

Abstract
Deletion 22q11.2 syndrome (Del22) (DiGeorge/Velo-Cardio-Facial syndrome) is characterized by congenital heart defect (CHD), palatal anomalies, facial dysmorphisms, neonatal hypocalcemia, immune deficit, speech and learning disabilities. CHD is present in 75% of patients with Del22. The most frequently seen cardiac malformations are “conotruncal” defects, including tetralogy of Fallot (TF), pulmonary atresia with ventricular septal defect (PA-VSD), truncus arteriosus (TA), interrupted aortic arch (IAA), and ventricular septal defect (VSD). The study of the specific “cardiac phenotype” in patients with Del22 shows that a particular cardiac anatomy can be identied in these subjects. In addition to CHD, various organ systems can be involved, so that a multidisciplinary approach is needed in the evaluation of patients with Del22.
MeSH: Deletion 22q11, DiGeorge syndrome, Velo-Cardio-Facial syndrome, Heart defects, congenital
Introduction
DiGeorge and Velo-Cardio-facial syndromes are genetic conditions with overlapping features, including congenital heart defect (CHD), facial anomalies, hypoplastic thymus with immune deficit, palatal anomalies, neonatal hypocalcemia, speech and learning disabilities.1,3 Cytogenetic and molecular studies have demonstrated that microdeletion of chromosome 22q11.2 (del22) is detectable in the majority of patients with DiGeorge/Velo-cardio-facial syndrome.4,5 Del22 syndrome is considered to be developmentally related to neural crest anomalies, influencing the differentiation of the branchial arches.6,7 It is estimated that del22 syndrome occurs in 1 in 4000-6000 live births, making this disorder a significant health concern in general population.8
Clinical features
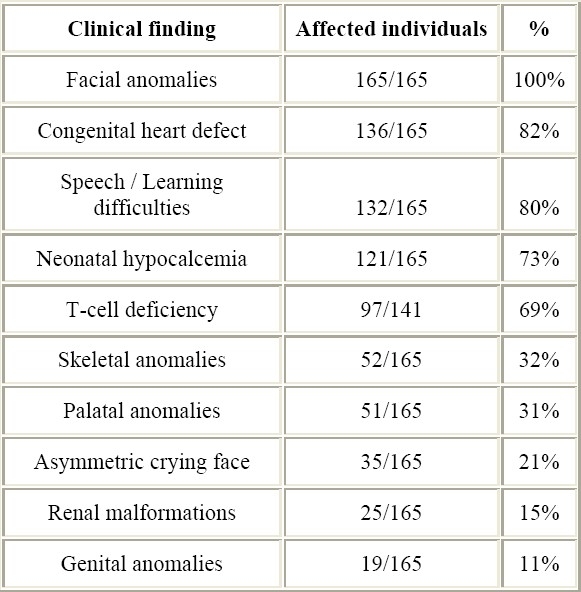
The variability in the clinical expression of del22 syndrome is extremely wide.9 Classical features of del22 syndrome include CHD, velopharyngeal insufficiency or cleft palate, facial anomalies, speech and learning disabilities, neonatal hypocalcemia, and T-cell immune deficit. Nevertheless, the spectrum of anomalies associated with del22 is becoming wider and wider.2 Inter-individual variability in del22 phenotype is characteristic, since subjects with full-blown clinical expression of the syndrome as well as mildly affected individuals can be found. The main clinical features of Del22 syndrome and their occurrence in our series of 165 patients are listed in Table 1.
Table 1.
Main clinical features of Del22 syndrome and their occurrence in this series
Facial anomalies
Accurate phenotypical evaluation of patients with Del22 demonstrates that facial anomalies, severely or mildly expressed, are detectable in all subjects. Characteristic facial features include periorbital fulness, narrow upslanted palpebral fissures, prominent nose with large tip and hypoplastic nares, small mouth with everted upper lip and small dysmorphic ears (Figures 1 and 2). Many children, particularly in neonatal age, may have only a “subtle” facial phenotype. The experience of physicians caring with children affected by del22 is fundamental in recognizing mild facial anomalies associated with the syndrome.
Figure 1.
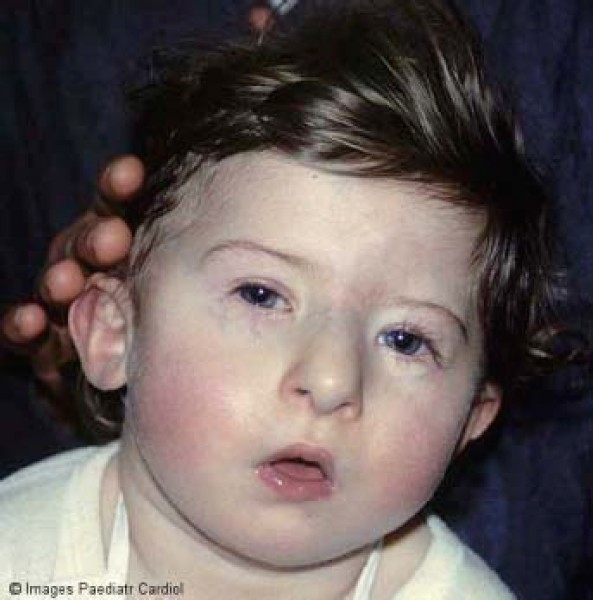
Facial appearance of patient with Del22 syndrome
Figure 2.

Facial appearance of patient with Del22 syndrome
Congenital heart defects
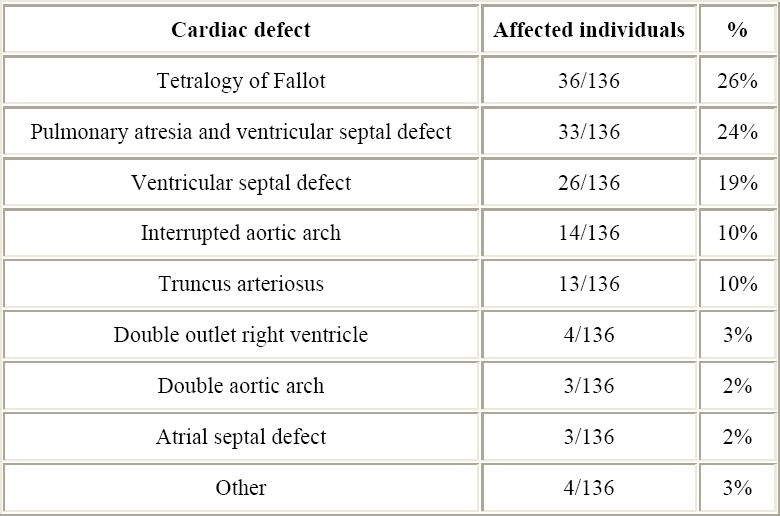
75% of patients with Del22 have symptomatic CHD.2 The most frequently seen cardiac malformations are “conotruncal” defects, including tetralogy of Fallot (TF), pulmonary atresia with ventricular septal defect (PA-VSD), truncus arteriosus (TA), and interrupted aortic arch (IAA).2,10–15 VSD, as well as asymptomatic aortic arch malformations are increasingly been diagnosed in patients with Del22 (Table 2).16
Table 2.
Cardiac anomalies in this series.
The study of the specific “cardiac phenotype” in patients with Del22 shows that a particular cardiac anatomy can be identified.15 In fact, patients with conotruncal heart defect and Del22 have often additional CHDs as a distinctive recognizable pattern.
Tetralogy of Fallot: Additional cardiac defects are found in the half of the patients with this TF and Del22.17,18 The associated cardiac defects include: 1) right or cervical aortic arch with or without aberrant left subclavian artery; 2) hypoplasia or absence of the infundibular septum; 3) absence of the pulmonary valve; 4) discontinuity and diffuse hypoplasia of the pulmonary arteries.
Pulmonary atresia with ventricular septal defect: Considering the pattern of pulmonary blood supply, among children with pulmonary atresia with ventricular septal defect two major groups of patients can be recognized: 1) children with a single ductus arteriosus that usually presents confluent and well formed pulmonary arteries (also called tetralogy of Fallot with pulmonary atresia), and 2) children with major aortopulmonary collateral arteries (MAPCA) frequently associated with discontinuity, hypoplasia or absence of the central pulmonary arteries. In children with pulmonary atresia with ventricular septal defect and Del22 the pattern of pulmonary blood supply provided by the MAPCA is prevalent.19–22 Additionally, a high prevalence of right aortic arch and of discontinuity and defects of arborization of the pulmonary arteries can be found.
Truncus arteriosus: Del22 is prevalent in patients with TA with nonconfluent pulmonary arteries (Type 3 of van Praagh), in which the right pulmonary artery arises from the TA near to the truncal valve, and the left pulmonary artery is supplied by the ductus arteriosus.23–25 Additional CHDs to TA are: 1) interruption or right aortic arch; 2) discontinuity, stenosis, or crossing of the pulmonary arteries; 3) severe dysplasia of the truncal valve; 4) origin of the TA from the right ventricle.
Interrupted aortic arch: Del 22 is particularly common in patients with IAA type B.26–30 In these cases the infundibular septum is often hypoplastic or absent and is deviated posteriorly and to the left; the VSD results in a subarterial position doubly committed with the pulmonary and aortic valves.
Ventricular septal defect: The type of VSD in patients with Del22 is prevalently subarterial doubly committed.16,31 Right or cervical aortic arch may be associated.
Palatal anomalies
Two thirds of the patients with Del22 are found to have palatal anomalies, and the spectrum of malformations is wide.2,3,40 The majority have velopharyngeal insufficiency in the absence of cleft palate, but overt clefts, bifid uvula and cleft lip may also be present. Palatal function plays an important role in the development of speech.
Immune deficit
The immunodeficiency in Del22 syndrome is due to poor formation of thymic tissue and impaired production of T-cells. The most common immunologic abnormality is low number of T-cell, and functional T-cell deficiency is found in a minority of all patients.41 Nevertheless, the spectrum of immunocompromise in the Del22 patient population is broad. Children with Del22 have a significant risk of infection due to anatomical effects such as CHD and cleft palate. The additional risk associated with immunodeficiency may represent a critical factor in the management of these patients. Fortunately, the T-cell number usually increases by time and the cell function is not altered. Usually, patients with del22 are at most risk for repeated infections in the first years of life, but older children and adults do generally not have recurrent infections.
Hypocalcemia
Neonatal hypocalcemia is recognized in most of children with Del22.2 This symptom is related to hypoparathyroidism due to absence or underdevelopment of parathyroid glands, which leads to low blood calcium levels.42,43 Hypocalcemia may cause tremors, seizures, and arrhythmia. Calcium supplementation leads to normalization of blood calcium levels. The development of hypoparathyroidism later in life is rarer.
Speech / Learning impairment
Developmental delays can be present in children with del22, including delays in the motor, linguistic and cognitive domains.44,45 Delayed motor development is mainly attributed to the hypotonia present in more than half of the patients with del22. Psychomotor therapy is therefore recommended from an early age. Difficulties in expressive language are generally evident in preschooler children. This delayed speech development is prevalently due to the concomitance of velopharyngeal insufficiency and developmental delay. Speech difficulties related to velo-palatal abnormalities may become clearly manifest late, particularly when speech has fully developed. During childhood, the majority of the children has learning problems, especially in the area of reading comprehension, arithmetic and problem-solving. Common problems seen in these patients are impulsivity, distractibility, perseveration, and hyperactivity. Several common behavioural and temperamental features have been observed in children and adolescents with del22, including a predisposition to show a withdrawn behavior, depression, anxiety and a tendency to engage in obsessive and/or compulsive behaviours. Therefore, good monitoring and follow up of the socio-emotional development is important in all ages.
Skeletal anomalies
Skeletal anomalies and deformations have been detected in patients with del22.46–48 In the first reports, a variety of hand malformations and clubfoot have been described. Thereafter, scoliosis and vertebral maformations, including butterfly vertebrae and vertebral coronal cleft, have been added to the list. As characteristic diagnostic marker, most of del22 patients have long tapered fingers (Figure 3).
Figure 3.
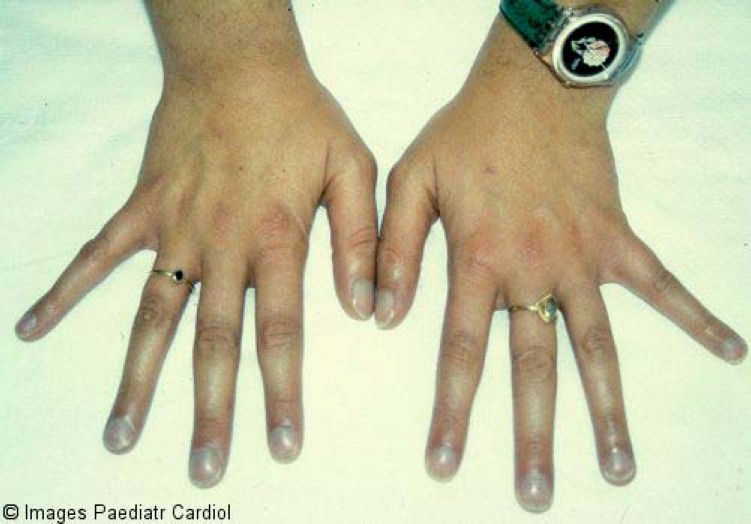
Long tapering fingers characteristics of Del22 syndrome
Asymmetric crying face
Unilateral partial facial palsy due to hypoplasia of the depressor anguli oris muscle (DAOM) results in asymmetry of the lower lip, especially evident in smiling and crying. The eye on the affected side usually closes normally. DAOM can be detected in about 20% of neonates with del22.49
Audiological anomalies
Hearing impairment is documented in 60% of patients with del22.2,50 Hearing loss is conductive in the majority of the cases, probably related to chronic otitis media with effusion and upper respiratory tract infections. Nevertheless, congenital sensorineural deafness is also diagnosed in some cases. Audiological evaluation is recommended in Del22 children, in order to reduce the risk of speech deficit.
Growth
A specific pattern of growth is identifiable in patients with Del22.51 Weight deficiency is frequent in the first years of age, being prevalently related to feeding difficulties. The growth in weight improves with time leading to overweight and obesity in adolescence. On the contrary, short stature is present in a minority of the patients. In fact, adolescent patients have prevalently normal height, with a small group of patients showing a constitutional delay in height growth.
Genetics
In 1992, a microdeletion of chromosome 22 at the q11.2 band was reported.52 The critical region has been subsequently defined (Figure 4).
Figure 4.
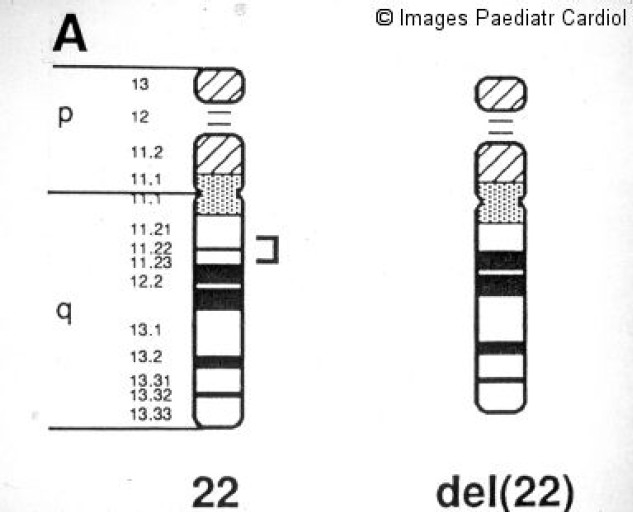
The 22q11 critical region commonly deleted in DiGeorge/velocardiofacial patients
The majority of the patients carry a common 3 Mb deletion, but smaller deletions have also been found. To date, the size of the deletion has not been correlated with the phenotypical expression of the syndrome. Fluorescent in situ hybridization (FISH) is used as genetic laboratory test for 22q11.2 microdeletion detection (Figure 5).
Figure 5.
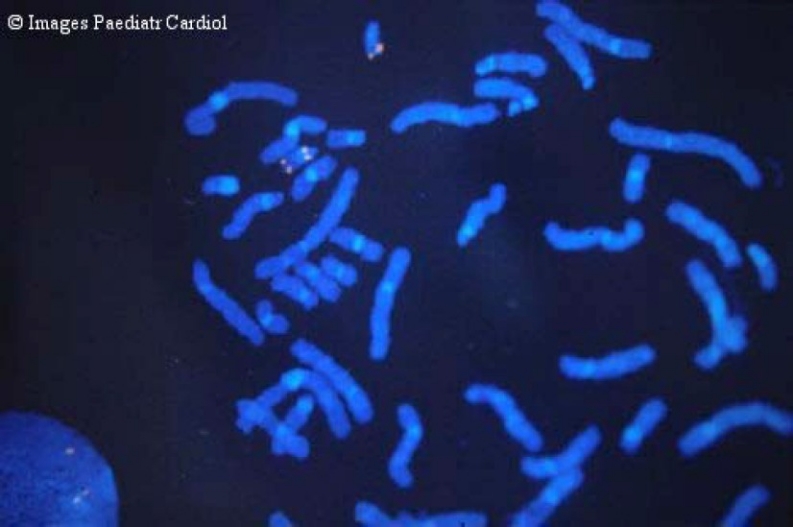
Fluorescent in situ hybridization showing Del22
At least 30 genes have been mapped in the typical deleted region. Among genes located in the “critical” chromosomal region, TUPLE1, UFD1L and TBX1 have been particularly studied.53–55 Although TBX1 mutations have been recently detected in rare patients with clinical features of DiGeorge/Velo-Cardio-Facial syndrome without an identifiable Del22,56 it remains unclear whether several genes must be haploinsufficient to cause a clinical phenotype or whether a single locus predominates.
Familial transmission
The Del22 is a “de novo” occurrence in the family in most of the cases. Nevertheless, inheritance of the microdeletions from one of the parents is possible, the frequency varying from 6% to 28% in different series.2,9,57–59 The affected parent often demonstrates a milder phenotype. Various genetic and non-genetic factors, including modifier genes, mosaicism, unstable mutations, allelic variations chance and environmental interactions, can be hypothesized to be involved in variable clinical expression of the syndrome in the same family.
Non-syndromic conotruncal heart defects
Several observations suggesting that del22 could be associated with “non-syndromic” CHDs can be found in the previous literature on this argument.32,33 However, 80% of these patients, reported as apparently “isolated” CHD, had in fact extracardiac features fitting within the del22 syndrome phenotype. Other investigations have shown that Del22 is virtually never found in non-syndromic patients with conotruncal CHDs.34–38 In a personal series of 305 patients with true non-syndromic CHD, we detected only one deleted patient.14 Thus, we believe that, in clinical practice, genetic tests searching for del22 are not indicated in all patients with conotruncal CHDs, but only in subjects with clinical anomalies of del22 syndrome. Classic or subtle facial anomalies are fundamental useful tools for selecting children who should be tested for Del22.14,39 In this regard, also patients with distinct anatomic conotruncal defect subtypes must be included.14,39
Conclusions
Clinical expression of Del22 syndrome can be extremely variable and the degree of various organ systems involved is wide. Early recognition of the deletion is important, so that the treatment of involved organ anomalies can be initiated, screening for associated malformations performed and prevention of neuropsychological problems provided. In fact, as occurrs in several multiple system clinical conditions, the care of patients with Del 22 can be very complex. A multidisciplinary approach is fundamental to ensure that the patient will be able to attain his or her maximal potential.
References
- 1.Lipson AH, Yuille D, Angel M, Thompson PG, Vanderwood KG, Beckenham EJ. Velocardiofacial (Shprintzen) syndrome: an important syndrome for the dysmorphologist to recognize. J Med Genet. 1991;28:596–604. doi: 10.1136/jmg.28.9.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Stewart F, Van Essen T, Patton M, Paterson J, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, LaRossa D, Emanuel BS, Zackai EH. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- 4.Scambler PJ, Carey AH, Wyse RKH, Roach S, Dumanski JP, Nordenskjold M, Williamson R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–206. doi: 10.1016/0888-7543(91)90501-5. [DOI] [PubMed] [Google Scholar]
- 5.Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai E, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: Implications for genetic counseling and prenatal diagnosis. J Med Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Mierop LHS, Kutsche LM. Cardiovascular anomalies in DiGeorge syndrome and importance of neural crest as a possible pathogenetic factor. Am J Cardiol. 1986;58:133–137. doi: 10.1016/0002-9149(86)90256-0. [DOI] [PubMed] [Google Scholar]
- 7.Kirby ML, Waldo KL. Role of neural crest in congenital heart disease. Circulation. 1990;82:332–340. doi: 10.1161/01.cir.82.2.332. [DOI] [PubMed] [Google Scholar]
- 8.Botto LD, May K, Ferhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O’Leary LA, Wong L-Y, Elixson M, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: Phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 9.Digilio MC, Angioni A, De Santis M, Lombardo A, Giannotti A, Dallapiccola B, Marino B. Spectrum of clinical variability in familial deletion 22q11.2: from full manifestation to extremely mild clinical anomalies. Clin Genet. 2003;63:308–313. doi: 10.1034/j.1399-0004.2003.00049.x. [DOI] [PubMed] [Google Scholar]
- 10.Young D, Shprintzen RJ, Goldberg RB. Cardiac malformation in the velocardiofacial syndrome. Am J Cardiol. 1980;46:643–648. doi: 10.1016/0002-9149(80)90515-9. [DOI] [PubMed] [Google Scholar]
- 11.Marmon LM, Balsara RK, Chen R, Duan JM. Congenital cardiac anomalies associated with DiGeorge syndrome: a neonatal experience. Ann Thorac Surg. 1984;38:146–150. doi: 10.1016/s0003-4975(10)62223-0. [DOI] [PubMed] [Google Scholar]
- 12.Momma K, Kondo C, Matsuoka R, Takao A. Cardiac anomalies associated witha chromosome 22q11 deletion in patients with conotruncal anomaly face syndrome. Am J Cardiol. 1996;78:591–594. doi: 10.1016/s0002-9149(96)00374-8. [DOI] [PubMed] [Google Scholar]
- 13.Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai EH, Emanuel BS, Driscoll DA. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 14.Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart defects in patients with DiGeorge/Velocardiofacial syndrome and del22q11. Genet Couns. 1999;10:25–33. [PubMed] [Google Scholar]
- 15.Marino B, Digilio MC, Toscano A, Anaclerio S, Giannotti A, Feltri C, de Ioris MA, Angioni A, Dallapiccola B. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. 2001;3:45–48. doi: 10.1097/00125817-200101000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Toscano A, Anaclerio S, Digilio MC, Giannotti A, Fariello G, Dallapiccola B, Marino B. Ventricular septal defect and deletion of chromosome 22q11: anatomical types and aortic arch anomalies. Eur J Pediatr. 2002;161:116–117. doi: 10.1007/s00431-001-0877-5. [DOI] [PubMed] [Google Scholar]
- 17.Marino B, Digilio MC, Grazioli S, Formigari R, Mingarelli R, Giannotti A, Dallapiccola B. Associated cardiac anomalies in isolated and syndromic patients with tetralogy of Fallot. Am J Cardiol. 1996;77:505–508. doi: 10.1016/s0002-9149(97)89345-9. [DOI] [PubMed] [Google Scholar]
- 18.Momma K, Kondo C, Ando M, Matsuoka R, Takao A. Tetralogy of Fallot associated with chromosome 22q11 deletion. Am J Cardiol. 1995;76:618–621. doi: 10.1016/s0002-9149(99)80170-2. [DOI] [PubMed] [Google Scholar]
- 19.Momma K, Kondo C, Matsuoka R. Tetralogy of Fallot with pulmonary atresia associated with chromosome 22q11 deletion. J Am Coll Cardiol. 1996;27:198–202. doi: 10.1016/0735-1097(95)00415-7. [DOI] [PubMed] [Google Scholar]
- 20.Digilio MC, Marino B, Grazioli S, Agostino D, Giannotti A, Dallapiccola B. Comparison of occurrence of genetic syndromes in ventricular septal defect with pulmonic stenosis (classic tetralogy of Fallot) versus ventricular septal defect with pulmonic atresia. Am J Cardiol. 1996;77:1375–1376. doi: 10.1016/s0002-9149(96)00212-3. [DOI] [PubMed] [Google Scholar]
- 21.Chessa M, Butera G, Bonhoeffer P, Iserin L, Kachaner J, Lyonnet S, Munnich A, Sidi D, Bonnet D. Relation of genotype 22q11 deletion to phenotype of pulmonary vessels in tetralogy of Fallot and pulmonary atresia-ventricular septal defect. Heart. 1998;79:186–190. doi: 10.1136/hrt.79.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofbeck M, Rauch A, Buheitel G, Leipold G, van der Emde J, Pteiffer R, Singer H. Monosomy 22q11 in patients with pulmonary atresia, ventricular septal defect, and major aortopulmonary arteries. Heart. 1998;79:180–185. doi: 10.1136/hrt.79.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Momma K, Ando M, Matsuoka R. Truncus arteriosus communis associated with chromosome 22q11 deletion. J Am Coll Cardiol. 1997;30:1067–1071. doi: 10.1016/s0735-1097(97)00240-4. [DOI] [PubMed] [Google Scholar]
- 24.Marino B, Digilio MC, Dallapiccola B. Severe truncal valve dysplasia: association with DiGeorge syndrome? Ann Thorac Surg. 1998;66:980. doi: 10.1016/s0003-4975(98)00680-8. [DOI] [PubMed] [Google Scholar]
- 25.Marino B, Digilio MC, Toscano A. Common arterial trunk, DiGeorge syndrome and microdeletion 22q11. Progress in Pediatric Cardiology. 2002;15:9–17. [Google Scholar]
- 26.Lewin MB, Lindsay EA, Jurecic V, Goytia B, Towbin JA, Baldini A. A genetic etiology for interruption of the aortic arch type B. Am J Cardiol. 1997;80:493–497. doi: 10.1016/s0002-9149(97)00401-3. [DOI] [PubMed] [Google Scholar]
- 27.Rauch A, Hofbeck M, Leipold G, Klinge J, Trautmann U, Kirsch M, Singer H, Pfeiffer RA. Incidence and significance of 22q11.2 hemizygosity in patients with interrupted aortic arch. Am J Med Genet. 1998;78:322–331. [PubMed] [Google Scholar]
- 28.Marino B, Digilio MC, Persiani M, Di Donato R, Toscano A, Giannotti A, Dallapiccola B. Deletion 22q11 in patients with interrupted aortic arch. Am J Cardiol. 1999;84:360–361. doi: 10.1016/s0002-9149(99)00297-0. [DOI] [PubMed] [Google Scholar]
- 29.Momma K, Ando M, Matsuoka R, Joo K. Interruption of the aortic arch associated with deletion of chromosome 22q11 is associated with a subarterial and doubly committed ventricular septal defect in Japanese patients. Cardiol Young. 1999;9:463–467. doi: 10.1017/s1047951100005357. [DOI] [PubMed] [Google Scholar]
- 30.Loffredo CA, Ferencz C, Wilson PD, Lurie IW. Interrupted aortic arch: an epidemiologic study. Teratology. 2000;61:368–375. doi: 10.1002/(SICI)1096-9926(200005)61:5<368::AID-TERA8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 31.Matsuoka R, Kimura M, Scambler PJ, Morrow BE, Imamura S, Minoshima S, Shimizu N, Yamagishi H, Joh-O K, Watanabe S, Oyama K, Saji T, Ando M, Takao A, Momma K. Molecular and clinical study of 183 patients with conotruncal anomaly face syndrome. Hum Genet. 1998;103:70–80. doi: 10.1007/s004390050786. [DOI] [PubMed] [Google Scholar]
- 32.Wilson DI, Goodship JA, Scambler PJ, Carey A, Cross I, Burn J. Is monosomy for the DiGeorge locus on chromosome 22 responsible for isolated heart malformation? Am J Hum Genet. 1991;49:901. [Google Scholar]
- 33.Goldmuntz E, Driscoll D, Budarf ML, Zackai EH, McDonald-McGinn DM, Biegel JA, Emanuel B. Microdeletions of chromosomal region 22q11 in patients with congenital conotruncal cardiac defects. J Med Genet. 1993;30:807–812. doi: 10.1136/jmg.30.10.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amati F, Mari A, Digilio MC, Mingarelli R, Marino B, Giannotti A, Novelli G, Dallapiccola B. 22q11 deletions in isolated and syndromic patients with tetralogy of Fallot. Hum Genet. 1995;95:479–482. doi: 10.1007/BF00223856. [DOI] [PubMed] [Google Scholar]
- 35.Digilio MC, Marino B, Giannotti A, Dallapiccola B. Search for 22q11 deletion in non-syndromic conotruncal cardiac defects. Eur J Pediatr. 1996;155:619–624. doi: 10.1007/BF01957919. [DOI] [PubMed] [Google Scholar]
- 36.Debrus S, Berger G, de Meeus A, Sauer U, Guillaumont S, Voisin M, Bozio A, Demczuk S, Aurias A, Bouvagnet P. Familial non-syndromic conotruncal defects are not associated with a 22q11 microdeletion. Hum Genet. 1996;97:138–144. doi: 10.1007/BF02265254. [DOI] [PubMed] [Google Scholar]
- 37.Digilio MC, Marino B, Giannotti A, Dallapiccola B. Chromosome 22q11 microdeletion and isolated conotruncal heart defects. Arch Dis Child. 1997;76:79–81. doi: 10.1136/adc.76.1.79a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Digilio MC, Marino B, Giannotti A, Novelli G, Dallapiccola B. Conotruncal heart defects and chromosome 22q11 microdeletion. J Pediatr. 1997;130:675–677. doi: 10.1016/s0022-3476(97)70260-9. [DOI] [PubMed] [Google Scholar]
- 39.Digilio MC, Marino B, Giannotti A, Mingarelli R, Dallapiccola B. Guidelines for 22q11 deletion screening of patients with conotruncal defects. J Am Coll Cardiol. 1999;33:1746–1747. doi: 10.1016/s0735-1097(99)00084-4. [DOI] [PubMed] [Google Scholar]
- 40.Shprintzen RJ, Goldberg R, Young D, Wolford L. The velo-cardio-facial syndrome: A clinical and genetic analysis. Pediatrics. 1981;67:167–172. [PubMed] [Google Scholar]
- 41.Sullivan KE, Jawad AF, Randall P, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin Immunol Immunopathol. 1998;86:141–146. doi: 10.1006/clin.1997.4463. [DOI] [PubMed] [Google Scholar]
- 42.Scirè G, Dallapiccola B, Iannetti P, Bonaiuto F, Galasso C, Mingarelli R, Boscherini B. Hypoparathyroidism as the major manifestation in two patients with 22q11 deletions. Am J Med Genet. 1994;52:478–482. doi: 10.1002/ajmg.1320520415. [DOI] [PubMed] [Google Scholar]
- 43.Brauner R, Le Harivel De Gonneville A, Kindermans C, Le Bidois J, Prieur M, Lyonnet S, Souberbielle J-C. Parathyroid function and growth in 22q11.2 deletion syndrome. J Pediatr. 2003;142:504–508. doi: 10.1067/mpd.2003.156. [DOI] [PubMed] [Google Scholar]
- 44.Swillen A, Devriendt K, Legius E, Eyskens B, Dumoulin M, Gewillig M, Fryns JP. Intelligence and psichosoacial adjustment in velo-cardio-facial syndrome: a study of 27 children and adolescents with VCFS. J Med Genet. 1997;34:453–458. doi: 10.1136/jmg.34.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swillen A, Vogels A, Devriendt K, Fryns JP. Chromosome 22q11 deletion syndrome: an update and a review of the clinical presentation, the cognitive-behavioral spectrum, and the psychiatric complications. Am J Med Genet. 2000;97:128–135. doi: 10.1002/1096-8628(200022)97:2<128::aid-ajmg4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 46.Cormier Daire V, Iserin L, Théophile D, Sidi D, Vervel C, Padovani JP, Vekemans M, Munnic A, Lyonnet S. Upper limb malformations in DiGeorge syndrome. Am J Med Genet. 1995;56:39–41. doi: 10.1002/ajmg.1320560111. [DOI] [PubMed] [Google Scholar]
- 47.Ming JE, McDonald-McGinn DM, Megerian TE, Driscoll DA, Elias ER, Russell BM, Irons M, Emanuel BS, Markowitz RI, Zackai EH. Skeletal anomalies and deformities in patients with deletions of 22q11. Am J Med Genet. 1997;72:210–215. doi: 10.1002/(sici)1096-8628(19971017)72:2<210::aid-ajmg16>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 48.Digilio MC, Giannotti A, Marino B, Guadagni AM, Orzalesi M, Dallapiccola B. Radial aplasia and chromosome 22q11 deletion. J Med Genet. 1997;34:942–944. doi: 10.1136/jmg.34.11.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giannotti A, Digilio MC, Marino B, Mingarelli R, Dallapiccola B. Cayler cardiofacial syndrome and del 22q11: Part of the CATCH22 phenotype. Am J Med Genet. 1994;53:303–304. doi: 10.1002/ajmg.1320530320. [DOI] [PubMed] [Google Scholar]
- 50.Digilio MC, Pacifico C, Tieri L, Marino B, Giannotti A, Dallapiccola B. Audiological findings in patients with microdeletion 22q11 (diGeorge/velocardiofacial syndrome) Br J Audiol. 1999;33:329–334. doi: 10.3109/03005369909090116. [DOI] [PubMed] [Google Scholar]
- 51.Digilio MC, Marino B, Cappa M, Cambiaso P, Giannotti A, Dallapiccola B. Auxological evaluation in patients with DiGeorge/velocardiofacial syndrome (deletion 22q11.2 syndrome) Genetics in Medicine. 2001;3:30–33. doi: 10.1097/00125817-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Scambler P, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, Wilson DI, Cross IE, Burn J. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339:1138–1139. doi: 10.1016/0140-6736(92)90734-k. [DOI] [PubMed] [Google Scholar]
- 53.Halford S, Wadey R, Roberts C, et al. Isolation of a putative transcriptional regulator from the region of 22q11 deleted in DiGeorge syndrome, Shprintzen syndrome and familial congenital heart disease. Hum Molec Genet. 1993;2:2099–2107. doi: 10.1093/hmg/2.12.2099. [DOI] [PubMed] [Google Scholar]
- 54.Pizzuti A, Novelli G, Ratti A, Amati F, Mari A, Calabrese G, Nicolis S, Silani V, Marino B, Scarlato G, Ottolenghi S, Dallapiccola B. UFD1L, a developmentally expressed ubiquitination gene, is deleted in CATCH22 syndrome. Hum Molec Genet. 1997;6:259–265. doi: 10.1093/hmg/6.2.259. [DOI] [PubMed] [Google Scholar]
- 55.Lindsay EA, Vitelli F, Su H, et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 56.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 57.Leana-Cox J, Pangkanon S, Eanet KR, Curtin MS, Wulfsberg EA. Familial DiGeorge/Velocardiofacial syndrome with deletions of chromosome area 22q11.2: report of five families with a review of the literature. Am J Med Genet. 1996;65:309–316. doi: 10.1002/(SICI)1096-8628(19961111)65:4<309::AID-AJMG12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 58.Thompson PW, Davies SJ. Frequency of inherited deletions of 22q11. J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative. Cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]