figure 1.
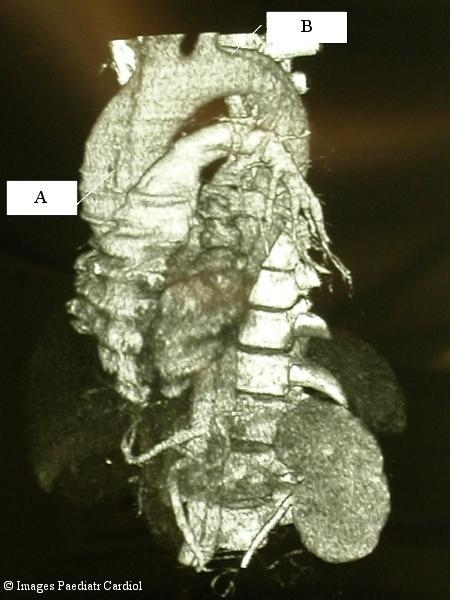
Three dimensional CT reconstruction of an acute aortic dissection type A extending until the ilac arteries. The primary entry tear in the ascending aorta is seen (A). The re-entry just distal the left subclavian artery is also seen (B).
Figure 3.
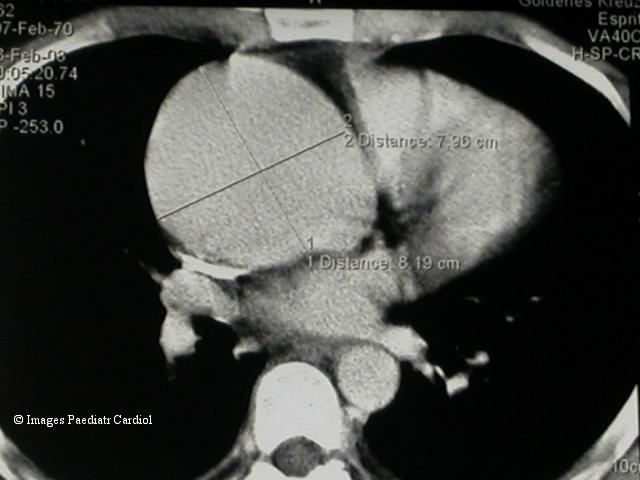
CT Scan showing a huge ascending aortic aneurysm with a maximum diameter of 8.2 cm.
Figure 2.
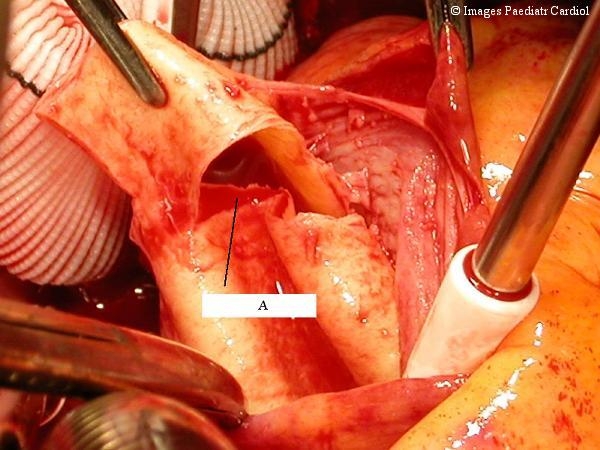
Intraoperative view of an acute type A dissection. One can clearly see the site of the entry tear in the intima 1.5 cm above the aortic valve (A).
The exact timing of operation is crucial in a Marfan patient. The following parameters should be taken into consideration when planning surgery:
Diameter of the ascending thoracic aorta: aneurysms larger than 6 cm have a 10% increased risk of rupture. Elective replacement indication for Marfan patients is suggested when the thoracic aorta approaches 5 cm or 6 cm in descending portions. Commonly a diameter approaching 5 cm is considered the upper limit for conservative treatment strategies.
Progression of enlargement: subsequent progression of aortic dimensions mandates early replacement.
Family history of dissection
Grade of aortic valve regurgitation. Operation should be performed before an increase in left ventricular end systolic dimension is noted, in order to prevent irreversible damage to the left ventricle and to improve long term prognosis.
Surgical techniques
A recent survey of 10 major Marfan surgical centres reported an overall operative mortality rate of 1.5% in patients undergoing elective root replacement. However, differences became apparent when emergency procedures were performed for acute dissection which resulted in an eight fold increased mortality rate (11.6%) in contrast to elective surgery.8
Ascending thoracic aorta
In 1968 the composite graft technique with replacement of the aortic valve, the sinus of Valsalva and the ascending aorta was introduced by Bentall and de Bono.9 Briefly, this technique consists of a composite graft consist of a mechanical valve inserted into a gelatine impregnated Dacron graft that comes preassembled. The coronary arteries are reimplanted as buttons on to the graft. However, in order to avoid life long anticoagulation, subsequent efforts were directed towards preserving the aortic valve and thereby obviating the need for continuous anticoagulation. Valve sparing aortic root replacements were subsequently introduced by Yacoub as remodelling techniques in 1979 and by David with the reimplantation procedure in 1988.10,11 Preservation of the aortic valve has many advantages namely preservation of the dynamic structure of the outflow tract which influences left ventricular function, coronary flow and cardiac output.12 A recent study by de Oliviera and colleagues compared aortic valve reimplantation versus remodelling of the aortic root technique; the reimplantation group emerged as more appropriate for prevention of further root dilatation.13 Even though some concern had been raised concerning the durability of this procedure as the aortic leaflets are involved in fibrillin deficiency, long term results have been encouraging (figures 4,5).13
Figure 4.
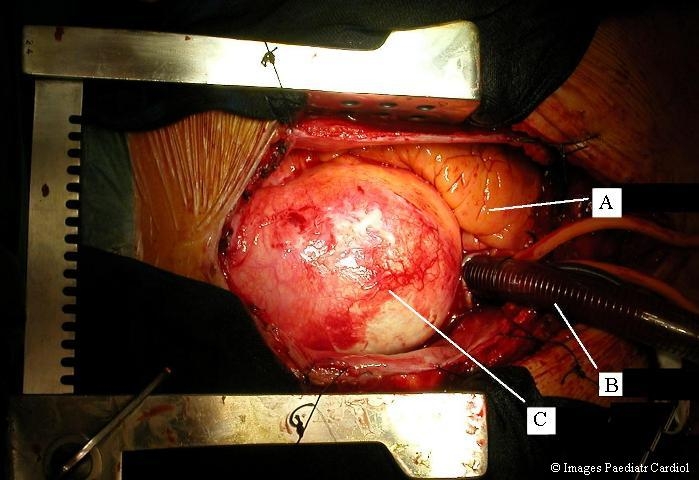
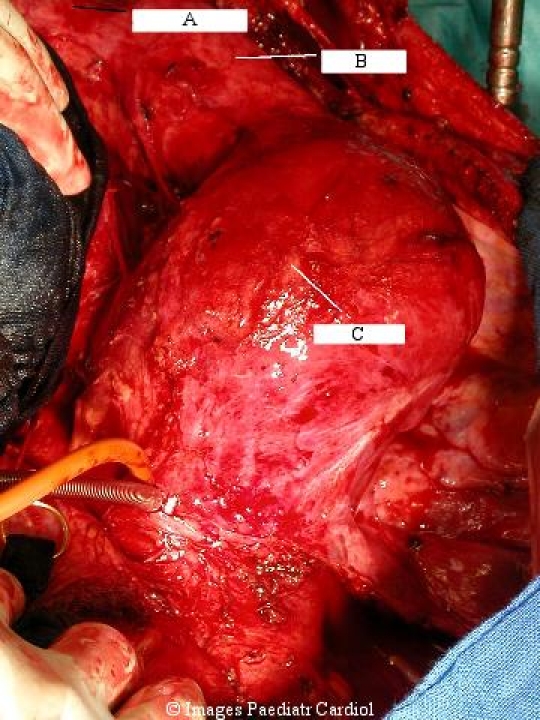
Ascending aortic aneurysm intraoperative view from the patient in figure 3. A=right ventricle, B=venous cannula in right atrium, C=ascending aortic aneurysm
Figure 5.
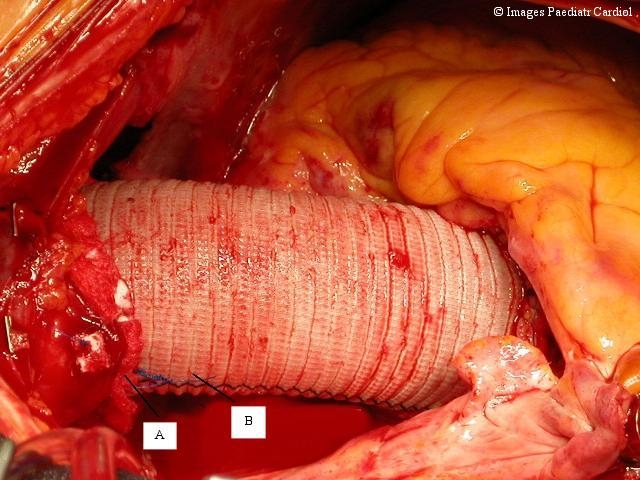
Intraoperative view of a replaced ascending aorta with a gelatine coated Dacron prosthesis (A). Anastomoses are usually augmented with Teflon felt strips (B).
Furthermore as most patients with MFS necessitate subsequent vascular or orthopaedic interventions, the avoidance of anticoagulation results in a diminished risk of bleeding complications.
Descending thoracic aorta
The risk of additional aneurysm formation after intervention on the ascending aorta is around 50%. The majority of patients need further surgery in the remaining aorta followed by the iliac and subclavian artery. In a previous study by Finkbohner and colleagues, it was determined that the presence of aortic dissection, hypertension and history of smoking are risk factors for the development of secondary aneurysms. Moreover, patients with a dissection at the time of first surgery were more likely to require subsequent surgery on the remaining aorta as outlined in the current study. The ascending thoracic aorta was the site of first operation in 84% of their patients, followed by the descending thoracic aorta and the abdominal aorta. Median age at the time of first operation was 32 years.14
A recent publication by Coselli et al reported a favourable experience of replacement of the ascending thoracic aorta with contemporary techniques in 50 Marfan patients with an overall complication rate of 5% and 96% 30 day survival.15
Endovascular stent grafting (ESG) of the descending aorta emerged as promising less invasive alternative for conventional open surgical repair.16 Particularly in patients with MFS conservative treatment as usually advocated in an uncomplicated acute aortic dissection type B seems less reliable because of the fragility of the aortic wall. Therefore more aggressive treatment methods are warranted. With the implementation of ESG it became feasible to treat those patients, who would otherwise have to undergo emergency surgery with an increased risk of complications. Moreover the presence of ESG does not preclude further surgery on the aorta, as the can be easily removed.
However, long term results for ESG are still limited. In our series of 80 patients treated with ESG for thoracic aortic disease 4 (5%) needed further open surgical repair. Particularly, we do not recommend ESG for descending aortic aneurysm in MFS as through the decreased elasticity of the aortic wall, the radial forces generated by the expanded stent graft may result in endoleak formation or potential rupture. Longer follow up is definitely needed to predict more accurately the long term durability of these devices (figure 6).
Figure 6.
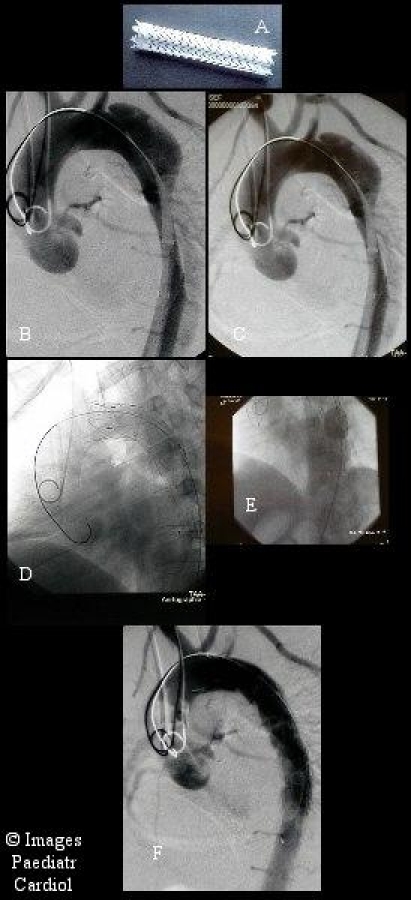
Acute type B Dissection beginning distal the left subclavian artery A. A typical stent-graft consists of a nitinol wire stent shaped in a zigzag formation, which is covered with extra-thin polyester (Dacron) A straight nitinol wire secures the length of the device and avoids twisting or kinking. This self-expandable stent-graft is compressed over a placement catheter. B. Both the stent-graft and the catheter are loaded into a polyurethane sheath for insertion. C. The endoluminal stent-graft system is passed over the guidewire and positioned at the desired location as determined by intraoperative angiography. D. After exact positioning, the stent-graft is released by removing the sheath. E. Exact modelling of the stent onto the aortic wall with a balloon. F. Successful exclusion of the entry tear with no detectable filling of the aneurysmal sack.
Open repair of the thoracoabdominal aorta with Left heart bypass and selective visceral perfusion
Aneurysms of the thoracoabdominal aorta (TAAA) are mainly the long term result of a chronic dissected aorta (figures 7,8).
Figure 7.
TAAA aneurysm with involvement of the entire descending thoracic and abdominal aorta. A: Proximal descending aorta. B: Distal descending aorta. C: Abdominal aorta
Figure 8.
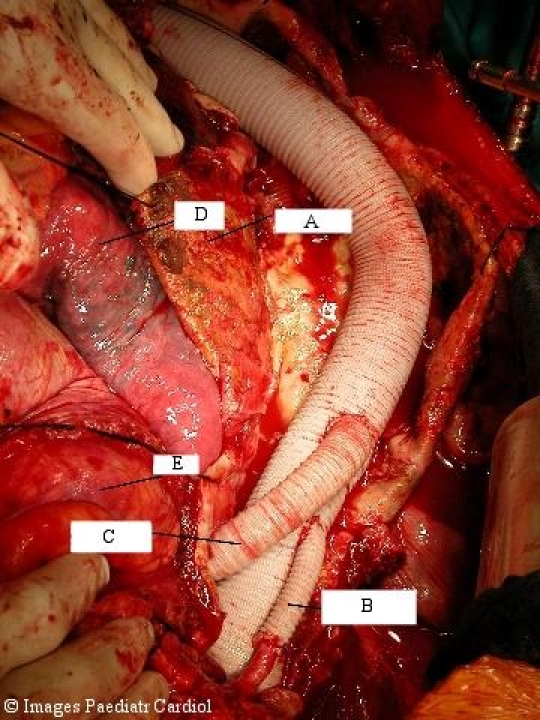
Same patient after replacement of the TAAA with a Dacron prosthesis. A=Aneurysm sac. The visceral and renal vessels have been reimplanted as patch. One renal artery was implanted via an 8 mm Dacron graft (B). Intercostal arteries, especially in the region from Th 8 to L2 were reimplanted to reduce the risk of postoperative spinal cord ischemia and paraplegia (C). D=Lung, E=Bowel.
Extended repair of a TAAA was associated with high mortality and complication rates until Coselli developed a technique with the aid of left heart bypass, sequential clamping and selective visceral perfusion to obviate the need for cardiopulmonary bypass and the associated bleeding problems and on the other hand improve end organ perfusion, especially of the spinal cord in order to reduce the incidence of postoperative paraplegia.15
Conclusions
The primary life threatening complication in MFS patients is rupture of an aortic aneurysm. In the era before cardiac surgery over 90% of patients died from aneurysm rupture. Through refinements, particular in cardiovascular surgery, life expectancy has substantially increased during the last 20 years. Whereas in 1972 the median life expectancy was 32 years, these numbers rose to 45 years in 1998.17 This was mainly accomplished through elective replacement of the aortic root which prolonged survival in MFS patients as reported previously.17 The presence of dissection and hypertension emerged to be a significant predictor of subsequent aneurysm operation. It has to be stressed that prophylactic surgery does not decrease the risk of sudden death from aneurysm rupture but decreases the risk of subsequent vascular complications. The presence of dissection is considered a risk factor for future vascular complications most probably through the underlying severe pathology.
A typical pattern of aneurysm formation has been observed, with the ascending thoracic aorta being the first vascular event followed by the descending thoracic aorta. Notably, peripheral vascular arteries are usually not affected. As more than 50% of Marfan patients require subsequent surgery on their vascular system, recurrent evaluation of the entire aorta by computed tomography or magnetic resonance imaging is crucial to the clinical follow up, before and after aortic root surgery.
References
- 1.Treasure T. Cardiovascular surgery for Marfan syndrome. Heart. 2000;84:674–678. doi: 10.1136/heart.84.6.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kainulainen K, Pulkkinen L, Savolainen A, Kaitila I, Peltonen L. Location on chromosome 15 of the gene defect causing Marfan syndrome. N Engl J Med. 1990;323:935–939. doi: 10.1056/NEJM199010043231402. [DOI] [PubMed] [Google Scholar]
- 3.Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet. 2000;37:9–25. doi: 10.1136/jmg.37.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beighton P, de Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA. International nosology of heritable disorders of connective tissue. Am J Med Genet. 1986;29:581–594. doi: 10.1002/ajmg.1320290316. [DOI] [PubMed] [Google Scholar]
- 5.De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 6.Lipscomb KJ, Clayton-Smith J, Harris R. Evolving phenotype of Marfans syndrome. Arch Dis Child. 1997;76:41–46. doi: 10.1136/adc.76.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, Bieber FR, Lin A, MacLeod P, Hall B, Graham JM., Jr Diagnosis and management of infantile Marfan syndrome. Pediatrics. 1990;86:888–895. [PubMed] [Google Scholar]
- 8.Gott VL, Greene PS, Alejo DE, Cameron DE, Naftel DC, Miller DC, Gillinov AM, Laschinger JC, Pyeritz RE. Replacement of the aortic root in patients with Marfan syndrome. N Engl J Med. 1999;340:1307–1313. doi: 10.1056/NEJM199904293401702. [DOI] [PubMed] [Google Scholar]
- 9.Bentall HH, DeBono A. A technique for complete replacement of the ascending aorta. Thorax. 1968;23:338–339. doi: 10.1136/thx.23.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yacoub MH, Gehle P, Chandrasekaran V, Birks EJ, Child A, Radley-Smith R. Late results of a valve sparing operation in patients with aneurysms of the ascending aorta and root. J Thorac Cardiovasc Surg. 1998;115:1080–1090. doi: 10.1016/S0022-5223(98)70408-8. [DOI] [PubMed] [Google Scholar]
- 11.David TE, Feindel CM. An aortic valve sparing operation for patients with aortic incompetence and aneurysm of the ascending aorta. J Thorac Cardiovasc Surg. 1992;103:617–622. [PubMed] [Google Scholar]
- 12.Birks EJ, Webb C, Child A, Radley-Smith R, Yacoub MH. Early and long term results of a valve sparing operation for Marfan syndrome. Circulation. 1999;100:II-29. doi: 10.1161/01.cir.100.suppl_2.ii-29. [DOI] [PubMed] [Google Scholar]
- 13.de Oliveira NC, David TE, Ivanov J, Armstrong S, Eriksson MJ, Rakowski H, Webb G. Results of surgery for aortic root aneurysm in patients with Marfan syndrome. J Thorac Cardiovasc Surg. 2003;125:789–796. doi: 10.1067/mtc.2003.57. [DOI] [PubMed] [Google Scholar]
- 14.Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM. Marfan syndrome: Long term survival and complications after aortic aneurysm repair. Circulation. 1995;91:728–733. doi: 10.1161/01.cir.91.3.728. [DOI] [PubMed] [Google Scholar]
- 15.Coselli JS, LeMaire SA. Current status of thoracoabdominal aortic aneurysm repair in Marfan syndrome. J Card Surg. 1997;12:167–172. [PubMed] [Google Scholar]
- 16.Gowda RM, Misra D, Tranbaugh RF, Ohki T, Khan IA. Endovascular stent grafting of descending aortic aneurysms. Chest. 2003;124:714–719. doi: 10.1378/chest.124.2.714. [DOI] [PubMed] [Google Scholar]
- 17.Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, Boxer M, Devereux RB, Tsipouras P. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995;75:157–160. doi: 10.1016/s0002-9149(00)80066-1. [DOI] [PubMed] [Google Scholar]