

Abstract
Context:
We report hereditary pituitary hyperplasia.
Objective:
The objective of the study was to describe the results of the clinical and laboratory analysis of this rare instance of hereditary pituitary hyperplasia.
Design:
The study is a retrospective analysis of three cases from one family.
Setting:
The study was conducted at the National Institutes of Health, a tertiary referral center.
Patients:
A mother and both her sons had very early-onset gigantism associated with high levels of serum GH and prolactin.
Interventions:
The condition was treated by total hypophysectomy.
Main Outcome Measure(s):
We performed clinical, pathological, and molecular evaluations, including evaluation basal and provocative endocrine testing, neuroradiological assessment, and assessment of the pituitary tissue by microscopic evaluation, immunohistochemistry, and electron microscopy.
Results:
All three family members had very early onset of gigantism associated with abnormally high serum levels of GH and prolactin. Serum GHRH levels were not elevated in either of the boys. The clinical, radiographic, surgical, and histological findings indicated mammosomatotroph hyperplasia. The pituitary gland of both boys revealed diffuse mammosomatotroph hyperplasia of the entire pituitary gland without evidence of adenoma. Prolactin and GH were secreted by the same cells within the same secretory granules. Western blot and immunohistochemistry demonstrated expression of GHRH in clusters of cells distributed throughout the hyperplastic pituitary of both boys.
Conclusions:
This hereditary condition seems to be a result of embryonic pituitary maldevelopment with retention and expansion of the mammosomatotrophs. The findings suggest that it is caused by paracrine or autocrine pituitary GHRH secretion during pituitary development.
GH excess in childhood causes gigantism with clinical manifestations that may include increased growth velocity with tall stature, enlargement of the hands and feet, excessive perspiration, coarsening of facial features, and headaches. Most cases are due to benign pituitary adenomas. Nonadenomatous GH excess due to somatotroph hyperplasia is exceptional but occasionally occurs in patients with multiple endocrine neoplasia syndrome type 1 (MEN 1), Carney complex (CNC), or McCune-Albright syndrome (MAS). Except for MAS, these syndromes are typically inherited in an autosomal dominant manner. Transgenic mice overexpressing GHRH also develop pituitary hyperplasia and later neoplasia (1–3). In humans, however, diffuse pituitary hyperplasia has been observed almost exclusively in a sporadic setting as a result of excess secretion of hypothalamic-releasing factors, usually arising from ectopic sources. We present a family in which a mother and both her sons exhibited similar clinical presentation with extraordinary early onset of pituitary gigantism caused by diffuse mammosomatotroph hyperplasia; describe the clinical, microscopic, ultrastructural, and molecular findings in the boys; and illustrate a possible pathogenetic mechanism.
Subjects and Methods
Case reports
Case 1
The older brother, whose prenatal and postnatal history had been unremarkable, came to medical attention because of rapid and excess growth beginning at 1 yr of age. By 18 months he exceeded the 97th percentile for height (Fig. 1A) and had increased perspiration, coarsening of facial features, and acral enlargement. Investigation confirmed grossly elevated serum GH (138 ng/ml), prolactin (PRL; 520–795 ng/ml) and IGF-I. Magnetic resonance (MR) imaging revealed a symmetrically enlarged pituitary gland (Fig. 2) without evidence of an adenoma. Octreotide and bromocriptine failed to control his levels of GH or PRL or his rapid pace of growth. A surgical procedure was performed and a portion of his anterior lobe was removed surgically. The histological features of the excised tissue were reported to be similar to those of his mother (case 2, below), a presumed GH-secreting pituitary adenoma with hyperplastic features (4). The elevated hormone levels did not respond, and at his peak growth rate, he grew 0.5–1.0 cm/wk. He was referred to the National Institutes of Health (NIH).
Fig. 1.
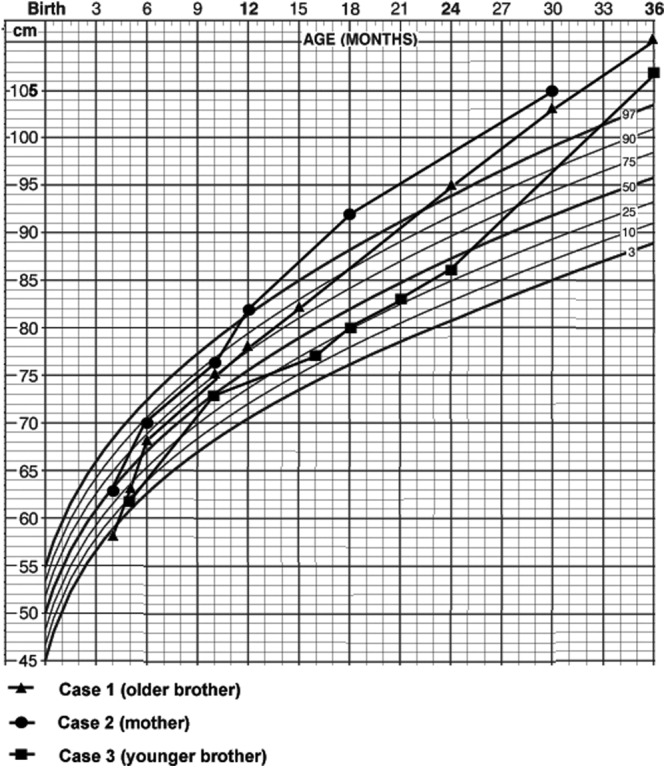
Growth curves of the mother and her sons.
Fig. 2.
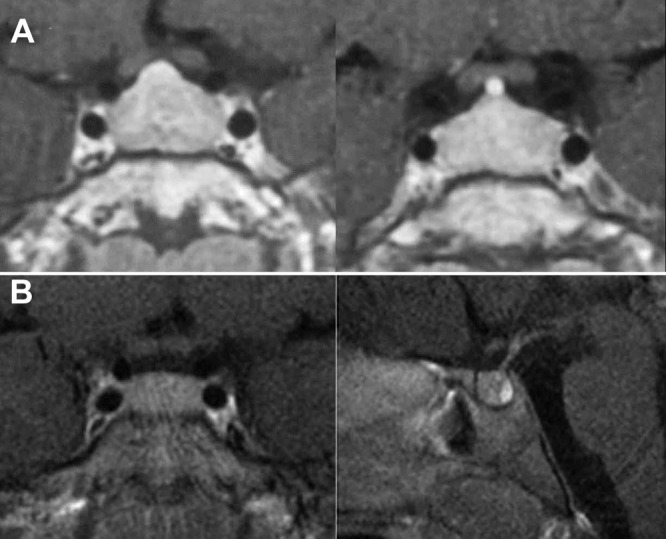
Preoperative contrast-enhanced MR imaging scans of the older brother (case 1) demonstrating symmetric enlargement of the pituitary (A) and the younger brother (case 3) demonstrating a slightly enlarged, symmetric gland (B). The posterior lobe is prominently seen. No focus suggestive of an adenoma is present in either scan.
At presentation to the NIH at age 46 months, he measured 121.5 cm, 11.5 cm above the 95th percentile; his weight was 31.0 kg, and his body mass index was 21.0 kg/m2. His bone age was approximately 60 months. Neurological examination was normal. There were no cutaneous stigmata of MEN 1, CNC, or MAS syndromes; radiographic survey was unremarkable except for his large size. Imaging of the chest, abdomen, and pelvis with MR failed to reveal an ectopic source for GH or GHRH. Serum chemistries, liver function values, and calcium, magnesium, and phosphorous levels were normal. Formal ophthalmological examination was unremarkable. Gastrin and parathyroid hormone levels were normal.
Random serum GH (70–120 ng/ml), serum IGF-I (300–350 ng/ml; normal age related 95th percentile, <105 ng/ml), and serum PRL (570–620 ng/ml; normal 0–11 ng/ml) levels were elevated. Results of TRH stimulation, iv insulin challenge, and glucose suppression tests are summarized in Fig. 3. There was evidence of stimulation of PRL and GH by TRH. GH failed to suppress during an oral glucose suppression test (1.75 g/kg). Next, basal GH levels and the response to GHRH were evaluated. GH and PRL levels were sampled at 20-min intervals over 6 h before and for 2 h after administration of GHRH (1 mg/kg). Discordant results were obtained for GH and PRL: basal GH and PRL levels were markedly elevated, with values 5- and 10-fold above the normal range, respectively. GHRH normally stimulates GH but not PRL secretion. However, after GHRH, both levels rose transiently, with a slightly greater response in GH than PRL, which suggests that pituitary cells were both GHRH responsive and made and secreted both hormones. The patient had suboptimal diurnal TSH variability, with a ratio of nocturnal to diurnal average of 1.4 (normal >1.5), although the TSH and free T4 levels were in the normal range. Diurnal cortisol secretion and ACTH-stimulated cortisol responses were normal. Serum GHRH levels ranged from 21 to 23 pg/ml (reference range <50 pg/ml) (5).
Fig. 3.
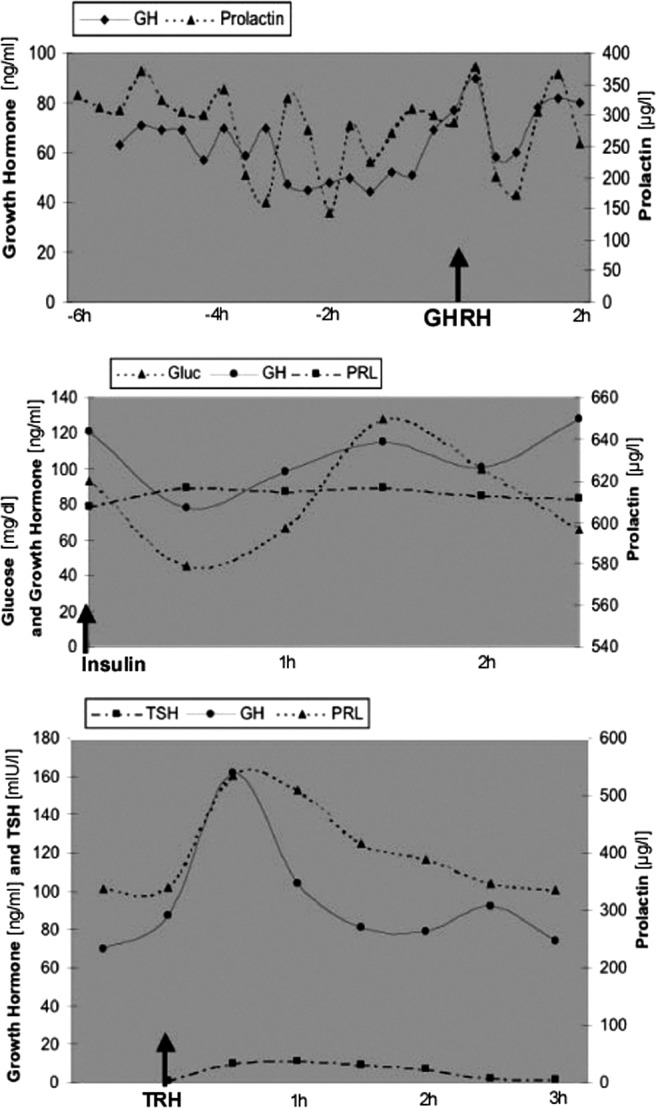
Results of basal and provocative biochemical testing in case 1. Top panel, GH and PRL levels increase immediately after GHRH stimulation. Middle panel, GH reaches its nadir of 80 ng/ml 30 min after the insulin challenge, when the glucose level reaches 40 mg/dl. Bottom panel, Serum GH and PRL peak approximately 30 min after TRH injection.
MR imaging of the brain and sella revealed a homogeneous and diffusely enlarged pituitary gland, which measured approximately 22 mm in width, 15 mm in height, and 12 mm in depth but no visible tumor; the gland enhanced modestly and homogeneously after gadolinium administration. MR imaging of the chest, abdomen, and pelvis failed to reveal an ectopic tumor, as did octreotide scanning. At a second transphenoidal surgery, when the dura was opened, the gland appeared diffusely and homogeneously enlarged with no evidence of tumor. Several vertical incisions were made in the gland, from side to side, extending deeper in stages from the anterior surface to the interface with the posterior pituitary; an adenoma was not identified. Because no tumor was seen, rather than performing a total hyposysectomy in a 4-yr-old child without a diagnosis and to permit analysis of the pituitary tissue to establish a diagnosis, the left half of the anterior lobe of the pituitary was removed. The patient recovered from surgery without difficulty.
Microscopic views of the tumor revealed that the pituitary architecture was grossly preserved in the presence of marked hyperplasia of GH- and PRL-secreting cells, which were interspersed with the other pituitary cell types (Fig. 4A). The somatotroph and mammotroph cell numbers were markedly increased. The expanded acini were composed of acidophilic cells. Reticulin staining demonstrated that the reticulin fibers surrounding each acinus were largely intact but markedly expanded by the proliferation of pituicytes. Immunohistochemical staining was positive for GH and PRL, with dual staining of most cells. On PRL staining, perinuclear inclusions were identified. Staining for ACTH, TSH, and LH showed rare positive cells scattered among the hyperplastic cells; staining for GHRH was positive (Fig. 4A and data not shown). Electron microscopy was used to examine these hyperplastic cells (Fig. 4B). Immunolabeling was performed with antibodies directed against GH and prolactin. Most cells were positive for PRL and GH: the majority of cells contained numerous neuroendocrine granules positive for both prolactin and GH; occasional granules contained prolactin or GH only (Fig. 4B).
Fig. 4.

A, Pathological features of case 1. Panels A and B, Hematoxylin and eosin sections of the pituitary lesion at medium (×200) and high-power (×400) magnifications, respectively. Enlarged acini are nearly entirely filled with acidophilic endocrine cells. Panel C, Reticulin stain (×200) demonstrates acinar expansion caused by pituitary hyperplasia. Panels D and E, Immunohistochemical staining with GH (×200 and ×400). Panels F and G, Immunohistochemical staining with PRL (×200 and 400, respectively), demonstrates perinuclear inclusion bodies (arrows in G). Panel H, Immunohistochemical staining with ACTH through the same sections as panels D–G shows rare positive cells scattered among the PRL- and ACTH-positive cells. B, Ultrastructural features of case 1. Immunolabeling of pituitary hyperplasia with antibodies directed against GH (small gold beads, ∼10 nm in diameter) and PRL (large gold beads, ∼20 nm in diameter). Most cells are positive for PRL and GH. Within the individual cells, numerous neuroendocrine granules contain both PRL and GH, and occasional granules contain PRL or GH only. In A, black bar indicates 5 μm distance; B, 1 μm distance; and C, 0.5 μm.
Molecular genetic analysis was performed. High-resolution spectral karyotypic (SKY) analysis of metaphase spreads of peripheral blood lymphocytes from cases 1 and 2 (below) were normal. Single-strand conformation polymorphism analysis of the promoter and coding sequence of the GHRH gene, as well as the coding sequence of both POU1F1(Pit-1) and PROP1 failed to demonstrate abnormalities in these genes. Comparative genomic hybridization (CGH) analysis of the hyperplastic pituicytes demonstrated chromosomal region loss on 11p and 14q, with smaller regions of loss on the distal arms of 2p and 2q; regions of heterochromatin near the pericentrosomal regions, which are not thought to be transcriptionally active, were excluded from the analysis. There were no alterations in the regions of 2p16, 11q13, or 17q22–24 or on chromosome 20.
After confirming the diagnosis and in the face of persistent elevation of GH, 2 wk later the remainder of the pituitary gland was surgically removed, leaving a small remnant of the pituitary stalk. The posterior pituitary was left intact. There were no complications and the postoperative recovery was uneventful. After surgery, both morning GH and PRL rapidly decreased to low normal levels by the first postoperative day (GH 1.6 ng/ml; PRL 6 ng/ml). IGF-I reached normal, age-adjusted levels by postoperative d 3. The patient was placed on hydrocortisone and thyroid replacement immediately after surgery.
Case 2
The mother was reported in 1981 as the “youngest example of pituitary gigantism on record” (4). She presented in 1970 at 21/2 yr age with a history of increased growth velocity beginning at 1 yr old (Fig. 1) associated with acral thickening, excess perspiration, and body odor. Random serum GH ranged from 60 to 109 μg/liter and did not suppress with an oral glucose load; the patient had an exaggerated insulin response to an intravenous glucose bolus. GH levels reached 200 μg/liter in response to insulin, with a glucose nadir of 40 mg/dl (2.2 mmol/liter). PRL levels were elevated 18-fold using the pigeon crop assay. The sella turcica was symmetrically enlarged on skull x-rays. GH increased from 81 to 200 ng/ml during hypoglycemia induced by iv insulin.
In 1971 at the age of 3 yr, an adenoma was removed by a near total hypophysectomy via frontal craniotomy, leaving a nubbin of pituitary gland attached to the pituitary stalk (4). Histopathological findings were interpreted as a mixed pituitary adenoma with eosinophilic features. Because reticulin stain and immunohistochemistry was not performed, hyperplasia cannot be excluded. However, most of the pituitary was removed because she developed panhypopituitarism requiring replacement therapy. Surgery reduced GH and PRL levels to slightly above the normal range. One year after surgery, she had a marked reduction of her growth rate, despite GH levels of 6–8 ng/ml and a normal IGF-I level, and at 5 yr of age, her height was within the 97th percentile. Reassessment at 5 yr after surgery revealed low growth velocity and low random plasma GH levels with no rise during electroencephalogram-verified stage IV sleep (4). GH replacement resulted in normalization of growth velocity. Since that time, she has required glucocorticoid, T4, and cyclical estrogen replacement. She also required pubertal induction. She married and conceived with assisted reproduction. She gave birth to her two boys in 1995 (case 1) and 2001 (case 3). She has had no evidence of recurrent excess GH or PRL secretion. In 1999, at age 33 yr, her basal GH levels were 1.5–2.3 ng/ml, with a peak of 5.9 ng/ml after TRH stimulation. Pituitary MR imaging at the NIH in 2005 at age 37 yr revealed no evidence of tumor, and serum GH and PRL were 1.3 and 24 ng/ml, respectively. GH and PRL suppressed upon oral glucose load.
Case 3
The second son had celiac disease diagnosed shortly after birth, which limited his growth initially. He was treated for this and then had normal development until 14 months of age, when rapid and excess growth occurred (Fig. 1). His random serum GH was 27.2 μg/liter, his IGF-I was 326 ng/ml, and his PRL was 55.5 ng/ml. Pituitary MR imaging revealed a symmetrical and slightly enlarged pituitary gland without evidence of an adenoma (Fig. 2B). He was referred to the NIH at the age of 4 yr for which there was a paradoxical increase of GH from 24.8 to 43.0 ng/ml during an oral glucose load test. Serum IGF-I was 462 ng/ml (normal age related 95th percentile, <105 ng/ml), and PRL was 48 ng/ml. Serum GHRH levels were within the normal range. Considering the prior findings in his mother and brother, we anticipated that he would exhibit primary mammosomatotroph hyperplasia of the pituitary gland, and at surgery, after no adenoma was identified, total hypophysectomy was performed. Histopathological examination demonstrated that the entire anterior pituitary gland was characterized by diffuse hyperplasia of mammosomatotrophs (Fig. 5). As had occurred in his brother, there was no normal pituitary in the specimen, and no foci of adenoma were observed on serial sections of the paraffin block. Furthermore, by 5 d after surgery, serum GH, PRL, and IGF-I levels were 0.1, less than 1, and 72 ng/ml, respectively.
Fig. 5.

A, The surgical specimen of case 3 consisted of homogeneous tissue with no evidence of adenoma. Immunohistochemistry for GH in ×1 (B) and ×40 (D) magnification revealed staining of the entire specimen with accentuation in the areas that look white on macroscopic examination (A). Immunohistochemistry for PRL (C and E) also revealed staining of the entire specimen; however, the areas that appeared pink on macroscopic examination were accentuated (C: ×1, E: ×40 magnification).
Clinical material and patient information
The boys (cases 1 and 3) and their mother (case 2) were evaluated at the NIH under NIH Institutional Review Board-approved protocols. No pathological material from the mother was available, but her old and current medical records were evaluated.
Endocrine evaluation
Assays for GHRH were performed by two separate clinical testing facilities: Inter Science Institute (Inglewood, CA; normal range for GHRH 5–18 pg/ml) and Quest Diagnostics (San Juan Capistrano, CA; normal range, <50 pg/ml). IGF-I levels were determined at the Mayo Medical Laboratories (Mayo Clinic, Rochester, MN); control values for a male, from 0 to 5 yr were: fifth percentile, 0 ng/ml; 50th percentile, 31 mg/ml; and 95th percentile, 105 ng/ml.
Tissue procurement
The largest part of the tissue of the two boys was fixed in formalin and oriented and stored in a tissue cassette permitting correlation of macroscopic and microscopic findings (Fig. 5). For Western blot analyses, tissue pieces were embedded in optimal cutting temperature compound, snap frozen. and stored at −80 C until use. Frozen sections were examined by hematoxylin and eosin staining and reticulin immunohistochemistry. Other portions of the tissue were embedded in glutaraldehyde for electron microscopy.
Microscopic evaluation and immunohistochemistry
Serial sections were taken from paraffin-embedded tissue blocks. Immunohistochemistry was performed following standard protocols as previously described in detail (6). GHRH primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used in a 1:200 dilution. The presence and intensity of antibody expression were examined in conjunction with hematoxylin and eosin sections.
Western blot analysis
Protein extraction was performed by three cycles of quick freezing and thawing. The concentrations of protein were measured with the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Forty micrograms of extracted protein were subjected to 4–20% SDS-polyacrylamide gradient gel (Invitrogen, Carlsbad, CA) electrophoresis, transferred to nitrocellulose (Invitrogen), and primarily probed primarily with GHRH antiserum (Active Motif, Inc., Carlsbad, CA) at a 1:200 dilution and horseradish peroxidase-conjugated second antibody (Santa Cruz Biotechnology) was applied at a 1:20,000 dilution. β-Actin monoclonal antibody (Sigma, St. Louis, MO) at a 1:500 dilution was used for internal protein quantity control. Horseradish peroxidase-conjugated second antibody (Santa Cruz Biotechnology) was applied at a 1:20,000 dilution. Signal was detected by enhanced chemiluminescence substrate (Pierce, Rockford, IL). Normal brain was used as negative control.
Molecular genetics
Comparative genomic hybridization
Genomic DNA from peripheral blood from case 2 and from the blood and pituitary tissue of case 1 as well as from a normal male donor was extracted by proteinase-K digestion using the QIAGEN Dneasy tissue kit, according to the manufacturer's instructions (QIAGEN, Valencia, CA). Metaphase chromosome spreads were prepared from peripheral blood leukocytes from the healthy male donor, as previously described (7).
Spectral karyotyping
SKY was performed on metaphase spreads of peripheral blood lymphocytes obtained from both case 1 and case 2, according to previously described protocols (8).
Mutation analysis
Blood and tissue DNA was investigated using single-strand conformation polymorphism analysis of the promoter and coding regions of the GHRH gene and coding regions of PROP1 and POU1F1 using standard techniques and previously described primers and conditions (9).
Cell culture and sequencing of GHRH receptor
Tissue obtained at the surgery of case 1 was established in tissue culture. Initial high GH levels became undetectable by 4 wk, coinciding with loss of immunostaining for GH and PRL. This is consistent with the observation that nonneoplastic pituitary cells are difficult to culture and that attempts to culture them frequently result in overgrowth of populations of supporting cells (9). An activating mutation of the GHRH receptor gene was also considered as a possible explanation for the clinical findings. To examine this possibility, total RNA was extracted from the pituitary tissue, purified, and reverse transcribed with appropriate controls. PCR was then used to amplify cDNA products, which were run on a 1.0% agarose gel, stained with ethidium bromide. The 1.3-kb DNA amplification product (corresponding to the size of the GHRH receptor) was purified. DNA products from two independent PCR were subcloned to control for nucleotide misincorporation resulting from PCR. Seven clones from two PCR were completely sequenced in both directions. Inserted fragments were sequenced using a LICOR 4000LD analyzer (LICOR, Lincoln, NE). These sequences were identical with the GHRH receptor sequence published by Gaylinn et al. (10). They all have an alanine residue at position 178 and therefore differ from the sequence published by Mayo et al., which has an arginine at position 178 (11).
Results and Discussion
We report hereditary pituitary hyperplasia in humans. The three patients had extraordinary early onset of pituitary gigantism associated with elevated GH, PRL, and IGF-I but no abnormality of the other pituitary axes. Examination of the pituitary of the two boys demonstrated diffuse mammosomatotroph hyperplasia of the entire anterior pituitary gland with cosecretion of GH and PRL by the same cells. Removal of the anterior pituitary in the mother and both boys led to rapid reversal of excess GH, PRL, and IGF-I secretion to normal or low normal levels, with deceleration of excessive growth, and, in the distant postoperative period, a more normal growth pattern.
Hypersecretion of GH during childhood, when open epiphyseal plates permit linear growth, results in gigantism (12). Children with gigantism may also have acromegalic physical features, such as enlargement of the hands and feet, coarse facies, prognathism, and soft tissue thickening (13). Biochemical features in children are similar to those of acromegalic adults and include elevations in serum GH levels and IGF-I, a failure to suppress GH after oral glucose administration, and a paradoxical rise in GH after infusion of TRH. Approximately 100 cases of children with gigantism have been described (13–15).
Excessive GH secretion may be considered to arise in one of two ways, from a primary pituitary source or as the result of increased GHRH secretion (12). Most cases of gigantism are caused by a pituitary adenoma or hyperplasia, arising sporadically or in association with MEN 1, CNC (16), or MAS (12, 17–19). Alternatively, most cases of secondary GH excess result from ectopic or hypothalamic GHRH excess (12, 20, 21).
Regardless of the etiology, the consequences of prolonged GH excess in childhood are similar: growth acceleration. Although sporadic cases of mammosomatotrope hyperplasia has been previously described [reviewed by Horvath (22)], this is the first report of familial gigantism presenting in infancy in two successive generations, most likely in a dominantly inherited fashion.
Disorders that may cause hyperplasia of GH-secreting cells include MAS (19), MEN, CNC (16), and lesions of the hypothalamus or elsewhere (ectopic) that secrete GHRH (12, 20, 21). MAS-causing gigantism has been described in approximately 20 children, although the exact nature of the pituitary disorder, hyperplasia or adenoma, is controversial (12, 18, 19, 23). MAS is the result of a specific mutation in GNAS1 on chromosome 20q13.2 (an Arg201 to His201 somatic mutation) and is characterized by polyostotic fibrous dysplasia, pigment patches of the skin, and endocrinological abnormalities, including precocious puberty, thyrotoxicosis, pituitary gigantism, and Cushing's syndrome (17). Our patients' clinical, endocrine, and histological features are similar to a child with infantile gigantism, mammosomatotroph hyperplasia, and normal serum levels of GHRH previously described by Moran et al. (24), although in that instance there was no family history and the potential etiology remained unclear. It is possible that that isolated, nonfamilial case could be caused by MAS in which the mosaic affected only the pituitary.
None of our patients had evidence of fibrous dysplasia of the skull base in the region of the pituitary, which is a consistent feature of patients with MAS. Thus, because our patients must have the mutant gene present in their germline, because none of the patients had other phenotypic features, and because the activating mutation in MAS occurs as a mosaic state and is believed to be embryonically lethal in the nonmosaic state, MAS can be eliminated as the cause of this inherited pituitary hyperplasia.
Although GH hypersecretion may occur in CNC and MEN 1, GH excess is a relatively minor feature of these disorders, usually occurring in the third to fourth decade. CNC is an autosomal dominant disorder in which patients commonly have atrial myxomas, spotty skin pigmentation, and Cushing's syndrome due to primary pigmented adrenocortical disease. Occasionally patients also, generally in their third or fourth decade, have pituitary tumors that produce GH and PRL associated with mammoosommatotroph hyperplasis. MEN 1 is commonly associated with pituitary tumors (usually prolactinomas), parathyroid tumors associated with hyperkalemia, and pancreatic tumors. In addition, the extraordinary early onset of disease, mammosomatotroph hyperplasia, not pituitary tumors, and the fact that the mother has reached her 30s with no other manifestations of either of these disorders (regular screening for thyroid and parathyroid disease and examination of skin for cutaneous stigmata) makes these diagnoses unlikely. Although in the absence of genetic sequencing we have not eliminated the possibility of mutations of the genes associated with these disorders, CGH failed to demonstrate evidence of genetic abnormalities involving either MEN 1 (chromosome 11q13) or in the two loci postulated to be involved in CNC (located on chromosomes 2p16 and 17q22–24), and the clinical picture is inconsistent with them.
Finally, although alterations in POU1F1 (located on chromosome 3p11) and PROP1 (located on 5q), which influence pituitary cell differentiation during embryonic development, have clinically been established only in states of pituitary insufficiency, analysis of the promoter regions of both genes failed to demonstrate alterations of any type. This is supported on the global level by the lack of CGH alterations involving either locus and by the absence of alterations in either case by spectral karyotyping of normal peripheral blood lymphocytes.
Loss of genetic material was detected reproducibly from the long arm of chromosome 14 and the short arm of chromosome 11. A review of known genes from 14q failed to identify a possible candidate. The short arm of chromosome 11 contains a large number of genes important in tumorigenesis. A minimum of 1500 genes have been identified to date along the length of chromosome 11p, several of which are of interest in relation to our patients, including GTF2H1, a general, nuclear transcriptional factor (25); RAG1 and RAG2, which are involved in recombination events (26); GAS2 (general arrest specific protein 2), involved in growth cycle arrest (27); WT1, the Wilm's tumor 1 protein that can act as a transcriptional activator or repressor depending on cellular or chromosomal context (28); BHC80 (BRAF/HDAC complex, 80 kDa subunit), which mediates gene repression in neuronal and neuroendocrine genes (29); DDB2, the damage-specific DNA binding protein 2 (30); and KAI1 (kangai 1), a tumor suppressor gene, which is activated by p53 (31).
Chromosome11p15.5, in particular, is a region of some interest. It is one of the two large imprinted domains in the human genome (the other being 15q11-q13). It is genetically defined proximally by marker D11S12 and distally by D11S1318. Genes in this region include insulin-like growth factor-II (IGF-II) (32), which is expressed when of paternal origin, and a series of other genes (ASCL2, KCNQ1, p57/KIP2), and two tumor-suppressing, subchromosomal transferable cDNA fragment (TSSC5 and TSSC3) regions, which are expressed from the maternal allele. All are thought to play a role in or to be the subject of epigenetic regulation and functional imprinting. Although most of the known genes acting as tumor suppressor genes in this region are ubiquitously expressed, it is possible that interaction of some proteins from genes on 11p interact with other proteins, which may confer tissue specificity in much the same way that some mutations of the ret oncogene result in isolated familial medullary thyroid carcinoma.
Certain abnormal circumstances, all of which are rare, are associated with pituitary somatotroph hyperplasia [reviewed by Horvath (22)]. Excessive GHRH secretion by hypothalamic or extracranial tumors is generally associated with abnormally high levels of serum GHRH, and when it arises in later life, it mainly affects somatotrophs. Secondary causes of mammosomatotroph hyperplasia were investigated. GHRH excess, from an ectopic source, was not identified. Serum GHRH levels were within the normal range in both boys. Mutational analysis of the promoter and coding regions of GHRH in case 1 was normal.
Altered pituitary differentiation probably underlies the features observed here. Mammosomatotroph cells are very rare in the adult pituitary gland but are abundant during development (33). The anterior pituitary undergoes rapid cellular differentiation during the first 12 wk, by which time the major secretory cell compartments are structurally and functionally intact, except lactotrophs. The differentiation of lactotrophs and somatotrophs is controlled by several factors, including regulated expression of the transcription factors prophet of POU1F1 (PROP1) and POU1F1. However, alterations in PROP1 and POU1F1 or their protein products have been established only in states of pituitary insufficiency. Furthermore, in our patients mutation analysis of the coding sequences of PROP1 and POU1F1 and the promoter regions of both genes failed to demonstrate abnormalities. This is supported by the lack of CGH alterations involving either locus and the absence of alterations in either case by spectral karyotyping of normal peripheral blood lymphocytes. Similarly, there were no alterations identified by either CGH or SKY that involved other genes of interest, including: 16p13 [pituitary adenoma predisposition, Mendelian Inheritance in Man (MIM) 102200], 11q13.3 (aryl hydrocarbon receptor-interacting protein, MIM 605555); 7p15 (GHRH receptor, MIM 139190), or 12p13 (multiple endocrine neoplasia, type IV, MIM 610755), which are involved in or thought to play a potential pathogenetic role in some GH-secreting pituitary adenomas (http://www.ncbi.nlm.nih.gov/omim).
The hyperplastic pituitary contained abnormal expression of GHRH (Fig. 6). GHRH is a hypothalamic-releasing factor initially isolated from a pancreatic islet tumor in a patient with acromegaly (34). Hormonal and metabolic factors control its expression [reviewed by Müller et al. (35)]. GHRH is not expressed in the pituitary gland under physiological conditions (36); however, it has been found in some somatotroph pituitary adenomas (37), and it functions as an autoendocrine loop in certain other types of human neoplasia (38). Postnatal GHRH stimulates mature somatotrophs only (35).
Fig. 6.

A, Western blot revealed that GHRH was expressed in two separate samples of anterior pituitary gland (lanes 1 and 3) from case 3. A small piece of posterior lobe, which had been attached to the surgical specimen, did not show expression of GHRH (lane 2). As negative controls we used a pituitary specimen from a patient with sporadic mammosomatotroph hyperplasia (McCune-Albright, lane 4) and a piece of normal brain (lane 5). B and C, Immunohistochemistry with anti-GHRH confirms the expression of GHRH by a subset of cells distributed within the tissue surrounded by the majority of the cells that do not express GHRH. The positive cells were assembled in groups (B, ×10; C, ×40 magnification).
The most likely circumstance in our patients is that the mammosomatotrophs, which are abundant during normal human embryogenesis, simply persisted and expanded due to adjacent GHRH-secreting cells. Mice transgenic for GHRH develop pituitary gigantism of very early onset associated with hyperplasia of mammosomatotrophs in which GH and PRL are expressed in the same cells and in the same secretory granules (1–3, 39). In these mice GHRH is expressed in the pituitary gland and in other tissues (2, 39). Furthermore, in humans excess secretion of GH and PRL associated with extensive mammosomatotroph hyperplasia occurs almost uniquely with congenital gigantism (12, 24, 40) and has been observed in very young children with gigantism associated with ectopic GHRH secretion (15, 41) but has not been a prominent feature in adults with ectopic GHRH secretion. These observations indicate that chronic exposure of the developing, but not the adult, pituitary to high levels of GHRH stimulates hyperplasia of mammosomatotrophs and excess secretion of GH and PRL.
Thus, we investigated frozen pituitary tissue of case 3 for GHRH expression. GHRH was expressed in two separate samples of anterior pituitary but not in control tissues; immunohistochemistry with anti-GHRH demonstrated expression of GHRH in the pituitary specimen of both boys, and they both had the same unusual distribution of GHRH staining; GHRH was expressed by only a subset of cells (Fig. 6, B and C), which were frequently assembled in clusters and which were observed throughout the pituitary, whereas the majority of cells expressed no GHRH. Our observations suggest that the hereditary mammosomatotroph hyperplasia that we describe here is a paracrine effect of GHRH emanating from clusters of GHRH-expressing cells.
Acknowledgments
We acknowledge Dr. Maria Tsokos and Mones Abu-Asab (Laboratory of Pathology, National Cancer Institute) for electron microscopy as part of the clinical diagnostics. We also acknowledge Dr. John Heiss for providing brain tissue removed during epilepsy surgery for control.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke. S.G. is recipient of a grant from the German Research Society.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CGH
- Comparative genomic hybridization
- CNC
- Carney complex
- MAS
- McCune-Albright syndrome
- MEN 1
- multiple endocrine neoplasia syndrome type 1
- MR
- magnetic resonance
- PRL
- prolactin
- SKY
- spectral karyotypic.
References
- 1. Lloyd RV, Jin L, Chang A, Kulig E, Camper SA, Ross BD, Downs TR, Frohman LA. 1992. Morphologic effects of hGRH gene expression on the pituitary, liver, and pancreas of MT-hGRH transgenic mice. An in situ hybridization analysis. Am J Pathol 141:895–906 [PMC free article] [PubMed] [Google Scholar]
- 2. Mayo KE, Hammer RE, Swanson LW, Brinster RL, Rosenfeld MG, Evans RM. 1988. Dramatic pituitary hyperplasia in transgenic mice expressing a human growth hormone-releasing factor gene. Mol Endocrinol 2:606–612 [DOI] [PubMed] [Google Scholar]
- 3. Asa SL, Kovacs K, Stefaneanu L, Horvath E, Billestrup N, Gonzalez-Manchon C, Vale W. 1990. Pituitary mammosomatotroph adenomas develop in old mice transgenic for growth hormone-releasing hormone. Proc Soc Exp Biol Med 193:232–235 [DOI] [PubMed] [Google Scholar]
- 4. Espiner EA, Carter TA, Abbott GD, Wrightson P. 1981. Pituitary gigantism in a 31 month old girl: endocrine studies and successful response to hypophysectomy. J Endocrinol Invest 4:445–450 [DOI] [PubMed] [Google Scholar]
- 5. Penny ES, Penman E, Price J, Rees LH, Sopwith AM, Wass JA, Lytras N, Besser GM. 1984. Circulating growth hormone releasing factor concentrations in normal subjects and patients with acromegaly. Br Med J (Clin Res Ed) 289:453–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gläsker S, Lonser RR, Tran MG, Ikejiri B, Butman JA, Zeng W, Maxwell PH, Zhuang Z, Oldfield EH, Vortmeyer AO. 2005. Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res 65:10847–10853 [DOI] [PubMed] [Google Scholar]
- 7. Pack SD, Qin LX, Pak E, Wang Y, Ault DO, Mannan P, Jaikumar S, Stratakis CA, Oldfield EH, Zhuang Z, Weil RJ. 2005. Common genetic changes in hereditary and sporadic pituitary adenomas detected by comparative genomic hybridization. Genes Chromosomes Cancer 43:72–82 [DOI] [PubMed] [Google Scholar]
- 8. Padilla-Nash HM, Barenboim-Stapleton L, Difilippantonio MJ, Ried T. 2006. Spectral karyotyping analysis of human and mouse chromosomes. Nat Protoc 1:3129–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weil RJ, Huang S, Pack S, Vortmeyer AO, Tsokos M, Lubensky IA, Oldfield EH, Zhuang Z. 1998. Pluripotent tumor cells in benign pituitary adenomas associated with multiple endocrine neoplasia type 1. Cancer Res 58:4715–4720 [PubMed] [Google Scholar]
- 10. Gaylinn BD, Harrison JK, Zysk JR, Lyons CE, Lynch KR, Thorner MO. 1993. Molecular cloning and expression of a human anterior pituitary receptor for growth hormone-releasing hormone. Mol Endocrinol 7:77–84 [DOI] [PubMed] [Google Scholar]
- 11. Mayo KE, Cerelli GM, Lebo RV, Bruce BD, Rosenfeld MG, Evans RM. 1985. Gene encoding human growth hormone-releasing factor precursor: structure, sequence, and chromosomal assignment. Proc Natl Acad Sci USA 82:63–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eugster EA, Pescovitz OH. 1999. Gigantism. J Clin Endocrinol Metab 84:4379–4384 [DOI] [PubMed] [Google Scholar]
- 13. Pandey P, Ojha BK, Mahapatra AK. 2005. Pediatric pituitary adenoma: a series of 42 patients. J Clin Neurosci 12:124–127 [DOI] [PubMed] [Google Scholar]
- 14. Abe T, Tara LA, Lüdecke DK. 1999. Growth hormone-secreting pituitary adenomas in childhood and adolescence: features and results of transnasal surgery. Neurosurgery 45:1–10 [DOI] [PubMed] [Google Scholar]
- 15. Gelber SJ, Heffez DS, Donohoue PA. 1992. Pituitary gigantism caused by growth hormone excess from infancy. J Pediatr 120:931–934 [DOI] [PubMed] [Google Scholar]
- 16. Boikos SA, Stratakis CA. 2006. Pituitary pathology in patients with Carney complex: growth-hormone producing hyperplasia or tumors and their association with other abnormalities. Pituitary 9:203–209 [DOI] [PubMed] [Google Scholar]
- 17. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. 1991. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325:1688–1695 [DOI] [PubMed] [Google Scholar]
- 18. Dötsch J, Kiess W, Hänze J, Repp R, Lüdecke D, Blum WF, Rascher W. 1996. Gs α mutation at codon 201 in pituitary adenoma causing gigantism in a 6-year-old boy with McCune-Albright syndrome. J Clin Endocrinol Metab 81:3839–3842 [DOI] [PubMed] [Google Scholar]
- 19. Akintoye SO, Chebli C, Booher S, Feuillan P, Kushner H, Leroith D, Cherman N, Bianco P, Wientroub S, Robey PG, Collins MT. 2002. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab 87:5104–5112 [DOI] [PubMed] [Google Scholar]
- 20. Sano T, Asa SL, Kovacs K. 1988. Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev 9:357–373 [DOI] [PubMed] [Google Scholar]
- 21. Asa SL, Scheithauer BW, Bilbao JM, Horvath E, Ryan N, Kovacs K, Randall RV, Laws ER, Jr, Singer W, Linfoot JA. 1984. A case for hypothalamic acromegaly: a clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab 58:796–803 [DOI] [PubMed] [Google Scholar]
- 22. Horvath E. 1988. Pituitary hyperplasia. Pathol Res Pract 183:623–625 [DOI] [PubMed] [Google Scholar]
- 23. Keil MF, Stratakis CA. 2008. Pituitary tumors in childhood: update of diagnosis, treatment and molecular genetics. Expert Rev Neurother 8:563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moran A, Asa SL, Kovacs K, Horvath E, Singer W, Sagman U, Reubi JC, Wilson CB, Larson R, Pescovitz OH. 1990. Gigantism due to pituitary mammosomatotroph hyperplasia. N Engl J Med 323:322–327 [DOI] [PubMed] [Google Scholar]
- 25. Fischer L, Gerard M, Chalut C, Lutz Y, Humbert S, Kanno M, Chambon P, Egly JM. 1992. Cloning of the 62-kilodalton component of basic transcription factor BTF2. Science 257:1392–1395 [DOI] [PubMed] [Google Scholar]
- 26. Nagawa F, Hirose S, Nishizumi H, Nishihara T, Sakano H. 2004. Joining mutants of RAG1 and RAG2 that demonstrate impaired interactions with the coding-end DNA. J Biol Chem 279:38360–38368 [DOI] [PubMed] [Google Scholar]
- 27. Brancolini C, Bottega S, Schneider C. 1992. Gas2, a growth arrest-specific protein, is a component of the microfilament network system. J Cell Biol 117:1251–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al. 1990. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell 60:509–520 [DOI] [PubMed] [Google Scholar]
- 29. Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. 2002. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA 99:7420–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S. 1995. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics 29:62–69 [DOI] [PubMed] [Google Scholar]
- 31. Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, Barrett JC. 1995. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 268:884–886 [DOI] [PubMed] [Google Scholar]
- 32. Jansen M, van Schaik FM, Ricker AT, Bullock B, Woods DE, Gabbay KH, Nussbaum AL, Sussenbach JS, Van den Brande JL. 1983. Sequence of cDNA encoding human insulin-like growth factor I precursor. Nature 306:609–611 [DOI] [PubMed] [Google Scholar]
- 33. Asa SL, Kovacs K, Horvath E, Losinski NE, Laszlo FA, Domokos I, Halliday WC. 1988. Human fetal adenohypophysis. Electron microscopic and ultrastructural immunocytochemical analysis. Neuroendocrinology 48:423–431 [DOI] [PubMed] [Google Scholar]
- 34. Rivier J, Spiess J, Thorner M, Vale W. 1982. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumour. Nature 300:276–278 [DOI] [PubMed] [Google Scholar]
- 35. Müller EE, Locatelli V, Cocchi D. 1999. Neuroendocrine control of growth hormone secretion. Physiol Rev 79:511–607 [DOI] [PubMed] [Google Scholar]
- 36. Bloch B, Brazeau P, Ling N, Bohlen P, Esch F, Wehrenberg WB, Benoit R, Bloom F, Guillemin R. 1983. Immunohistochemical detection of growth hormone-releasing factor in brain. Nature 301:607–608 [DOI] [PubMed] [Google Scholar]
- 37. Levy A, Lightman SL. 1992. Growth hormone-releasing hormone transcripts in human pituitary adenomas. J Clin Endocrinol Metab 74:1474–1476 [DOI] [PubMed] [Google Scholar]
- 38. Chatzistamou I, Schally AV, Pafiti A, Kiaris H, Koutselini H. 2002. Expression of growth hormone-releasing hormone in human primary endometrial carcinomas. Eur J Endocrinol 147:381–386 [DOI] [PubMed] [Google Scholar]
- 39. Stefaneanu L, Kovacs K, Horvath E, Asa SL, Losinski NE, Billestrup N, Price J, Vale W. 1989. Adenohypophysial changes in mice transgenic for human growth hormone-releasing factor: a histological, immunocytochemical, and electron microscopic investigation. Endocrinology 125:2710–2718 [DOI] [PubMed] [Google Scholar]
- 40. Felix IA, Horvath E, Kovacs K, Smyth HS, Killinger DW, Vale J. 1986. Mammosomatotroph adenoma of the pituitary associated with gigantism and hyperprolactinemia. A morphological study including immunoelectron microscopy. Acta Neuropathol 71:76–82 [DOI] [PubMed] [Google Scholar]
- 41. Zimmerman D, Young WF, Jr, Ebersold MJ, Scheithauer BW, Kovacs K, Horvath E, Whitaker MD, Eberhardt NL, Downs TR, Frohman LA. 1993. Congenital gigantism due to growth hormone-releasing hormone excess and pituitary hyperplasia with adenomatous transformation. J Clin Endocrinol Metab 76:216–222 [DOI] [PubMed] [Google Scholar]