

Abstract
A neonate presented with mucopolysaccharidosis-like phenotypic expression and typical signs of dysostosis multiplex but without urinary excretion of glycosaminoglycans. Investigations of lysosomal enzymes in cultured fibroblasts revealed a mucolipidosis type 2, known as I-cell disease. We describe the fatal course of the patient due to complications of an uncommon dilated cardiomyopathy in this rare disease and discuss the pathogenesis.
MeSH: Mucolipidosis; Infant, Newborn; Dilated cardiomyopathy
Introduction
Mucolipidosis type 2 (ML-2; McKusick 252500) is a lysosomal storage disorder and is caused by an autosomal recessive inherited defect of N-acetylglucosamine-1-phosphotransferase (E.C. 2.7.8.17). This cytosolic enzyme is involved in the processing of various hydrolases to permit their recognition and subsequent internalisation into lysosomes. Defective enzyme function ends in general in misrouting of newly synthesised enzymes. Vacuolised lymphocytes and the appearance of inclusion bodies in cultured fibroblasts resulted in naming this disorder „I-cell-disease”. Affected cells are mostly of mesenchymal origin. Characteristic and revealing clinical symptoms are Hurler-like features without mucopolysacchariduria. The disorder is slowly progressive with onset at birth and fatal outcome in early childhood due to bronchopneumonia or congestive heart failure. Cardiac manifestations in systemic storage disorders are common but especially in mucolipidosis type 2 there are few reports about involvements of cardiovascular system during the first weeks of life. The majority of described patients suffers from hypertrophic cardiomyopathy.1
We report a case with mucolipidosis type 2 complicated by severe dilative cardiomyopathy (DCM) which is very rare associated with this lysosomal storage disorder and we discuss possible mechanisms of pathogenesis.
Case report
The female patient of healthy German couples was the first child after an uneventful pregnancy and delivery. The newborn was hypotrophic with a birth weight of 2370 g. She presented with the following signs and dysmorphisms which implicated infantile onset heteroglycanosis: coarse face, gum hyperplasia, rough voice, joint contractions and hip luxations. Dysostosis multiplex and a globular heart were X-rays findings at the age of 8 weeks (Fig. 1). The roentgenographic appearance of the ribs was specific, as well as short and stubby bones of the upper extremities or thoracolumbar gibbus of the spine. ECG showed a dilated left ventricle with marginal reduced systolic parameters of cardiac function (Tab. 1).
Figure 1.
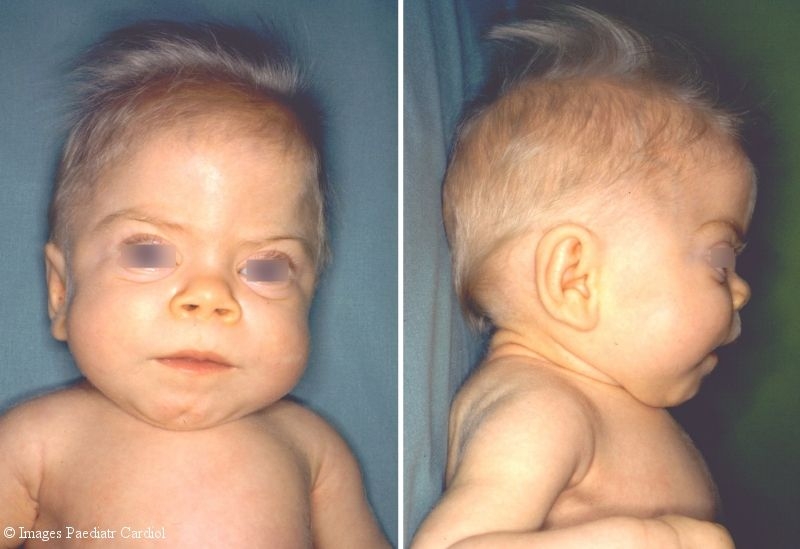
Patient with mucolipidosis type 2 at the age of 9 months
Table 1.
Echocardiographic findings in the girl with ML-2 before and during symptomatic therapy. Normal ranges are given in brackets. Normal values for LVEDD were interpolated.8 Abbrevations: FS - fraction shortening, EF – ejection fraction, LVEDD - left ventricular end diastolic diameter
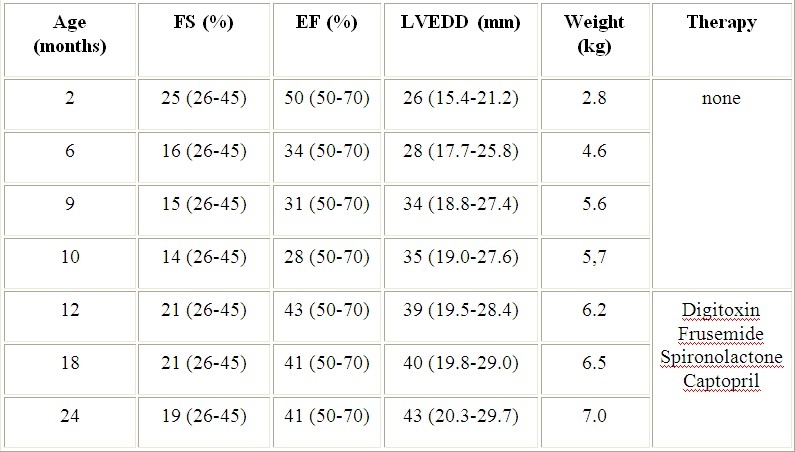
Laboratory findings of liver and renal functions including chromosomes showed no abnormalities, however there were vacuoles in peripherial white blood cells pointing to lysosmal storage disorder. A mucopolysaccharidosis of Hurler-type was presumed but several examinations detected no excess of mucopolysaccharides in urine. The patient was admitted to outside hospital due to airway infections, failure to thrive and poor development for several times during the next 4 months. Regular cardiac examinations revealed an increasing tendency of disturbed systolic function with dilation of the heart muscle (Tab. 1). At the age of 9 months the patient's condition was severely restricted due to signs of cardiac insufficiency with poor feeding, sweating, hepatomegaly (2-3 cm) and perioral cyanosis. No specific therapy was initiated until transfer to centres for specialised cardiac and metabolic investigations. Chest X-ray investigation showed a cardiomegaly (Fig. 2).
Figure 2.
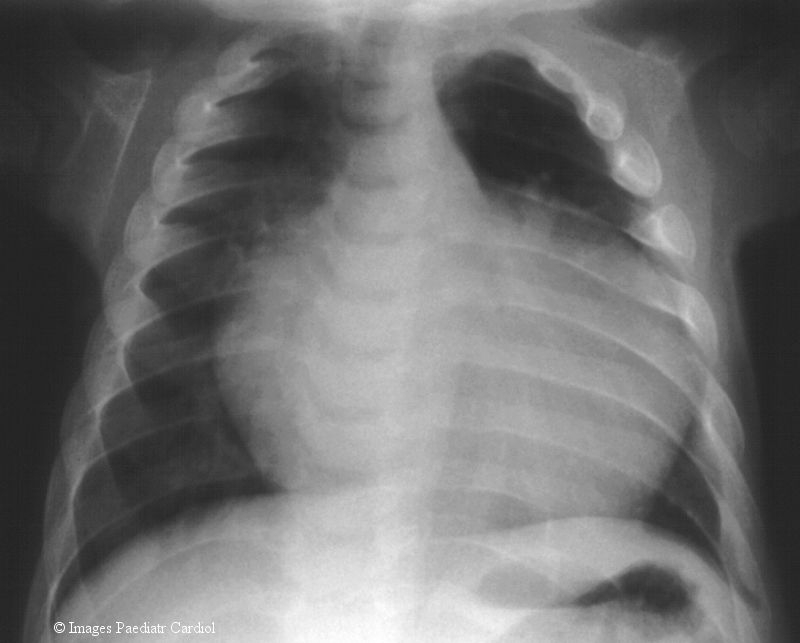
Chest X-ray illustrating the cardiomegaly and the broadened spatulate ribs at the age of 9months
ECG clarified marked signs of biventricular hypertrophy with 1st degree atrioventricular blockade and disturbed re-excitations. Echocardiographic findings were interpreted as a dilative cardiomyopathy with poorly contracting action and extremely systolic dysfunction of the left ventricle (Tab. 1). There were no structural anomalies in the heart including the valves. Atypic origins of coronary arteries (Bland-White-Garland-syndrome) as the cause of dilated ventricles were excluded by coronarography. Treatment with digitalis, angiotensin-converting-enzyme inhibitor and diuretics was started at the age of ten months. Clinical signs of cardiac insufficiency disappeared within two weeks and the patient stabilised. But pathological echocardiographic findings of cardiac function persisted (Tab. 1). A skin biopsy and fibroblast culture was initiated at age of 10 months. Cells grew atypically and slowly and showed characteristic granular inclusions suggesting ML-2 (Fig. 3).
Figure 3.
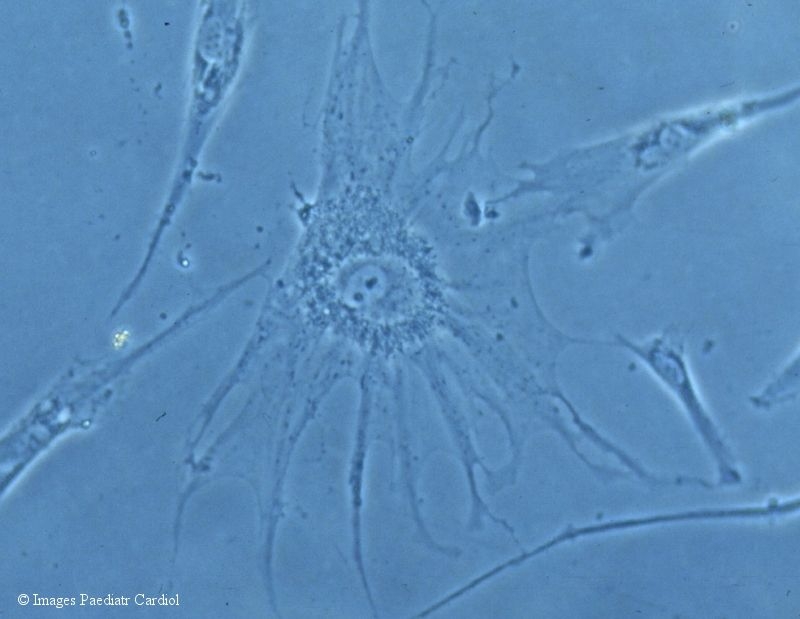
Light microscopy of N-acetylglucosamine-1-phosphotransferase deficient fibroblasts with characteristic inclusion bodies (enlargement 250-fold)
Assays of lysosomal hydrolases in fibroblasts confirmed the diagnosis with low enzyme activities between 5 and 10 % of normal range for β -hexosaminidase, β -galactosidase and arylsulphatase A contrary to 10 to 50-fold increased enzyme activities in plasma. Despite of medication and physical therapy the patient's condition remained poor due to cardiac dystrophy and resulted in delayed psychomotoric development (DQ 55). During an episode of recurrent pulmonary infections the girl died unexpected from an atypical pneumonia, when she was 30 months old.
Discussion
Dilated cardiomyopathies (DCM) are the most common type of cardiomyopathy in early childhood and about 50 % have to be classified as idiopathic variants. In about 20 % sequals of myocarditis or endocardial fibroelastosis, anomalies of coronary arteries or previous therapy with doxorubicin (adriblastin®) lead to secondary DCM.2 Approximately 30 % are genetic cardiomyopathies of autosomal dominant or X-linked inheritance pattern.3,4 DCM is characterised by dilation and impaired contraction of the left or both ventricles, whereas hypertrophic forms commonly caused by metabolic cardiomyopathies appearing with diastolic dysfunction. Most of the patients with ML-2 suffer from hypertrophic cardiomyopathy (HCM) and / or valvular thickening and prolapse due to mucolipid storage.5 Only one patient was described with dilative cardiomyopathy and endocardial fibroelastosis without signs of cardiac insufficiency at the age of 10 weeks.6 Our patient showed classical signs and symptoms of Mucolipidosis type 2 with exception of early onset DCM. Diagnosis was certainly confirmed with enzyme assays. Data for mutational analysis of the deficient enzyme N-acetylglucosamine-1-phosphotransferase are currently not available so far. To our knowledge, there are also no further reports of I-cell disease where a HCM decompensated and resulted in DCM. Pathomechanisms of this atypical clinical phenotype are not fully understood. Mannose-6-phosphates are recognition sites of enzymes addressed to lysosomes. Almost all mammalian cells express insulin-like growth factor-II / mannose-6-phosphate (IGF-II/Man-6-P) receptors. There is a specific developmental pattern and tissue distribution known with highest expression rate in the heart and decreasing rates in lung, muscle, liver and lowest rate in the brain. This rank order is only consistent with organ manifestations in mucolipidosis type 2 with regard to the moderate involvement of the brain which reflects a milder psychomotor delay in these patients in relation to other lysosomal storage disorders. In the heart, the bifunctional IGF-II/Man-6-P receptor amounts to 1.7 % of total protein in extracted tissue. Normally, the binding of IGF-II to the receptor is inhibited by acid lysosomal enzymes. This may be understood as a principle of modulation between cellular metabolic build up and break down. Because of reduced binding of correctly phosphorylated hydrolases an increased binding of IGF-II to the receptor is postulated. IGF-II stimulates certain cellular biologic effects: Beside actions on glucose metabolism there are influences known on the calcium gating system.7 It remains speculative if an excess of IGF-II may lead to an overgrowth of the heart coupled with disturbed calcium channeling, mechanoenergetic changes and resulting dilation of the heart muscle. From the metabolic point of view it is not clear in detail admittedly, why only a few of patients develop this cardiac complication. Thus, we suggest that dilated cardiomyopathy presented in coincidental occurrence with ML-2 by chance in these two patients described up to now.6
Specific treatments (enzyme replacement therapy) have not been developed whereas procedures for prenatal diagnosis are available and genetic counselling can be offered in this lysosomal storage disorder. Because of progressive complications in nearly all organ systems these patients are still no candidates for heart transplants. In order to get prognostic information we advise to perform detailed cardiac examinations as essential components of the diagnostic work-up in patients with mucolipidosis type 2.
Acknowledgments
The authors gratefully acknowledge Hans Kresse, MD, PhD (University of Münster, Germany) for enzyme assays of lysosomal hydrolases in fibroblasts.
References
- 1.Nolan CM, Sly WS. I-cell-disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Disease. McGraw-Hill; 1989. pp. 1589–1601. [Google Scholar]
- 2.Kasper EK, Agema WRP, Hutchins GM, Deckers JW, Hare JM, Baughman KM. The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll Cardiol. 1994;23:586–590. doi: 10.1016/0735-1097(94)90740-4. [DOI] [PubMed] [Google Scholar]
- 3.Michels VV, Moll PP, Miller FA, Taijk AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HE. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 4.Barth PG, Wanders RJA, Vreken P, Janssen EA, Lam J, Baas F. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) J Inher Metab Dis. 1999;22:555–567. doi: 10.1023/a:1005568609936. [DOI] [PubMed] [Google Scholar]
- 5.Okada S, Owada M, Sakiyama T, Yutaka T, Ogawa M. I-cell disease: Clinical studies of 21 Japanese cases. Clin Genet. 1985;28:207–215. doi: 10.1111/j.1399-0004.1985.tb00388.x. [DOI] [PubMed] [Google Scholar]
- 6.Schulz R, Vogt J, Voss W, Hanefeld F. Mucolipidosis type II (I-cell disease) with severe cardiac involvement. Monatsschr Kinderheilkd. 1987;135:708–711. [PubMed] [Google Scholar]
- 7.Kiess W. Molecular biology of the IGF-II/Mannose-6-phosphate receptor. In: Rosenfeld R, Roberts C Jr, editors. Contemporary Endocrinology: The IGF System. Totowa: Humana Press; 1999. pp. 89–109. [Google Scholar]
- 8.First T, Skovranek J. Normal values of M-mode echocardiographic parameters in children. Cesk Pediatr. 1984;39:699–708. [PubMed] [Google Scholar]