

Abstract
Shigella dysenteriae serotype 1 (S. dysenteriae 1) is unique among the Shigella species and serotypes in the expression of Shiga toxin which contributes to more severe disease sequelae and the ability to cause explosive outbreaks and pandemics. S. dysenteriae 1 shares characteristics with other Shigella species, including the capability of causing clinical illness with a very low inoculum (10 to 100 CFU) and resistance to multiple antibiotics, underscoring the need for efficacious vaccines and therapeutics. Following the demonstration of the successful attenuating capacity of deletion mutations in the guaBA operon in S. flexneri 2a vaccine strains in clinical studies, we developed a series of S. dysenteriae 1 vaccine candidates containing the fundamental attenuating mutation in guaBA. All strains are devoid of Shiga toxin activity by specific deletion of the gene encoding the StxA subunit, which encodes enzymatic activity. The StxB subunit was overexpressed in several derivatives by either plasmid-based constructs or chromosomal manipulation to include a strong promoter. All strains are attenuated for growth in vitro in the HeLa cell assay and for plaque formation and were safe in the Serény test and immunogenic in the guinea pigs. Each strain induced robust serum and mucosal anti-S. dysenteriae 1 lipopolysaccharide (LPS) responses and protected against wild-type challenge. Two strains engineered to overexpress StxB induced high titers of Shiga toxin neutralizing antibodies. These candidates demonstrate the potential for a live attenuated vaccine to protect against disease caused by S. dysenteriae 1 and potentially to protect against the toxic effects of other Shiga toxin 1-expressing pathogens.
INTRODUCTION
Within the genus Shigella, four species, including S. dysenteriae, S. flexneri, S. sonnei, and S. boydii, cause diarrheal disease and dysentery in humans. It is estimated that more than 160 million cases of shigellosis and 1.1 million deaths occur per year (24). Each species except S. sonnei contains multiple serotypes. S. dysenteriae serotype 1 (S. dysenteriae 1), the first member of the genus Shigella identified and named the Shiga bacillus, is unique among all of the Shigella in causing the most severe form of disease with the highest case fatality rates (25, 50). Like all Shigella, S. dysenteriae 1 causes disease following ingestion of contaminated food or water. The extremely low infectious dose (10 to 100 CFU) also facilitates person-to-person transmission (13). Shigella bacteria invade the intestinal epithelium through M cells and proceed to spread from cell to cell, causing death and sloughing of contiguously invaded epithelial cells and inducing a potent inflammatory response resulting in the characteristic dysentery syndrome (45). In addition to this series of pathogenic events, only S. dysenteriae 1 has the ability to elaborate the potent Shiga toxin that inhibits protein synthesis in eukaryotic cells and that may lead to extraintestinal complications, including hemolytic-uremic syndrome and death (4, 19).
S. dysenteriae 1 has caused explosive epidemics and pandemics in Central America in the 1960s and 1970s and more recently in Africa and Asia (5, 17, 29, 35, 47). Epidemics occur in regions where there is a low background level of immunity and in situations where there is crowding combined with poor hygiene, such as refugee camps. Under these conditions, S. dysenteriae 1 exhibits high attack rates in all age groups. The problem is compounded by the fact that epidemic strains are typically resistant to multiple antibiotics, thus limiting therapeutic options. The development of an S. dysenteriae 1 vaccine is a priority for the protection of individuals in less industrialized countries where S. dysenteriae 1 exerts its most significant burden and for the protection of the U.S. population which is considered a vulnerable target population if this pathogen were to be used as a biological weapon. Indeed, based on its characteristics of high virulence, low infectious dose, high person-to-person transmissibility, resistance to drug therapy, and the immunological susceptibility of the U.S. population, S. dysenteriae 1 was designated the highest priority category B pathogen on the NIAID list of priority pathogens (38).
Epidemiological and experimental evidence suggest that protective immunity to Shigella is directed to the O somatic antigen of the Shigella lipopolysaccharide (LPS) and is serotype specific (12, 16). Among the various vaccine strategies currently being pursued, live attenuated vaccines hold much promise in engendering serum and mucosal O-antigen-specific protective responses (26, 54). Recent clinical trials with S. flexneri 2a live attenuated vaccine strains demonstrate that targeted mutations in the guaBA operon, encoding critical enzymes in the guanine nucleotide biosynthetic pathway, markedly attenuated virulence yet preserved immunogenicity in volunteers (22, 23). These studies showed that clinical acceptability was increased with the deletion of genes encoding Shigella enterotoxins 1 and 2 (ShET-1 and ShET-2, respectively) from the guaBA mutant. Accordingly, we have constructed derivatives of S. dysenteriae 1 with deletions in the stxAB genes, the guaBA genes, as well as in sen, the gene encoding ShET-2. In vitro and in vivo preclinical studies demonstrated that these strains were attenuated for virulence compared to the wild type (WT) and safe, immunogenic, and protective in the guinea pig model.
In order to maximize the usefulness of these vaccine strains, we evaluated the overexpression of the Shiga toxin B subunit within the background of the ΔstxA ΔguaBA Δsen deletion strain. Shiga toxin is composed of a single A subunit that is enzymatically active and five B subunits that form a pentamer which is responsible for target cell binding. While the A subunit must be deleted in a live vaccine strain to completely abolish toxin enzymatic activity, the B subunit has been demonstrated to elicit antibodies that can neutralize toxin activity (1, 6, 8, 27). Here, a series of vaccine candidates were evaluated in the guinea pig model and found to protect against challenge with wild-type S. dysenteriae 1 and induce Shiga toxin neutralizing antibody responses.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Table 1 lists the Shigella strains used in these studies. Shigella strains were grown on tryptic soy agar (TSA) (Becton Dickinson, Sparks, MD) or on animal product-free medium, high-soy (Sigma-Aldrich, St. Louis, MO), Hy-Yest 412 yeast extract (TEKnova, Hollister, CA) or soytone (AthenaES, Baltimore, MD), with Congo red (0.02% solution) and guanine (0.005%). Escherichia coli was grown on Luria-Bertani (LB) agar. Carbenicillin and kanamycin were used at concentrations of 100 μg/ml and 50 μg/ml, respectively, where indicated. Growth curve studies were performed using half-LB medium containing 5 g Bacto tryptone, 2.5 g yeast, and 5 g sodium chloride in 1 liter and supplemented with 0.005% guanine where indicated or chloramphenicol (40 μg/ml).
Table 1.
S. dysenteriae serotype 1 strains used in these studies
| Strain | Genotype |
|---|---|
| 1617 | Wild type |
| CVD 1251 | 1617 ΔguaBA |
| CVD 1254 | 1617 ΔstxAB |
| CVD 1255 | 1617 ΔguaBA ΔsenΔstxAB |
| CVD 1256 | 1617 ΔguaBAΔsenΔstxA::mLpp-stxB |
| CVD 1257 | 1617 ΔguaBAΔsenΔstxA::trc-stxB |
| CVD 1258 | CVD 1255(pOmpC-StxB) |
| CVD 1259 | CVD 1255(pTrc-StxB) |
Molecular genetic techniques.
All primers used for PCR were generated at the University of Maryland Biopolymer core facility and are listed in Table 2. Restriction enzymes, ligase, and polymerase were purchased from New England BioLabs (Beverly, MA) and used according to the manufacturers' instructions.
Table 2.
Primers used in these studies
| Primer | Sequence |
|---|---|
| CVOL-13 | CTGCAGTATTCCCACTCAATGGTAGC |
| CVOL-41 | GAAGGAGTATTGCCCATGCTACGTATCG |
| EB1 | TAGCCTTGCATAATGGACGTGTA |
| F-10 | CATAAAAAACTTTGTGTTATACTT |
| R-10 | TAGCGTTGCTGGAGCAACCTG |
| F-35 | TTGTATATCGAAGCGCCCCT |
| R-35 | CACAAAGTTTTTTATGTTGTCAAT |
| F-Lpp | TAAGGATCCCTAAAACCCAGCGTTCGATGC |
| R-Lpp | TAAGGATCCGCGGCCGCATTAATACCCTCTAGATTG |
| Gua311 | ATGCTACGTATCGCTAAAGAAG |
| Gua3226 | CATTACGCCAACGGAACGTAC |
| LB-1 | CCCGGGCGCTGCGGTAACCACAC |
| LB-2 | AAGCTTGCAGTGAGCCCAACGCA |
| Red-senF | TTGGATTAGCTCAGTCAATCCCATACGCATCGCGTGATAAATAACTTTGGGGTGAAGTAAATTATCAGGCGTGTAGGCTGGAGCTGCTTC |
| Red-senR | CAACAACACTAAGTCTGCGTCACAACCCATCAATGAAAGGAATATATACATATGCCATCAGTAAATTTAACATATGAATATCCTCCTTA |
| StxB-1 | GGGCCCAGTTGAGGGGGTAAAATGAAAAAAACATTATTAATAGCTG |
| StxB-1 | GGATCCTCAACGAAAAATAACTTCGCTGAATCC |
| StxB-2409 | ATGCCCGGGAGTTGAGGGGGTAAAATGAAAAAAACATTA |
| StxB-2693 | ATCGAGCGGGATCCTCAACGAAAAATAACTTCGCTGAATCC |
| Trc-1 | GAATTCGATCCCGATCCTCGCATGGATGTTTTATAAAAAACATGATTGACAT |
| Trc-2 | CCCGGGGGCGGCCGCCCACACATTATAACCTATATGCAACATGATGTCAATCATGTTT |
| Wu016 | TAAAAGCTTGTGTAGGCTGGAGCTGCTTC |
| Wu017 | TAAGAATTCCCGGGCGGCCGCGGATCCATATGAATATCCTCCTTA |
| Wu029 | ATCCACCACTCTGATCGAGGTTC |
| Wu030 | GCTGAAGTCCGGAAGTTACAGG |
| Wu033 | TAGGCTAGCATGAAAAAAACATTATTAATAGC |
| Wu035 | TAGGCTAGCTCAACGAAAAATAACTTCGCTG |
| Wu036 | ATGAAAATAATTATTTTTAGAGTGC |
| Wu045 | ACTGCTAATAGTTCTGCGCATCAG |
| Wu046 | TAAAAGCTTTCGCATGAGATCTGACCAGAT |
| Wu047 | TAAGGATCCCCGGGTTACACAATACTCCTTGAGC |
| Wu048 | GTGAAGGTGAAGCCCGTGAAGT |
| Wu049 | TGCAGCAGCATTGCGGTTACG |
| Wu050 | TAAGGATCCAACAGGTCGTCATCCCACGT |
| Wu051 | TAATCTAGACCTGATATCGCCATCATGATTGC |
| Wu052 | TAATCTAGACTCAGAATAGCTCAGTGA |
| Wu053 | TAAAAGCTTCCCGGGTCCTGCACATCATGCAGAC |
| Wu061 | CGTTGTAGCGGTAGTCGAGCTGC |
| Wu064 | CGTAAATGAGTTGCTGTATGGTTGG |
| Wu065 | GTTACCGATCACTCTGGAAGACT |
| Wu125 | GAAGTTAGGGCTCGGATTATACCTGA |
| Wu126 | ATCTCTACTGAACGCGAGAAGCAGA |
| StxB-RTF | CGCCTGATTGTGTAACTGGA |
| StxB-RTR | TGCGCACTGAGAAGAAGAGA |
| rpoB-F | GCGTCTGGTGATCTATGAGCG |
| rpoB-R | CGTTGTCTGTCATGAGCGGA |
| gyrB-F | ACATTCGTACGCTGCTGTTGA |
| gyrB-R | CTTCACTTTGTACAGCGGCG |
Construction of strain CVD 1251.
S. dysenteriae 1 strain 1617 was the wild-type (WT) strain used to create all mutant derivative strains. The guaBA deletion was introduced by allelic exchange by the method of Noriega et al. for S. flexneri 2a (42). Suicide plasmid pFM726a contains regions flanking the guaBA genes that result in a 918-bp deletion in the operon following allelic exchange. Plasmid pFM726a is a pSC101-based suicide plasmid with a temperature-sensitive origin of replication and a sacB gene for counterselection. Plasmid pFM726a was electroporated into strain 1617. Colonies were selected at 37°C on medium with kanamycin to select for bacteria containing the integrated plasmid. These colonies were next grown on medium with sucrose to select those bacteria that lost the plasmid. The resulting colonies were screened for kanamycin sensitivity and an inability to grow on medium without guanine. The guaBA deletion was confirmed by diagnostic PCR and sequencing of the chromosomal region. This strain was named CVD 1251.
Complementation of guanine auxotrophy was tested with pGuaBA, a low-copy-number plasmid containing a minimal fragment with the guaBA operon under the control of the lactose promoter (PlacZ). First, a low-copy-number plasmid containing the lacZ gene and a multicloning site (MCS) was created. The lacZ gene and MCS were amplified from plasmid pGem5Z (Promega, Madison, WI) using primers LB-1 and LB-2. This fragment was cloned into HindIII-AvaI-digested pACYC184 to create pLoBlu. A 3.1-kb fragment containing the guaBA operon with an optimized ribosome binding site (GAAGGAG) 8 bp upstream of the start codon was amplified from genomic DNA using primers CVOL-13 and CVOL-41 and ligated as a NotI fragment into pLoBlu to create pGuaBA.
Construction of CVD1251ΔstxAB.
The stxAB genes were deleted in S. dysenteriae 1 CVD 1251 using the lambda red recombination system with plasmids kindly provided by Barry Wanner (10). A 171-bp fragment upstream of stxA was amplified using primers Wu046 and Wu047. A 374-bp fragment downstream of stxB was amplified using primers Wu026 and Wu027. The upstream fragment was cloned into pUC19 as a HindIII-BamHI fragment forming pUC19-13. Next, the fragment downstream of stxB was cloned as a BamHI-EcoRI fragment. The cml gene with flanking FLP recombination target (FRT) sites was then amplified from pKD3 with primers Wu016 and Wu017 and cloned as a HindIII-blunted/BamHI fragment and ligated between the flanking fragments, forming pUC19-14. Digestion of pUC19-14 with HindIII and EcoRI released the fragment including up- and downstream stx regions with the cml marker between these two regions. This fragment was electroporated into strain CVD 1251(pKD46). Plasmid pKD46 encodes the lambda red recombinase and catalyzes recombination between homologous sequences. The resulting chloramphenicol-resistant colonies were grown at 37°C to facilitate loss of pKD46. Next, plasmid pCP20, encoding the Flip recombinase was electroporated into the strain to catalyze recombination at FRT sites and excision of the cml gene. The resultant strain, CVD1251ΔstxAB, contains a deletion of the stxAB operon.
Construction of strains CVD1251ΔstxA::Ptrc-stxB and CVD1250ΔstxA::PmLpp-stxB.
In a single lambda red-mediated allelic exchange reaction, the stxA gene was deleted simultaneously with the insertion of the stxB gene driven by the constitutive trc promoter (Ptrc) or mutant Lpp (mLpp) promoter (PmLpp). These promoters were selected to drive high-level expression of stxB. The mutagenic fragments used for allelic exchange were constructed in a series of steps in the plasmid pUC19-14 described above containing a region upstream of stxA and the cml gene with FRT-flanking sequences. First, an expression plasmid containing the modified trc promoter was constructed. Ptrc is a derivative of the tac promoter containing 17 bp between the −10 and −35 regions of the promoter (7). An artificial promoter region containing the −35 and −10 sequences of the trc promoter with 17-bp intervening DNA was constructed using overlapping PCR with the oligonucleotides Trc-1 and Trc-2. This fragment was cloned as an EcoRI-SmaI fragment into the pGA2 plasmid (2) replacing the ompC promoter (POmpC) and named pGA2-trc. Next, the stxB gene was amplified from genomic DNA using the primers StxB-1 and StxB-2 and ligated into pGA2-trc as an SmaI-BamHI fragment to create pGA2-trc-StxB. The Ptrc-stxB fragment was excised with BamHI and EcoRI and cloned into pUC19-14 to create pUC19-15. Digestion with HindIII and EcoRI released a fragment containing the stxA upstream region followed by the cml gene with FRT sequences, followed by Ptrc-stxB. This fragment was electroporated in strain CVD 1251(pKD46), and recombinant colonies were selected on chloramphenicol. Plasmid pCP20 was then introduced by electroporation to catalyze recombination at the FRT sites, resulting in excision of the cml gene. The resultant strain, CVD1251ΔstxA::Ptrc-stxB, contains a complete deletion of stxA and insertion of Ptrc driving expression of stxB.
The Lpp promoter (PLpp) is one of the most efficient promoters in E. coli (36). One mutant derivative of PLpp containing 2 nucleotide changes in the −35 sequence and 1 nucleotide change in the −10 sequence demonstrated higher-level activity than the native sequence did (20). We used this mutated promoter sequence to drive very high expression of stxB in strain CVD 1256. The mutagenic primers F-35 and R-35 were used to amplify the −35 region of the Lpp promoter and introduce the 2-nucleotide mutation. The mutagenic primers F-10 and R-10 were used to amplify the −10 region and introduce the 1-bp mutation. The primers were designed to produce products that overlap. The products of the two reactions were used in a third PCR with primers F-Lpp and R-Lpp to produce a single product with PmLpp. The PmLpp fragment was ligated into pUC19-15 as a BamHI-NotI fragment to replace Ptrc upstream of stxB and create pUC19-16. As described above for Ptrc, digestion with HindIII and EcoRI released a fragment containing the stxA upstream region followed by the cml gene with FRT sequences, followed by PmLpp-stxB. This fragment was electroporated in CVD 1251(pKD46), and recombinant colonies were selected with chloramphenicol. Plasmid pCP20 was then introduced by electroporation to catalyze recombination at the FRT sites, resulting in deletion of the cml gene. The resultant strain, CVD1251ΔstxA::PmLpp-stxB, contains a complete deletion of stxA and insertion of PmLpp driving expression of stxB. The stx chromosomal region was amplified by PCR and sequenced to ensure accuracy.
Next, a deletion in the sen gene was introduced into each of the 3 stx modified strains using the lambda red recombination system. The kanamycin resistance gene plus FRT sequences from pKD46 were amplified using primers Red-senF and Red-senR. These primers contain tails that correspond to downstream (nucleotides 73530 to 73599 [GenBank accession no. AF348706]) and upstream (nucleotides 74279 to 74348 [GenBank accession no. AF348706]) sequences flanking the sen gene. The amplified product was electroporated into CVD1251ΔstxAB(pKD46), CVD1251ΔstxA::Ptrc-stxB(pKD46), and CVD1251ΔstxA::PmLpp-stxB(pKD46). Kanamycin-resistant colonies were selected and grown at 37°C to facilitate the loss of pKD46. Next, plasmid pCP20, encoding the Flip recombinase was electroporated into each strain to catalyze recombination at FRT sites and excision of the kanamycin resistance gene. Kanamycin-sensitive colonies were confirmed for deletion of sen by a series of diagnostic PCRs. The sen deletion corresponds to a 678-bp deletion. The resultant three strains were named CVD 1255, CVD 1257, and CVD 1256, respectively.
Construction of StxB plasmids.
The stxB gene was cloned into the stabilized expression plasmid pGA2 (2) and driven by Ptrc or POmpC. Plasmid pGA2-trc-StxB is described above and named pTrc-StxB for simplicity. Plasmid pGA2-StxB contains POmpC driving the expression of stxB and was constructed as follows. The stxB gene was amplified using primers StxB-2409 and StxB-2693 and cloned into pGA2 as an SmaI-BamHI fragment and named pOmpC-StxB. These plasmids were electroporated into S. dysenteriae 1 strain CVD 1255 to create CVD 1255(pTrc-StxB) and CVD 1255(pOmpC-StxB).
Construction of strain CVD 1254.
Complete deletion of stxAB was accomplished in the wild-type S. dysenteriae 1 strain 1617 using the suicide plasmid pCVD442 for allelic exchange. Primers Wu050 and Wu051 amplified a 1,020-bp region upstream of stxA, and primers Wu052 and Wu053 amplified a 830-bp fragment downstream of stxB. The two PCR products were cloned into suicide plasmid pCVD442 (11), which was electroporated into S. dysenteriae 1 strain 1617. Ampicillin-resistant colonies were selected as those containing the integrated plasmid. Following growth without ampicillin in the presence of sucrose, the colonies that had lost the plasmid were selected. Screening by PCR identified colonies that contained the stxAB deletion, and this strain was named CVD 1254.
PCR analysis.
Diagnostic PCR assays were performed to confirm the genetic integrity of each strain. Primers used for confirmation of each gene in the CVD 1256 master cell bank (MCB) are as follows. Primers Wu029 and Wu030 bind to sequences upstream of stxA and downstream of stxB, respectively, and amplify a 2.2-kb product from the WT and a 1.5-kb product due to the deletion of stxA in CVD 1256. Primers Wu029 and Wu035 bind sequences upstream of stxA and downstream of stxB, respectively, and amplify a 1.8-kb product in the WT and a 1.1-kb product due to the deletion of stxA in CVD 1256. In Fig. 1D, lanes 5 and 6, the templates were amplified with primers F-Lpp and Wu030. Primer F-Lpp binds to a region within the mLpp promoter which is present only in strain CVD 1256 and not present in the WT. Primers Wu048 and Wu049 bind sequences flanking the guaBA operon and amplify a 1.79-kb product in the WT and a 872-bp product in CVD 1256. Primers Wu061 and EB1 bind to regions upstream and downstream of sen, respectively, and amplify a 1.6-kb product in the WT and a 947-bp fragment in CVD 1256, reflecting the 679-bp deletion. Template DNA consisted of a single bacterial colony added to the reaction mixture. Reactions were performed on a MJ Research minicycler (Waltham, MA) with specific parameters based on the composition of the primers and the length of the amplified product.
Fig. 1.

(A to C) Genetic map of the stxAB locus in S. dysenteriae 1 derivative strains. (A) Genetic map of the wild-type stxAB locus. Solid-line arrows indicate upstream and downstream regions used in recombination to generate the derivative strains. Broken-line arrows indicate the primers used in PCR. (B) Genetic map of the resultant stxAB deletion in strain CVD 1255. (C) Genetic map showing the deletion of stxA and insertion of a promoter (PmLpp or Ptrc) to drive expression of stxB in strains CVD 1256 and CVD 1257. (D) Results of amplification of the stxAB locus in derivative strains with primers Wu029 and Wu030. Template genomic DNA was from the following strains: 1617 (lane 2), CVD 1251 (lane 3), CVD 1255 (lane 4), CVD 1256 (lane 5), and CVD 1257 (lane 6). The positions (in kilobases) of molecular size markers (lane 1) are shown to the left of the gel.
RNA extraction and cDNA production.
For in vitro analysis, bacterial cells were grown to log phase (optical density at 600 nm [OD600] of 0.6) in broth, then double volumes of RNAprotect (Qiagen, Valencia, CA) were added to each sample. RNA was extracted using the RNeasy minikit (Qiagen, Valencia, CA) following the manufacturer's instructions, including DNase treatment on the column. RNA was also isolated from bacteria within HeLa cells. The HeLa cell invasion protocol was followed, and a multiplicity of infection (MOI) of 1,000:1 was used. At 0-h and 4-h time points after invasion, the samples were harvested in TRIzol (Invitrogen) and processed with the PureLink RNA minikit (Invitrogen, Carlsbad, CA) following the manufacturer's instructions including DNase treatment on the column. cDNA was produced using 1 μg of total RNA and the qScript cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD) following the manufacturer's instructions.
Real-time reverse transcription-PCR (RT-PCR).
Expression of the stxB gene was examined by real-time PCR. PCR was performed using the iQ5 optical real-time machine and SsoFast EvaGreen supermix (both from Bio-Rad, Hercules, CA). Amplification was performed in triplicate by the following thermal cycling conditions: (i) one cycle of 1 min at 95°C; (ii) 35 cycles, with 1 cycle consisting of 10 s at 95°C and 20 s at 58°C; and (iii) one cycle of 5 min at 72°C. The sequences of primers StxB-RTF, StxB-RTR, rpoB-F (F stands for forward), rpoB-R (R stands for reverse), gyrB-F, and gyrB-R are given in Table 2. Transcript levels were normalized to those of rpoB and gyrB to account for variability in the amount of cDNA in each sample, and relative expression levels were calculated using the ΔΔCT (CT stands for threshold cycle) method with the Bio-Rad iQ5 analysis software.
Western blot analysis.
Protein expression was analyzed in whole-cell lysates or periplasmic fractions. Pelleted whole-cell bacteria or periplasmic fraction samples were mixed 1:1 (vol/vol) with Laemmli sample buffer and 5% β-mercaptoethanol and then boiled for 7 min. Aliquots of 5 μl of each sample were electrophoresed on SDS-15% polyacrylamide gels. The gels were stained with GelCode blue stain reagent (Thermo Scientific, Barrington, IL) for visualization of protein bands or transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corp., Bedford, MA) for Western immunoblot analysis. The membranes were probed with rabbit or mouse anti-StxB (BEI Resources, Manassas, VA). Western immunoblots were developed using the Odyssey system (Li-Cor Bioscience, Lincoln, NE).
Southern blot analysis.
Genomic DNA was isolated from strain CVD 1256 or 1617 (WT) using Qiagen genomic-tip 20 (Qiagen, Valencia, CA) and digested with a combination of restriction enzymes, including BamHI, EcoRI, EcoRV, PstI, and BglII (NEB, Ipswich, MA). Digested DNA was separated on an agarose gel and transferred to Hybond-N+ membrane (Amersham/GE Healthcare, Pittsburg, PA) where it was probed with a labeled DNA fragment corresponding to the stxA gene generated by PCR with primers Wu036 and Wu045 or the stxB gene generated by PCR with primers Wu033 and Wu035. Probe labeling and Southern blot detection were performed with the digoxigenin (DIG) high prime DNA labeling and detection starter kit II (Roche, Indianapolis, Indiana) according to the manufacturer's instructions.
HeLa cell invasion assays.
HeLa cells were cultured in Eagle minimal essential medium (MEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. For the invasion assay, HeLa cells were seeded at a density of 105 cells per well in a 24-well plate and incubated overnight. A 100-μl suspension of 108 bacteria was added to the HeLa cells in triplicate and incubated at 37°C with 5% CO2 for 90 min. The cells were washed and incubated with medium containing 100 μg/ml gentamicin for 30 min (0-h time point) or 4 hours (4-h time point). Following incubation, the monolayers were lysed with 1 ml/well of 1% Triton X-100 at the 0- and 4-h time points. Serial dilutions were plated to determine the number of viable intracellular bacteria.
Plaque assays.
Plaque assays were performed essentially as described previously (43) with some modifications. Monolayers of L929 cells (kindly provided by Anthony Maurelli, Uniformed Services University of the Health Services [USUHS]) were grown to confluence in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS in 60-mm petri dishes. Serial 10-fold dilutions of bacterial samples were prepared in DMEM, and 100-μl samples were added to each monolayer and incubated at 37°C with 5% CO2 for 2 h. Six milliliters of prewarmed overlay medium (3 ml of 2× DMEM containing 20% FBS and 50 μg/ml gentamicin, plus 3 ml of 1% agarose in water) was added to each monolayer. The agarose was allowed to solidify at room temperature for approximately 15 min, and the dishes were transferred back to the 37°C CO2 incubator for 2 or 3 days. The plaques were then stained with 2 ml Giemsa stain (1 ml of 10× Giemsa solution [0.5 g Giemsa in 33 ml of glycerol and 33 ml of methanol] diluted in 9 ml working solution containing 0.05924 g Na2HPO4 and 0.0364 g NaH2PO4 in 100 ml of dH2O) for visualization and enumeration.
T84 cell assays.
The inverted, polarized T84 cell assay was performed by the method of McCormick and colleagues (30, 31) with modifications. T84 human colonic intestinal cells (ATCC, Manassas, VA) were cultured in DMEM with nutrient mixture F-12 (DMEM:F12) (Invitrogen, Carlsbad, CA). Monolayers were grown on collagen-coated 12-well inverted 3μm Transwell inserts (Corning, Lowell, MA). The Transwells were inverted, seeded with 6 × 105 cells in 200 μl, incubated for 3 h, then inverted back into the well, and incubated for 7 days. The transepithelial electrical resistance (TEER) was measured using the epithelial voltohmmeter from World Precision Instruments (Sarasota, FL) over time, and only wells that reached between 600 and 3,000 Ω·cm2 were used in the assay. A 500-μl aliquot containing 109 bacteria/ml was added to the basolateral surface of the Transwell and incubated at 37°C with 5% CO2 for 90 min. The cells were then washed three times with Dulbecco's phosphate-buffered saline (DPBS) containing 50 μg/ml gentamicin and incubated with medium containing 50 μg/ml gentamicin. After 24 h, the transepithelial resistance was recorded, and the medium from the basolateral and apical surfaces was harvested separately and stored at −20°C for interleukin-8 (IL-8) quantification. The IL-8 enzyme-linked immunosorbent assay (ELISA) was performed in duplicate using the human IL-8 quantikine kit (R&D Systems, Minneapolis, MN) following the manufacturer's instructions.
Serény test.
The Serény (guinea pig keratoconjunctivitis) test was performed as described previously (49) to evaluate the level of attenuation of each vaccine strain versus wild-type S. dysenteriae 1. A 10-μl aliquot of each test strain at the designated concentration was instilled into the conjunctival sacculus of the right eye. Animals were observed daily for 5 days for signs of infection and inflammation in their eyes and assigned scores as follows: 0 for normal eye indistinguishable from the contralateral uninoculated eye, 1 for lacrimation or eyelid edema, 2 for lacrimation or eyelid edema plus mild conjunctival hyperemia, 3 for lacrimation or eyelid edema plus mild conjunctival hyperemia plus slight exudates, and 4 for full-blown purulent keratoconjunctivitis. All animal procedures were approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee.
Immunizations and sample collection.
After sedation with a 1:1 solution of ketamine-xylazine, randomized, female, Hartley guinea pigs (8 weeks old) were immunized intranasally with 100 μl of the bacterial suspension containing 109 CFU on day 0. An identical booster dose was administered 14 days later. Tears, elicited from guinea pigs with Capsicum baccatum flakes as previously described, were collected in 50-μl capillary tubes (41). Blood samples were obtained from anesthetized guinea pigs via the anterior vena cava. Tears and blood samples were collected on days 0, 14, and 28. Two weeks following the second immunization, the animals were challenged with a designated concentration of the wild-type strain 1617 following intraocular administration in the Serény test. Animals were scored for signs of inflammation for 5 days as described above.
ELISAs.
Antigens used in enzyme-linked immunosorbent assay included hot water-phenol-extracted S. dysenteriae 1 LPS from strain 1617. Purified Shiga toxin B subunit was a kind gift from Alison O'Brien's laboratory, USUHS. The specific IgG and IgA anti-LPS and anti-B-subunit titers were determined for serum and tears. To determine the endpoint titer, 3-fold dilutions of sera or tears in 10% milk powder were added to the coated plates and incubated for 1 hour at 37°C. Peroxidase-labeled secondary antibodies were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate for 15 min at room temperature. The reaction was stopped by the addition of 100 μl H2PO4, and the OD450 was determined in an ELISA microplate reader (Multiscan Ascent; Thermo Labsystems, Helsinki, Finland). Sera and tears were run in duplicate. Linear regression curves were plotted for each sample, and titers were calculated (through equation parameters) as the inverse of the serum dilution that produces an OD450 of 0.2 above the blank.
Shiga toxin assays.
In order to assess the neutralization activity of guinea pig serum, a modification of the Vero toxin assay was performed as previously described (28). Ninety-six-well flat-bottomed tissue culture treated plates (Corning, Corning, NY) were seeded with 1.4 × 104 Vero cells/well in complete medium (MEM with 10% FBS) and incubated overnight at 37°C in 5% CO2. Test samples were prepared as follows. A 50-μl aliquot containing 0.005 ng of purified Shiga toxin (BEI Resources, Manassas, VA) was mixed with an equal volume of serial dilutions of guinea pig sera and incubated for 1 h at 37°C. A 100-μl aliquot of each mixture was added to duplicate wells in the 96-well plate seeded with Vero cells. The plate was incubated at 37°C in 5% CO2for 48 h. The cells were fixed with 2% formalin for 5 min at room temperature and then stained with 0.3% crystal violet for 30 min at room temperature. The plates were read on a Labsystems Multiskan Ascent microtiter dish reader at 620 nm. The neutralization titer was defined as the dilution of guinea pig serum that neutralized 50% of the cytotoxic effect of a standard amount of Shiga toxin.
RESULTS
Attenuated S. dysenteriae 1 strain construction.
The guaBA deletion attenuating strategy that diminished the virulence of S. flexneri 2a (22) was applied to S. dysenteriae 1 strain 1617, resulting in S. dysenteriae 1 ΔguaBA strain CVD 1251. Since guaB and guaA form an operon, a single deletion inactivates both genes. The resultant derivative was auxotrophic for guanine, but growth could be complemented by adding exogenous guanine to the medium or providing a plasmid, pGua, containing wild-type guaBA in trans (data not shown).
Shigella enterotoxin 2 (ShET-2) is encoded by sen located on the invasion plasmid (37). The sen gene, found in all Shigella strains, was deleted from the S. dysenteriae 1 ΔguaBA derivatives as a secondary attenuating mutation. The set operon which is present only in S. flexneri 2a did not serve as a target for mutation in S. dysenteriae 1 (40).
Shiga toxin is encoded by the stxAB operon located within defective phage sequences in the S. dysenteriae 1 chromosome. It is essential that Shiga toxin activity be eliminated in a vaccine strain. An S. dysenteriae 1 ΔguaBA Δsen ΔstxAB strain, CVD 1255, was constructed with a complete deletion of the stxAB operon. PCR assays confirmed the 1.1-kb deletion in strain CVD 1255 (Fig. 1). Since antibodies to StxB can neutralize Shiga toxin activity in vitro and in vivo (1, 6, 8, 27), a series of strains was constructed with deletions in stxA but with directed high-level expression of stxB from either the native chromosomal site or from a plasmid. All candidate vaccine strains contained the guaBA and sen deletions.
A one-step process based on the lambda red recombination system was employed to catalyze deletion of the stxA gene with concomitant insertion of a promoter to drive high-level stxB expression from the chromosomal locus. Strains CVD 1256 and CVD 1257 harbor a complete deletion of stxA and insertion of a mutant Lpp promoter, PmLpp, or a trc promoter, Ptrc, strong constitutive promoters, upstream of stxB. The mutagenic fragments used for allelic exchange were constructed in a series of steps as described in Materials and Methods using a region upstream of stxA and the cml gene with FRT-flanking sequences. Each resultant strain was confirmed for genetic integrity at the stx locus using PCR assays (Fig. 1). Primers Wu029 and Wu030 amplify a 2.2-kb fragment containing stxAB in WT strain 1617 and in CVD 1251, which contains only the guaBA deletion. In contrast, with DNA from strain CVD 1255 which contains the complete stxAB deletion, a 1.1-kb fragment is amplified. Amplicons with sizes of 1.7 and 1.6 kb were generated from strains CVD 1256 and CVD 1257, respectively, confirming the deletion of stxA with the presence of stxB and a new promoter sequence. The locus in CVD 1256 is slightly larger than that in CVD 1257 due to the increased size of the PmLpp region.
High-level expression of StxB was also evaluated using a stabilized plasmid platform. The stxB gene driven by either the constitutive trc or OmpC promoter was cloned into this plasmid to create pTrc-StxB and pOmpC-StxB. Each plasmid was electroporated into S. dysenteriae 1 strain CVD 1255 and named CVD 1259 and CVD 1258, respectively.
One final derivative was constructed; this derivative contains only a single mutation, the complete deletion of the stxAB locus. S. dysenteriae 1 1617ΔstxAB, designated CVD 1254 maintains the full virulence capacity of Shigella except for the ability to manifest Shiga toxin activity.
StxB is expressed from chromosomal and plasmid locations in Shigella vaccine strains.
Quantitative real-time PCR was used to measure the expression of stxB in the vaccine strains (Fig. 2). Strain CVD 1255 with its complete deletion of stxAB was employed as a negative control; no expression was detected in this strain. Strain CVD 1251 has an intact stxAB locus that is regulated by iron in the wild-type background and is induced in low-iron environments. The strains were grown in LB, a rich medium not limited for iron, and stxB expression was low as expected. The expression level of stxB in all other strains is expressed relative to that of CVD 1251. Strains CVD 1256 and CVD 1257 containing chromosomal copies of stxB driven by PmLpp or Ptrc promoters, respectively, express stxB 63 and 14 times higher than CVD 1251. The two plasmid-containing constructs driving expression of stxB from the Ptrc or POmpC promoters expressed stxB 171 and 100 times higher, respectively, than CVD 1251.
Fig. 2.

stxB gene and protein expression in S. dysenteriae 1 vaccine strains. (A) qRT-PCR was used to measure stxB expression in each vaccine strain. Values shown are fold change in the CT value relative to the value for strain CVD 1251. (B) Western blot analysis of periplasmic fractions isolated from each strain probed with a combination of anti-StxA and anti-StxB antibodies. Bars or lanes: A, CVD 1251; B, CVD 1255; C, CVD 1256; D, CVD 1257; E, CVD 1255(pTrc-StxB); F, CVD 1255(pOmpC-StxB). The positions of the StxA and StxB subunits are indicated by the arrowheads to the right of the gel. The positions of molecular mass (in kilodaltons) in the unlabeled leftmost lane are indicated to the left of the gel.
Western blot analysis assessed StxB protein expression in periplasmic fractions of the S. dysenteriae 1 derivatives (Fig. 2). The protein levels mirrored gene transcription levels measured by quantitative reverse transcription-PCR (qRT-PCR). Both StxA and StxB subunits were detected when periplasmic fractions of strain CVD 1251 (which contains an intact stxAB locus) was probed with anti-StxA and anti-StxB antibodies. As expected, a low level of StxB was expressed due to the noninducing growth conditions. Neither the A subunit nor the B subunit is present in strain CVD 1255 due to the deletion of the entire locus (Fig. 2B, lane B). Insertion of a strong promoter in strains CVD 1256 and CVD 1257 resulted in increased StxB expression (Fig. 2B, lanes C and D). In vitro, PmLpp drove higher-level expression of StxB than Ptrc did. Attenuated S. dysenteriae 1 derivatives containing plasmids encoding StxB produced the highest levels of StxB protein (Fig. 2B, lanes E and F).
Attenuated Shigella derivatives display reduced virulence phenotypes.
The attenuated derivatives were evaluated for invasion and replication in HeLa cells, cell-to-cell spread in plaque assays, and IL-8 production in polarized T84 cells. Wild-type S. dysenteriae 1 strain 1617 invades cells at high efficiency within 90 min. The bacteria thereupon undergo intracellular replication with approximately 5 doublings in the subsequent 4 h (Fig. 3). As expected, introduction of a deletion in stxAB in strain CVD 1254 did not affect invasiveness or intracellular replication. In contrast, any mutant derivative containing the guaBA deletion was able to efficiently invade HeLa cells at levels comparable to that of the wild type but was unable to replicate within the subsequent 4-h incubation period (Fig. 3). Moreover, while the wild-type strain and stxAB-deleted S. dysenteriae 1 strains formed plaques in a dose-dependent manner, the guaBA-deleted strains were completely unable to form plaques (data not shown).
Fig. 3.
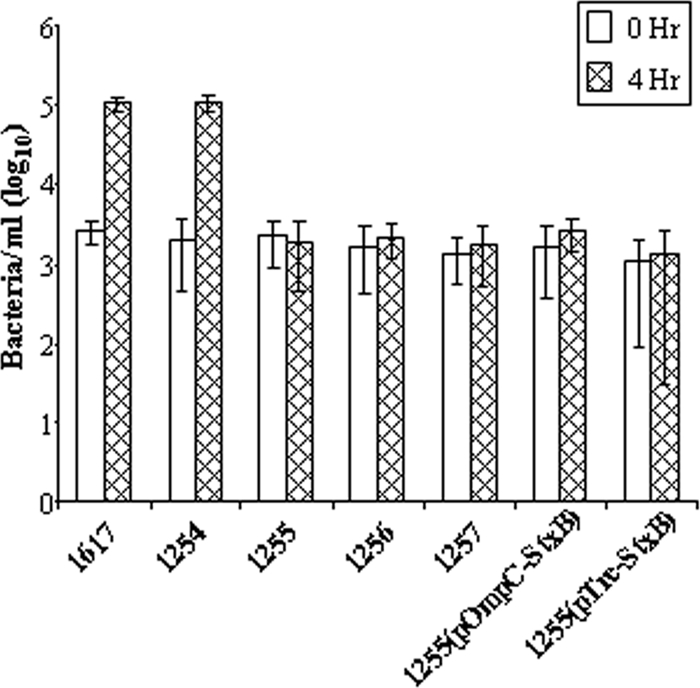
HeLa cell invasion by S. dysenteriae vaccine strains. Bacteria (strains 1617, CVD 1254, CVD 1255, etc.) were incubated with HeLa cell monolayers at an MOI of 100 for 90 min and washed with phosphate-buffered saline (PBS) containing gentamicin. The cells were lysed to enumerate intracellular bacteria at 0 h and 4 h postinfection.
A hallmark of Shigella infections is the induction of a robust inflammatory response characterized by production of the proinflammatory cytokine IL-8. The inverted T-84 cell polarized monolayer system was used to measure IL-8 induction by wild-type and attenuated vaccine strains. Wild-type strain 1617 and stxAB-deleted S. dysenteriae 1 strain CVD 1254 exhibited strong IL-8 responses within 24 hours (Fig. 4). In contrast, while not reaching statistical significance, the guaBA-deleted mutants tended to induce lower levels of IL-8 in this system. Transepithelial electrical resistance (TEER) was recorded in the polarized cells as a measure of the integrity of the monolayer. Wild-type S. dysenteriae 1 causes a notable drop in TEER over the 24-hour period following addition of the bacteria (Fig. 4A). CVD 1254 showed a comparable decrease. The derivative strains containing the guaBA deletion did not significantly diminish TEER over the 24-hour period. Instead, TEER continued to increase, as seen with uninfected cells.
Fig. 4.

IL-8 production and TEER in inverted polarized T-84 cells infected with S. dysenteriae 1 derivatives. T-84 cells were grown on inverted transwells, and bacteria were applied to the basolateral surface. IL-8 secretion was measured in basolateral supernatants after 24 hours of incubation. TEER was measured prior to infection and at 24 h postinfection and expressed as a percentage of the initial TEER. Bars: BL, blank; A, WT 1617; B, CVD 1254; C, CVD 1255; D, CVD 1256; E, CVD 1257; F, CVD 1255(pOmpC-StxB); G, CVD 1255(pTrc-StxB). The TEER values for strains 1617 and CVD 1254 were significantly less than all other strains and are indicated with an asterisk (P < 0.01, using an unpaired Student's t test).
Attenuated vaccine strains are safe and immunogenic in the guinea pig model.
Each vaccine candidate was tested for reduced virulence in the guinea pig Serény test. A dose of 108 CFU was inoculated into the guinea pig eye. The degree of inflammation was graded daily for 5 days. Wild-type S. dysenteriae 1 caused a profound inflammatory response that progressed to full-blown keratoconjunctivitis within 48 to 72 h postinoculation; all 18 animals reached the maximum score of 4 by 72 h postinfection (Table 3). The same response was observed in animals inoculated with the Shiga toxin-deleted mutant CVD 1254. This was expected, since the Serény test is a test of the invasive and inflammatory induction capabilities of Shigella. In contrast, all guaBA-deleted vaccine strains showed decreases in this composite measurement of virulence and did not cause inflammation in the guinea pig eye; the scores for all eyes were 0 at each observation point postchallenge. The overexpression of Shiga toxin in strains CVD 1256 and CVD 1257 and the plasmid-containing strains did not influence the attenuation of the strains.
Table 3.
Serény test results
| Test strain | Challenge dose (CFU) | No. of animals exhibiting keratoconjunctivitis/total no. of animals |
|---|---|---|
| 1617 (wild type) | 108 | 18/18 |
| CVD 1254 | 108 | 9/9 |
| CVD 1255 | 108 | 0/8 |
| CVD 1256 | 107 | 0/10 |
| CVD 1256 | 108 | 0/14 |
| CVD 1256 | 109 | 0/10 |
| CVD 1256 | 1010 | 0/10 |
The immunogenicity of the vaccine candidates was assessed in the guinea pig mucosal immunization model. The primary readout for immunity to Shigella is O-antigen-specific anti-LPS responses. Animals immunized with two 109 CFU doses of any attenuated S. dysenteriae 1 vaccine strain responded with significant rises in anti-S. dysenteriae 1 LPS serum IgG and mucosal IgA anti-LPS following the first dose (Fig. 5). These titers were boosted to higher levels following the second dose. Consistent with the high S. dysenteriae 1-specific titers, all immunized animals were protected against keratoconjunctivitis when challenged with wild-type S. dysenteriae 1 strain 1617 2 weeks following the second immunization (Table 4). All nine unimmunized controls succumbed to infection, attaining eye scores of 4 by 72 h postinfection, while no immunized animal exhibited keratoconjunctivitis following challenge. The eye of one animal immunized with strain CVD 1256 had a score of 1 at 72 hours postchallenge which resolved to 0 the following day (minimal evidence of fleeting inflammation).
Fig. 5.

Immunogenicity of attenuated S. dysenteriae 1 vaccine strains. (A and B) Anti-S. dysenteriae 1 LPS IgG in serum (A) and mucosal IgA in tears (B) were measured prior to immunization (bleed 1) and 2 weeks after each dose (bleeds 2 and 3). Geometric mean titers (GMTs) are presented for each immunization group. (C) Anti-Stx1B titers in serum were measured, and the titer for pooled samples in each group is shown for bleeds 1 and 2 and the GMT for individual animals is shown for bleed 3. (D) Stx1 toxin neutralization titers are shown for pooled samples from each group. Vaccine strains are as follows: A, CVD 1251; B, CVD 1255; C, CVD 1256; D, CVD 1257; E, CVD 1255(pOmpC-StxB); F, CVD 1255(pTrc-StxB).
Table 4.
Vaccine-induced protection against wild-type S. dysenteriae 1 challenge
| Vaccine strain | Vaccine dose (CFU) | Challenge dose (CFU) | No. of animals protecteda/total no. of animals |
|---|---|---|---|
| CVD 1255 | 109 | 108 | 4/4 |
| CVD 1256 | 109 | 107 | 7/7 |
| 109 | 108 | 13/13 | |
| 109 | 109 | 3/4 | |
| CVD 1257 | 109 | 108 | 8/8 |
| CVD 1255(pOmpC-StxB) | 109 | 108 | 8/8 |
| CVD 1255(pTrc-StxB) | 109 | 108 | 6/6 |
| None (PBS) (control) | 108 | 0/9 |
Protection against full-blown keratoconjunctivitis (score of 4) at any time point postchallenge.
A secondary endpoint was the elicitation of antibodies against the Shiga toxin B subunit. Strain CVD 1255(pTrc-StxB) induced the highest levels of serum IgG anti-StxB (Fig. 5). This is consistent with the results of Western blot analysis which demonstrated the highest level of StxB expression in this strain. The other plasmid-containing strain, CVD 1255(pOmpC-StxB), did not induce anti-StxB titers in vaccinated animals. This was unexpected, given Western blot analysis demonstrating expression of the antigen in this strain in vitro. CVD 1256, the vaccine strain with the constitutive mLpp promoter driving stxB expression from the chromosomal site elicited a robust anti-StxB antibody response. CVD 1257, with chromosomal StxB expression driven by the trc promoter, was unable to induce anti-StxB antibodies. The capacity of these antibodies to neutralize Shiga toxin in the Vero cell assay was evaluated. Results showed that neutralization titers corresponded to ELISA titers; the vaccine strain CVD 1255(pTrc-StxB) expressing StxB from a plasmid induced the highest toxin neutralizing titers, while chromosomally expressed StxB strain CVD 1256 induced lower neutralizing titers (Fig. 5).
Expanded preclinical studies with strain CVD 1256.
Because of the promising immune responses elicited by strain CVD 1256 to both S. dysenteriae 1 LPS and Shiga toxin B subunit, combined with the stability of the chromosomal location for StxB antigen expression, this strain was identified for advancement to clinical trials. A master cell bank (MCB) was prepared by the CVD Inoculum Preparation and Clinical Microbiology Unit. The CVD 1256 MCB underwent extended characterization and testing. The genotype of each of the 3 mutations was confirmed with a series of PCR assays and by sequencing each chromosomal locus, including 1-kb flanking regions on both ends to ensure integrity (data not shown).
An expanded dose range was tested for safety in the Serény test where inoculation of CVD 1256 MCB at 107 to 1010 CFU was found to be nonreactogenic (Table 3). The efficacy of vaccination with strain CVD 1256 to protect against an expanded range of challenge doses was established in the Serény test (Table 4). Immunization of guinea pigs with two doses of CVD 1256 was able to protect against wild-type S. dysenteriae 1 challenge at doses of 107 and 108 CFU. One of four animals immunized with CVD 1256 and challenged with the higher dose of 109 CFU of WT strain 1617 was not protected. The eye of this animal reached a score of 4 at 72 hours postinfection and represents one vaccine breakthrough compared to all 9 unvaccinated animals which succumbed to full-blown keratoconjunctivitis following challenge with a lower dose of 108 CFU of strain 1617.
DISCUSSION
Our interest in developing a vaccine to prevent Shiga dysentery is driven by two motivations: (i) to have a practical public health tool that can help limit morbidity and mortality among underprivileged populations in developing countries that are cyclically affected by epidemics and pandemics caused by S. dysenteriae 1 (17, 19, 35, 47) and (ii) to provide a vaccine for reactive intervention to protect U.S. populations in the face of a deliberate bioterror release of S. dysenteriae 1 by nefarious individuals, recognizing that the immunological susceptibility of the U.S. population, the high transmissibility and multiple antibiotic resistance of current strains of this pathogen, and the severity of the disease it causes (that can be fatal even for healthy adults) make it a fearsome category B bioterror pathogen.
We pursued a strategy to develop live oral vaccine candidates following the proof of principle provided by the streptomycin-dependent live oral vaccines (of other serotypes) that conferred a high level of protection against natural disease in controlled field trials (33, 34) and as a result of our experience in genetic manipulation and engineering of other serotypes of Shigella. The two specific tactics we applied were to construct a series of S. dysenteriae 1 vaccine candidates containing a fundamental attenuating mutation in the guaBA locus, in conjunction with deletions in the stx and sen loci to detoxify the vaccine strains. These deletions were introduced into antibiotic-sensitive S. dysenteriae 1 strain 1617, which was originally isolated from a patient early in the pandemic in Guatemala in the 1960s, and that was shown to cause Shiga dysentery in human volunteers and in rhesus macaques (25). The guaBA mutation, which rendered the vaccine candidates auxotrophic for guanine, did not impair their ability to invade cells but made them incapable of intracellular replication. This set of characteristics makes the strains nonreactogenic in the guinea pig keratoconjunctivitis test yet able to elicit protective immune responses.
With respect to enterotoxins elaborated by Shigella, the set operon encoding ShET-1 is present only in the chromosome of the S. flexneri 2a serotype (15, 40), so it is not relevant for the engineering of S. dysenteriae 1 vaccines. In contrast, sen, which encodes ShET-2, and is present on the invasiveness plasmid of all Shigella serotypes (37, 52), was targeted for deletion in the S. dysenteriae 1 derivatives. ShET-2 was originally identified as an enterotoxin involved in the early phase of watery diarrhea associated with Shigella and enteroinvasive E. coli infections (37). Recent reports demonstrate that ShET-2 is secreted via a type III secretion system and may also play a role in Shigella-induced inflammation of epithelial cells (14, 44).
S. dysenteriae 1 is unique among the Shigella in its expression of Shiga toxin, which is responsible for hemolytic-uremic syndrome, one of the most feared complications of Shiga dysentery, as it can lead to renal shutdown and death. Activity of this potent toxin was eliminated from the vaccine strains by deleting the entire stxAB operon in strains CVD 1254 and CVD 1255 or by deleting stxA (that encodes the enzymatically active A subunit) in CVD 1256 and CVD 1257. Earlier investigators isolated S. dysenteriae 1 strains that lacked Shiga toxin activity following anaerobic growth on medium containing chlorate, which selects for bacteria that have lost the fnr locus via deletion of a 20-kb chromosomal region that includes stxAB (39, 53). While this method was effective for isolating Shiga toxin-negative strains, the large chromosomal deletion consequent to the chlorate treatment includes the loss of other genes besides stxAB. Employing advanced molecular biological techniques, we were able to engineer precise defined deletions limited to the stx locus in these vaccine candidates. S. dysenteriae 1 strain CVD 1255 containing a precise deletion in stxAB, as well as in guaBA and sen, was attenuated for virulence in the Serény test and elicited serum IgG and mucosal IgA anti-S. dysenteriae 1 LPS responses in guinea pigs following mucosal (intranasal [i.n.]) immunization. Moreover, vaccinated animals were protected against challenge with virulent wild-type S. dysenteriae 1 that induced purulent keratoconjunctivitis in the unvaccinated control animals.
Four other vaccine candidates that overexpressed StxB from plasmid or chromosomal sites were constructed in order to elicit Shiga toxin neutralizing antibodies. Shiga toxin from S. dysenteriae 1 is essentially identical to Shiga-like toxin 1 (SLT-1) produced by Shiga toxin-expressing E. coli (STEC) with only a single amino acid difference in the A subunit. A vaccine capable of eliciting Shiga toxin neutralizing antibodies has the additional potential value of providing protection against the severe complications following infection with other SLT-1-expressing enteropathogens. However, protection would not be offered against strains that elaborate SLT-2.
As expected, stxB transcript and protein expression was highest in the plasmid-encoded strains with Ptrc driving the highest levels. CVD 1256 and CVD 1257, the strains harboring chromosomal stxB constructions, also expressed stxB transcript and protein, albeit at lower levels. The various candidates also allowed us to compare the effects of two different strong constitutive promoters and revealed that PmLpp was clearly superior to Ptrc for directing StxB expression from a chromosomal site, as evidenced by qRT-PCR and Western blot analysis. Interestingly, the level of expression of StxB in vitro did not accurately predict the level of anti-StxB antibodies elicited in vaccinated guinea pigs. While CVD 1255(pTrc-StxB) induced the highest anti-StxB ELISA titers, as well as the highest toxin neutralizing activity, the other plasmid-containing strain, CVD 1255(pOmpC-StxB), did not induce anti-StxB serum IgG. We hypothesize that POmpC did not drive a high level of StxB expression in vivo. However, POmpC has been used previously in a plasmid construct to drive expression of the enterotoxigenic E. coli (ETEC) colonization factor antigen I (CFA/I) operon which elicited robust antifimbrial responses in the guinea pig model (3, 21). In the latter case, the live vector was an attenuated S. flexneri 2a strain, whereas in the current studies, the vector is S. dysenteriae 1. It is possible that POmpC might function differently in vivo in different live vector backgrounds. Alternatively, characteristics of the antigens themselves may be responsible for differential responses. The chromosomally encoded stxB gene driven by PmLpp in strain CVD 1256 was also able to induce robust anti-StxB binding and neutralizing titers. It is noteworthy that Ptrc at the chromosomal site was ineffective in driving StxB expression in vitro and was also unable to induce anti-StxB responses following mucosal immunization of guinea pigs.
All the S. dysenteriae 1 vaccine candidates were able to elicit strong serum IgG and mucosal IgA anti-LPS responses at equivalent levels and protected guinea pigs against virulent wild-type bacterium challenge. The overexpression of StxB did not diminish the immunogenicity of the live vector. The ability of vaccine candidate CVD 1256 to induce protective anti-S. dysenteriae 1 responses and to induce Shiga toxin neutralizing antibodies from a stable chromosomal site led to its selection for advancement. While one plasmid-containing strain, CVD 1259, induced higher anti-StxB responses, the greater stability of the chromosomal StxB expression site in CVD 1256 was deemed more advantageous overall as a future human vaccine strain. Expanded Serény and challenge studies confirmed the safety and immunogenicity of the CVD 1256 MCB. These data will be included in studies required for advancement to clinical trials.
Another S. dysenteriae 1 derivative, strain CVD 1254, contained only the precise stxAB deletion. This strain remained fully invasive in HeLa cells and virulent in the Serény test equivalent to the wild-type parent. These results were anticipated, since Shiga toxin is not involved in the invasive phenotype associated with all Shigella. Shiga toxin-negative strains have been shown to cause dysentery in monkeys and humans (18, 25, 39). The CVD 1254 strain was generated to serve as a challenge strain for future volunteer studies to assess the efficacy of the Shiga vaccine candidates, while avoiding the potential dangers of clinical complications (hemolytic-uremic syndrome [HUS]) that might accompany the use of a fully virulent Shiga toxin-expressing challenge organism. Challenge with CVD 1254 can be pivotal in testing the efficacy of S. dysenteriae 1 vaccines, recognizing that it is difficult to assess the efficacy of Shiga vaccines due to the spotty occurrence of the disease globally during interpandemic periods.
Other strategies for the development of vaccines to prevent S. dysenteriae 1 disease that have advanced to clinical trials include an S. dysenteriae type 1 O-antigen LPS polysaccharide-carrier protein conjugate that was immunogenic in volunteers (9, 46, 51) and two live attenuated S. dysenteriae 1 vaccine strains, including strains SC599 (with mutations in icsA, ent, fep, and stxA::hgR) and WRSd1 (harboring mutations in virG and stxAB) (32, 48). Both of these live vaccines were well tolerated and moderately immunogenic, inducing anti-S. dysenteriae 1 LPS IgA antigen-secreting cell (ASC) responses in approximately 50% and 60% of volunteers, respectively. The SC599 vaccine strain retained the stxB gene; however, anti-StxB responses were not measured (48). These studies have paved the way for the evaluation of a new generation of live attenuated S. dysenteriae 1 vaccines. Clinical studies with strain CVD 1256 will confirm the attenuating capacity of the guaBA deletion in a second species of Shigella and its ability to induce Shiga toxin neutralizing responses. An efficacious S. dysenteriae 1 vaccine would serve as an important public health tool for impoverished populations in regions of the world that have been historically prone to S. dysenteriae 1 explosive epidemics and pandemics. Such a vaccine would also constitute a countermeasure that provides preparedness against the possibility of a bioterror attack involving the deliberate release of virulent S. dysenteriae 1.
ACKNOWLEDGMENTS
This work was supported by grants AI059223 and AI57168 from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.
We thank Alison O'Brien at USUHS for Shiga toxin reagents.
Footnotes
Published ahead of print on 3 October 2011.
REFERENCES
- 1. Acheson D. W., Levine M. M., Kaper J. B., Keusch G. T. 1996. Protective immunity to Shiga-like toxin I following oral immunization with Shiga-like toxin I B-subunit-producing Vibrio cholerae CVD 103-HgR. Infect. Immun. 64:355–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altboum Z., Levine M. M., Galen J. E., Barry E. M. 2003. Genetic characterization and immunogenicity of coli surface antigen 4 from enterotoxigenic Escherichia coli when it is expressed in a Shigella live-vector strain. Infect. Immun. 71:1352–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barry E. M., Wang J., Wu T., Davis T., Levine M. M. 2006. Immunogenicity of multivalent Shigella-ETEC candidate vaccine strains in a guinea pig model. Vaccine 24:3727–3734 [DOI] [PubMed] [Google Scholar]
- 4. Bloom P. D., et al. 1994. Haemolytic-uraemic syndrome in adults with resistant Shigella dysenteriae type I. Lancet 344:206. [DOI] [PubMed] [Google Scholar]
- 5. Bogaerts J., et al. 1985. Shigella and Salmonella species from Kigali (Rwanda) (1976-1982). Ann. Soc. Belg. Med. Trop. 65:281–292 [PubMed] [Google Scholar]
- 6. Boyd B., Richardson S., Gariepy J. 1991. Serological responses to the B subunit of Shiga-like toxin 1 and its peptide fragments indicate that the B subunit is a vaccine candidate to counter action of the toxin. Infect. Immun. 59:750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brosius J., Erfle M., Storella J. 1985. Spacing of the −10 and −35 regions in the tac promoter. Effect on its in vivo activity. J. Biol. Chem. 260:3539–3541 [PubMed] [Google Scholar]
- 8. Butterton J. R., Ryan E. T., Acheson D. W., Calderwood S. B. 1997. Coexpression of the B subunit of Shiga toxin 1 and EaeA from enterohemorrhagic Escherichia coli in Vibrio cholerae vaccine strains. Infect. Immun. 65:2127–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen D., et al. 1996. Safety and immunogenicity of investigational Shigella conjugate vaccines in Israeli volunteers. Infect. Immun. 64:4074–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donnenberg M. S., Kaper J. B. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DuPont H. L., et al. 1972. Immunity in shigellosis. II. Protection induced by oral live vaccine or primary infection. J. Infect. Dis. 125:12–16 [DOI] [PubMed] [Google Scholar]
- 13. DuPont H. L., Levine M. M., Hornick R. B., Formal S. B. 1989. Inoculum size in shigellosis and implications for expected mode of transmission. J. Infect. Dis. 159:1126–1128 [DOI] [PubMed] [Google Scholar]
- 14. Farfan M. J., Toro C. S., Barry E. M., Nataro J. P. 2011. Shigella enterotoxin-2 is a type III effector that participates in Shigella-induced interleukin 8 secretion by epithelial cells. FEMS Immunol. Med. Microbiol. 61:332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fasano A., et al. 1995. Shigella enterotoxin 1: an enterotoxin of Shigella flexneri 2a active in rabbit small intestine in vivo and in vitro. J. Clin. Invest. 95:2853–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferreccio C., et al. 1991. Epidemiologic patterns of acute diarrhea and endemic Shigella infections in children in a poor periurban setting in Santiago, Chile. Am. J. Epidemiol. 134:614–627 [DOI] [PubMed] [Google Scholar]
- 17. Gangarosa E. J., et al. 1970. Epidemic Shiga bacillus dysentery in Central America. II. Epidemiologic studies in 1969. J. Infect. Dis. 122:181–190 [DOI] [PubMed] [Google Scholar]
- 18. Gemski P., Jr., Takeuchi A., Washington O., Formal S. B. 1972. Shigellosis due to Shigella dysenteriae. 1. Relative importance of mucosal invasion versus toxin production in pathogenesis. J. Infect. Dis. 126:523–530 [DOI] [PubMed] [Google Scholar]
- 19. Houdouin V., et al. 2004. A pediatric cluster of Shigella dysenteriae serotype 1 diarrhea with hemolytic uremic syndrome in 2 families from France. Clin. Infect. Dis. 38:e96–e99 [DOI] [PubMed] [Google Scholar]
- 20. Inouye S., Inouye M. 1985. Up-promoter mutations in the lpp gene of Escherichia coli. Nucleic Acids Res. 13:3101–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koprowski H., et al. 2000. Attenuated Shigella flexneri 2a vaccine strain CVD 1204 expressing colonization factor antigen I and mutant heat-labile enterotoxin of enterotoxigenic Escherichia coli. Infect. Immun. 68:4884–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kotloff K. L., et al. 2004. Deletion in the Shigella enterotoxin genes further attenuates Shigella flexneri 2a bearing guanine auxotrophy in a phase 1 trial of CVD 1204 and CVD 1208. J. Infect. Dis. 190:1745–1754 [DOI] [PubMed] [Google Scholar]
- 23. Kotloff K. L., et al. 2007. Safety and immunogenicity of CVD 1208S, a live, oral ΔguaBA Δsen Δset Shigella flexneri 2a vaccine grown on animal-free media. Hum. Vaccin. 3:268–275 [DOI] [PubMed] [Google Scholar]
- 24. Kotloff K. L., et al. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 77:651–666 [PMC free article] [PubMed] [Google Scholar]
- 25. Levine M. M., et al. 1973. Pathogenesis of Shigella dysenteriae 1 (Shiga) dysentery. J. Infect. Dis. 127:261–270 [DOI] [PubMed] [Google Scholar]
- 26. Levine M. M., Kotloff K. L., Barry E. M., Pasetti M. F., Sztein M. B. 2007. Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat. Rev. Microbiol. 5:540–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marcato P., Griener T. P., Mulvey G. L., Armstrong G. D. 2005. Recombinant Shiga toxin B-subunit-keyhole limpet hemocyanin conjugate vaccine protects mice from shigatoxemia. Infect. Immun. 73:6523–6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marques L. R., Moore M. A., Wells J. G., Wachsmuth I. K., O'Brien A. D. 1986. Production of Shiga-like toxin by Escherichia coli. J. Infect. Dis. 154:338–341 [DOI] [PubMed] [Google Scholar]
- 29. Mata L. J., Gangarosa E. J., Caceres A., Perera D. R., Mejicanos M. L. 1970. Epidemic Shiga bacillus dysentery in Central America. I. Etiologic investigations in Guatemala, 1969. J. Infect. Dis. 122:170–180 [DOI] [PubMed] [Google Scholar]
- 30. McCormick B. A., Colgan S. P., Delp-Archer C., Miller S. I., Madara J. L. 1993. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J. Cell Biol. 123:895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCormick B. A., Fernandez M. I., Siber A. M., Maurelli A. T. 1999. Inhibition of Shigella flexneri-induced transepithelial migration of polymorphonuclear leucocytes by cadaverine. Cell. Microbiol. 1:143–155 [DOI] [PubMed] [Google Scholar]
- 32. McKenzie R., et al. 2008. Safety and immunogenicity of WRSd1, a live attenuated Shigella dysenteriae type 1 vaccine candidate. Vaccine 26:3291–3296 [DOI] [PubMed] [Google Scholar]
- 33. Mel D., Gangarosa E. J., Radovanovic M. L., Arsic B. L., Litvinjenko S. 1971. Studies on vaccination against bacillary dysentery. 6. Protection of children by oral immunization with streptomycin-dependent Shigella strains. Bull. World Health Organ. 45:457–464 [PMC free article] [PubMed] [Google Scholar]
- 34. Mel D. M., Terzin A. L., Vuksic L. 1965. Studies on vaccination against bacillary dysentery. 3. Effective oral immunization against Shigella flexneri 2a in a field trial. Bull. World Health Organ. 32:647–655 [PMC free article] [PubMed] [Google Scholar]
- 35. Mhalu F. S., Moshi W. K., Mbaga I. 1984. A bacillary dysentery epidemic in Dar es Salaam, Tanzania. J. Diarrhoeal Dis. Res. 2:217–222 [PubMed] [Google Scholar]
- 36. Nakamura K., Inouye M. 1979. DNA sequence of the gene for the outer membrane lipoprotein of E. coli: an extremely AT-rich promoter. Cell 18:1109–1117 [DOI] [PubMed] [Google Scholar]
- 37. Nataro J. P., et al. 1995. Identification and cloning of a novel plasmid-encoded enterotoxin of enteroinvasive Escherichia coli and Shigella strains. Infect. Immun. 63:4721–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. National Institute of Allergy and Infectious Diseases 2003. NIAID biodefense research agenda for category B and C priority pathogens. NIH publication no. 03-5315. National Institute of Allergy and Infectious Diseases, National Institutes of Health, U.S. Department of Health and Human Services, Bethesda, MD [Google Scholar]
- 39. Neill R. J., Gemski P., Formal S. B., Newland J. W. 1988. Deletion of the Shiga toxin gene in a chlorate-resistant derivative of Shigella dysenteriae type 1 that retains virulence. J. Infect. Dis. 158:737–741 [DOI] [PubMed] [Google Scholar]
- 40. Noriega F. R., Liao F. M., Formal S. B., Fasano A., Levine M. M. 1995. Prevalence of Shigella enterotoxin 1 among Shigella clinical isolates of diverse serotypes. J. Infect. Dis. 172:1408–1410 [DOI] [PubMed] [Google Scholar]
- 41. Noriega F. R., et al. 1996. Engineered ΔguaB-A ΔvirG Shigella flexneri 2a strain CVD 1205: construction, safety, immunogenicity, and potential efficacy as a mucosal vaccine. Infect. Immun. 64:3055–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noriega F. R., Losonsky G., Wang J. Y., Formal S. B., Levine M. M. 1996. Further characterization of ΔaroA ΔvirG Shigella flexneri 2a strain CVD 1203 as a mucosal Shigella vaccine and as a live-vector vaccine for delivering antigens of enterotoxigenic Escherichia coli. Infect. Immun. 64:23–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oaks E. V., Wingfield M. E., Formal S. B. 1985. Plaque formation by virulent Shigella flexneri. Infect. Immun. 48:124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parsot C. 2009. Shigella type III secretion effectors: how, where, when, for what purposes? Curr. Opin. Microbiol. 12:110–116 [DOI] [PubMed] [Google Scholar]
- 45. Phalipon A., Sansonetti P. J. 2007. Shigella's ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol. Cell Biol. 85:119–129 [DOI] [PubMed] [Google Scholar]
- 46. Pozsgay V., et al. 1999. Protein conjugates of synthetic saccharides elicit higher levels of serum IgG lipopolysaccharide antibodies in mice than do those of the O-specific polysaccharide from Shigella dysenteriae type 1. Proc. Natl. Acad. Sci. U. S. A. 96:5194–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rahaman M. M., Khan M. M., Aziz K. M., Islam M. S., Kibriya A. K. 1975. An outbreak of dysentery caused by Shigella dysenteriae type 1 on a coral island in the Bay of Bengal. J. Infect. Dis. 132:15–19 [DOI] [PubMed] [Google Scholar]
- 48. Sadorge C., et al. 2008. Phase 1 clinical trial of live attenuated Shigella dysenteriae type-1 ΔicsA Δent Δfep ΔstxA:HgR oral vaccine SC599 in healthy human adult volunteers. Vaccine 26:978–987 [DOI] [PubMed] [Google Scholar]
- 49. Sereny B. 1955. Experimental shigella keratoconjunctivitis: a preliminary report. Acta Microbiol. Acad. Sci. Hung. 2:293–296 [PubMed] [Google Scholar]
- 50. Shiga K. 1898. On the cause of dysentery in Japan. Zentralbl. Bakteriol. Parasitenkd. Abt. 1 23:599 [Google Scholar]
- 51. Taylor D. N., et al. 1993. Synthesis, characterization, and clinical evaluation of conjugate vaccines composed of the O-specific polysaccharides of Shigella dysenteriae type 1, Shigella flexneri type 2a, and Shigella sonnei (Plesiomonas shigelloides) bound to bacterial toxoids. Infect. Immun. 61:3678–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vargas M., Gascon J., Jimenez De Anta M. T., Vila J. 1999. Prevalence of Shigella enterotoxins 1 and 2 among Shigella strains isolated from patients with traveler's diarrhea. J. Clin. Microbiol. 37:3608–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Venkatesan M. M., et al. 2002. Construction, characterization, and animal testing of WRSd1, a Shigella dysenteriae 1 vaccine. Infect. Immun. 70:2950–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Venkatesan M. M., Ranallo R. T. 2006. Live-attenuated Shigella vaccines. Expert Rev. Vaccines 5:669–686 [DOI] [PubMed] [Google Scholar]