

Abstract
CCR8 immunity is generally associated with Th2 responses in allergic diseases. In this study, we demonstrate for the first time a pronounced attenuated influx of macrophages in ovalbumin (OVA)-challenged CCR8 knockout mice. To explore whether macrophages in human inflamed lung tissue also were CCR8 positive, human lung tissue from patients with chronic obstructive pulmonary disease (COPD) was evaluated. Indeed, CCR8 expression was pronounced in invading monocytes/macrophages from lungs of patients with Global Initiative for Obstructive Lung Disease (GOLD) stage IV COPD. Given this expression pattern, the functional role of CCR8 on human macrophages was evaluated in vitro. Human peripheral blood monocytes expressed low levels of CCR8, while macrophage colony-stimulating factor (M-CSF)-derived human macrophages expressed significantly elevated surface levels of CCR8. Importantly, CCL1 directly regulated the expression of CD18 and CD49b and hence influenced the adhesion capacity of human macrophages. CCL1 drives chemotaxis in M-CSF-derived macrophages, and this could be completely inhibited by lipopolysaccharide (LPS). Whereas both CCL1 and LPS monotreatment inhibited spontaneous superoxide release in macrophages, CCL1 significantly induced superoxide release in the presence of LPS in a dose-dependent manner. Finally, CCL1 induced production of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) and could inhibit LPS-induced cytokine production in a dose-dependent manner. Our data demonstrate, for the first time, the presence of CCR8 on inflammatory macrophages in human COPD lung tissue. Importantly, the functional data from human macrophages suggest a potential cross talk between the CCR8 and the Toll-like receptor 4 (TLR4) pathways, both of which are present in COPD patients.
INTRODUCTION
Inflammation is the common host defensive response to invading pathogens, tissue damage, and allergens. It is characterized by enhanced local blood flow, leakage of blood plasma, and, most importantly, recruitment, accumulation, and activation of leukocytes (22). These inflammatory cells are recruited to the sites of inflammation in a strictly coordinated manner through directed migration from the bloodstream to the tissues. This migration is mediated by a group of structurally related chemotactic cytokines called chemokines. Upon binding to their cognate seven transmembrane G-coupled receptors, they initiate a cascade of events ending in binding of GTP to the receptor, which becomes activated (33). Based on their molecular structures, four groups of chemokines have so far been established, represented by the CC, CXC, C, and CX3C subfamilies (25). The various chemokine subfamilies are induced at various time points during the inflammatory response and exert their recruitment function on a limited set of cell types. Whereas CXC chemokines are associated with early rapid responses on neutrophils, CC chemokines are considered to play a role at later stages in the inflammatory process and in other cell types such as monocytes, lymphocytes, and eosinophils (32). A characteristic feature of the chemokine receptor families is a promiscuous binding of several ligands to one receptor (21). Different chemokines can bind with distinct affinities to a receptor which activates a variety of signaling pathways ending in the same functional response for the given cell. This promiscuity illustrates the complexity of the chemokine system and has led to an idea that each ligand/receptor interaction is nonredundant for complex disease progression (32). In contrast to the majority of chemokines, the CCL1 (I-309/TCA-3) ligand has been shown to have only one high-affinity mammalian receptor, CCR8 (30). Likewise, CCR8 has only one high-affinity mammalian ligand, CCL1. This makes this chemokine/ligand pair unique in the large chemokine family.
Allergic diseases such as asthma and atopic dermatitis are associated with a significant influx of inflammatory cells such as eosinophils and T lymphocytes. In these human diseases a clear upregulation of CCR8 in the infiltrating cells has been demonstrated (11). In fact, the number of CCR8-positive activated T cells has been correlated with a decline in FEV1 and thus has been suggested to play a role in asthma (12, 29). Likewise, the level of CCL1 is enhanced in asthmatic patients (12, 24). In animal models of allergic airway disease, it has been more difficult to conclusively show a role for CCR8 due to variable results from a number of groups (4–6, 12, 13). In other inflammatory models, such as experimental autoimmune encephalomyelitis (EAE), sepsis, and type I diabetes, CCR8 has been more clearly demonstrated to play an essential role in disease progression (3, 9, 23). In these diseases the CCR8-expressing cell type that drives progression is considered to be monocytes/macrophages rather than T cells (3, 9, 23).
Macrophages are heterogeneous cells that, depending on the microenvironment, can be divided broadly into two groups: M1 and M2 macrophages. Generally, the classically activated M1 macrophages are described as proinflammatory, while the alternatively activated M2 macrophages have been characterized as immunomodulating. M1 and M2 macrophages differ in terms of receptors, cytokine and chemokine expression, and effector functions. Treatment of human monocytes with granulocyte-macrophage colony-stimulating factor (GM-CSF) has been shown to differentiate the cells toward an M1 phenotype, while culture with M-CSF differentiates the cells toward an M2 phenotype, which allows evaluation of the cellular functions in vitro (10).
Here we demonstrate that in human lung tissue samples from patients with chronic obstructive pulmonary disease (COPD), the CCR8-positive cells are of monocytic lineage; i.e., they are macrophages rather than T cells. Our functional data for human macrophages in vitro indicate a potential novel role for CCR8 signaling in macrophages. The data suggest that CCR8 signaling may be influenced by and can influence the Toll-like receptor 4 (TLR4) pathway of innate immunity, which under high bacterial load could alter the progression of the disease.
MATERIALS AND METHODS
OVA-induced allergic airway disease.
Allergic airway inflammation was induced in C57BL/6 mice (Taconic, Denmark) to evaluate the expression of CCR8 and its ligand CCL1 and in CCR8 knockout (KO) mice (Deltagen Inc., San Mateo, CA) to investigate the role of CCR8 in allergic airway disease. The CCR8 KO mice were generated by standard gene-targeting techniques in 129/Ola × C57BL/6 backgrounds and backcrossing onto C57BL/6 (N4) mice. All experiments were conducted with approval of an ethics committee.
Briefly, animals were sensitized using 0.3 ml/animal ovalbumin (OVA) in alum, given on day 0 and day 7 via intraperitoneal (i.p.) injection, and challenged on days 14, 21 and 22 using an aerosol of OVA (10 mg/ml) generated by a Bird nebulizer for 1 h. The animals were killed at 2, 6, 12, and 24 h after the last OVA challenge. For histological analysis, lungs were inflated using 0.3 ml OCT (Sakura, Japan) diluted 2:1 in phosphate-buffered saline (PBS), dissected, submerged in OCT, frozen in isopentane (Sigma, Sweden) on dry ice, and stored at −70°C. For mRNA analysis, tissue RNA was isolated using phenol-chloroform extraction followed by determination of sample concentrations using a NanoDrop spectrometer (Thermo Scientific, Waltham, MA). RNA samples were treated with DNase I and RNase inhibitor (Fermentas, Germany) to remove any genomic DNA and to prevent RNA degradation. Upon determination of the RNA concentration, 3 cDNA reactions were performed using a SuperScript III First-Strand Synthesis SuperMix for quantitative reverse transcription-PCR (qRT-PCR) kit (Invitrogen catalog no. 11752). Following reverse transcription, residual RNA was removed through Escherichia coli RNase H treatment and the cDNA concentrations normalized. qPCR was performed using an Applied Biosystems 7500 real-time PCR system and a SYBR GreenER qPCR SuperMix Universal kit (Invitrogen, Carlsbad, CA). Relative quantification values were normalized to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) control gene.
Immunohistochemistry and immunofluorescence.
Eosinophil peroxidase (EPO) in frozen lung sections from mice was visualized by diaminobenzidine staining in cyanide-containing buffer. Primary antibodies against CCR8 (Enzo Life Sciences, PA) and CCL1 (R&D Systems, Minneapolis, MN) were detected with secondary mouse anti-goat IgG-biotin antibodies (Jackson ImmunoResearch, West Grove, PA) followed by Alexa 488-conjugated streptavidin (Molecular Probes, Carlsbad, CA). Colocalization of CCR8 with CD4 (BD Biosciences, Franklin Lakes, NJ) was detected with secondary goat anti-rat IgG-Alexa 568 (Molecular Probes, Invitrogen, Carlsbad, CA) and counterstaining with DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen, Carlsbad, CA). Lung tissues sections were viewed under a fluorescence microscope under similar conditions. By comparing consecutive sections, a qualitative assessment of EPO or CD107b (Serotec, Oxford, United Kingdom) and CCR8 staining was performed.
Expression of CCR8 in explanted human lung tissue from patients with very severe COPD (Global Initiative for Obstructive Lung Disease [GOLD] stage IV), acquired in association with lung transplantation, was evaluated. All subjects had given their written informed consent to participate in the study, which was approved by the local ethics committee in Lund, Sweden (no. 91/2006). Briefly, after collagenase treatment, cell aggregates were removed and single-cell suspensions were enriched by magnetic cell sorting (MACS). Human tissue sections were deparaffinized, rehydrated, blocked in 20% donkey normal serum (DNS) in PBS plus 0.1% Tween 20 at room temperature, and then incubated with CCR8 antibody (Enzo Life Sciences, PA) at 4°C overnight. After washing, the sections were incubated with secondary donkey anti-goat IgG-Alexa 488, donkey anti-goat IgG-Alexa 568, and donkey anti-mouse IgG-Alexa 488 for 45 min at room temperature and then counterstained with DAPI and viewed with a fluorescence microscope.
Monocyte purification and generation of monocyte-derived macrophages.
Human mononuclear cells were isolated from EDTA-anticoagulated blood of healthy volunteers by using Ficoll density centrifugation. Briefly, the remaining thrombocytes were removed by placing the purified monocytes over 2 ml plasma and centrifuged at 600 rpm for 5 min. Monocytes were separated from other peripheral blood mononuclear cells (PBMCs) by MACS using the monocyte isolation kit II (Miltenyi Biotec, Auburn, CA) and MidiMACS separation columns according to the manufacturers' instructions. Routinely >90% pure CD14 (BD Biosciences, Franklin Lakes, NJ)-positive cells were obtained as determined by flow cytometric analysis.
Monocytes were differentiated into macrophages in RPMI 1640 with GlutaMAX and HEPES supplemented with 10% heat-inactivated fetal calf serum, 50 μg/ml penicillin-streptomycin (PEST), 1 mM Na-pyruvate, 1 mM nonessential amino acids (Gibco BRL Life Technologies, Carlsbad, CA), and 50 μM β-mercaptoethanol (Sigma, St. Louis, MO). Briefly, monocytes were grown in the presence of either 50 ng/ml recombinant human M-CSF or 10 ng/ml recombinant human GM-CSF (R&D Systems, Minneapolis, MN) at 37°C in a humidified 5% CO2 incubator for 1 to 8 days.
Flow cytometric analysis.
Human monocytes and M-CSF- and GM-CSF-derived macrophages were resuspended and stimulated for 24 h with 10 ng/ml LPS (L-4516; Sigma-Aldrich, St. Louis, MO) and/or 1 nM CCL1. After that, they were resuspended in PBS–0.5% bovine serum albumin (BSA) and unspecific binding was blocked with CD16 and CD32 antibodies (BD Biosciences, San Jose, CA), followed by incubation with antibodies toward CD14, CD11b, CD18, CD29, CD49a, CD49b, CD49d, and CD49f (all from BD Biosciences, San Jose, CA) and CCR8 (R&D Systems, Minneapolis, MN) at 4°C for 30 min. Viability was monitored and dead cells excluded by 7-aminoactinomycin D (7-AAD) staining. Surface expression was monitored on a FACSCalibur using standard settings and analyzed with CellQuest Pro software. Data from the macrophage experiments are presented as geometric mean fluorescence intensity.
Single-cell mouse lung suspensions were prepared from lavaged mouse lung. Briefly, the right lung lobe was removed from the chest, placed in RPMI 1640 supplemented with 10% fetal calf serum (FCS), cut into small tissue pieces, treated with digest solution containing 1.33 mg/ml collagenase (Roche Diagnostics GmbH, Penzberg, Germany) and 0.1 kU/ml DNase (Sigma-Aldrich, St. Louis, MO) in a water bath for 60 min at 37°C, and finally filtered through a 70-μm sieve, and the number of total cells was counted. The single-cell suspension was blocked with antibodies against CD16/CD32, stained with CD3 and CD4 (both from BD Biosciences, San Jose, CA), CCR3 (R&D Systems, Minneapolis, MN), and F4/80 (eBioscience, Inc., San Diego, CA) antibodies, and analyzed with a FACSAria using standard settings and Diva software. Data from in vivo experiments are presented as the number of cells in the lung tissue expressing T cell markers (CD3 and CD4), eosinophil markers (CCR3 and high side scatter), or monocyte/macrophage markers (F4/80 and medium scatter).
Superoxide assay.
Measurement of extracellular superoxide was performed using a standard procedure. Briefly, M-CSF- or GM-CSF-derived macrophages were grown in unsupplemented medium for 24 h to let the cells go into a resting state. The macrophages were then incubated with medium supplemented with either 10 ng/ml LPS, 1 nM CCL1, or a combination of 10 ng/ml LPS and increasing concentrations of CCL1, all diluted in 0.1% Hanks balanced salt solution (HBSS)–BSA–10-mg/ml ferricytochrome c (Sigma, Sweden), for 30 min. Superoxide was then measured in the supernatants in a Spectramax at 550 nm as a function of the reduction of ferricytochrome c. Percent inhibition was calculated as 100 × [1 − (value with CCL1/value with medium alone)].
Chemotaxis assay.
Chemotaxis of M-CSF- or GM-CSF-derived macrophages was monitored by standard techniques. Briefly, cells were grown either in medium alone or in the presence of 10 ng/ml LPS for 24 h. Cells were then resuspended in RPMI 1640 supplemented with 0.5% BSA, and cell suspensions (4 × 106 cells/ml) were added to 5-μm-pore-size microchambers (Neuro Probe Inc., Gaithersburg, MD) while CCL1, medium (spontaneous migration), or labeled cells (maximum response) was added to the bottom chamber. After 2 h of incubation at 37°C, the filter was removed and alamarBlue (Invitrogen, Carlsbad, CA) was added and incubated for an additional 20 h. Fluorescence intensity was measured with a Spectramax Gemini at 550 nm, and the obtained value was used as a measurement for migration. The CCL1-specific migration was calculated migration to CCL1 − migration to medium alone.
Cytokine measurement.
Supernatants from human macrophages treated with CCL1 alone or in the presence of 1 ng/ml LPS for 24 h were harvested. interleukin-1β (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α) were measured in the supernatants by using a Lincoplex kit (LINCO Research Inc., St. Charles, MO) according to manufacturer's instructions. Percent inhibition was calculated as 100 × {1 − [(value with CCL1 − value with medium alone)/(value with LPS − value with medium alone)]}.
Statistical analyses.
The significance of differences between groups was calculated using one-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test, using GraphPad Prism. Statistical power is described as follows: not significant (NS), P > 0.05; *, P ≤ 0.05 to P ≥ 0.01; **, P ≤ 0.01 to P ≥ 0.001; and ***, P < 0.001. EC50s were generated by standard algorithm in GraphPad Prism.
RESULTS
Expression of CCR8 and CCL1 in mouse lung tissue.
To determine how CCR8 and CCL1 were regulated during inflammation, we evaluated their expression patterns in an OVA challenge model in C57BL/6 mice. OVA challenge significantly induced both CCL1 (2 h) and CCR8 mRNA (6 h) (Fig. 1A and B). While only individual cells expressed CCL1 in naïve animals, vascular endothelial cells were found to be a pronounced source of CCL1 in OVA-challenged mice (Fig. 1C and E). Whereas only few individual cells expressed CCR8 in naïve mice (Fig. 1D), a clear induction was noted in OVA-challenged mice, with high expression on structural cells such as bronchial epithelial cells and vascular endothelial cells (Fig. 1F). Importantly, while only a minority of the infiltrating eosinophils (EPO-positive cells) expressed CCR8 (Fig. 1G and H), approximately 30% of the CD4-positive T cells and the majority of phagocytotic CD107b-positive monocytes expressed CCR8 (Fig. 1I and data not shown). Taken together, the data suggest a coordinated upregulation of CCL1 and CCR8 in response to OVA challenge in mouse airways.
Fig. 1.

CCR8 and CCL1 expression patterns are regulated in OVA-challenged mice. (A and B) mRNA for CCL1 (A) or CCR8 (B) in mouse lung tissue is upregulated in response to OVA challenge compared to PBS. (C to F) The numbers of CCL1 (C and E)- and CCR8 (D and F)-expressing cells are upregulated in response to OVA challenge (E and F) compared to PBS (C and D). (G and H) EPO staining (eosinophils) of lung tissue at 24 h postchallenge (G) and fluorescent staining with CCR8 (H) show that there is no overlap in expression. (I) Costaining of CCR8 with CD4 shows association of CCR8 with a number of the CD4-positive cells. Each group had 5 animals per experiment, and tissue was evaluated at several locations with similar results. Representative images from one of three experiments with similar results are shown.
CCR8 is essential for accumulation of inflammatory cells in inflamed lung tissue.
With the demonstration of enhanced accumulation of CCR8-positive cells in OVA-challenged mouse lung tissue, we quantitatively evaluated the cellular dynamics in the lung tissue in CCR8 KO and wild-type mice in response to OVA challenge. Flow cytometric analysis confirmed the immunohistochemical analysis, with only few inflammatory cells in naïve animals but significant influx upon OVA challenge (Fig. 2A to C). There was an attenuated influx of eosinophils (P = 0.022), CD4-positive T cells (P = 0.016), and macrophages (P = 0.013) in CCR8 KO compared to wild-type OVA-challenged mice (Fig. 2A to C). In contrast, no changes in CD8 T cells or neutrophil count were seen (data not shown). Taken together, these data show that the absence of CCR8 reduces the numbers of several inflammatory cell types in the lung tissue, while no significant reduction of lung weight was evident, in a model of airway inflammation.
Fig. 2.
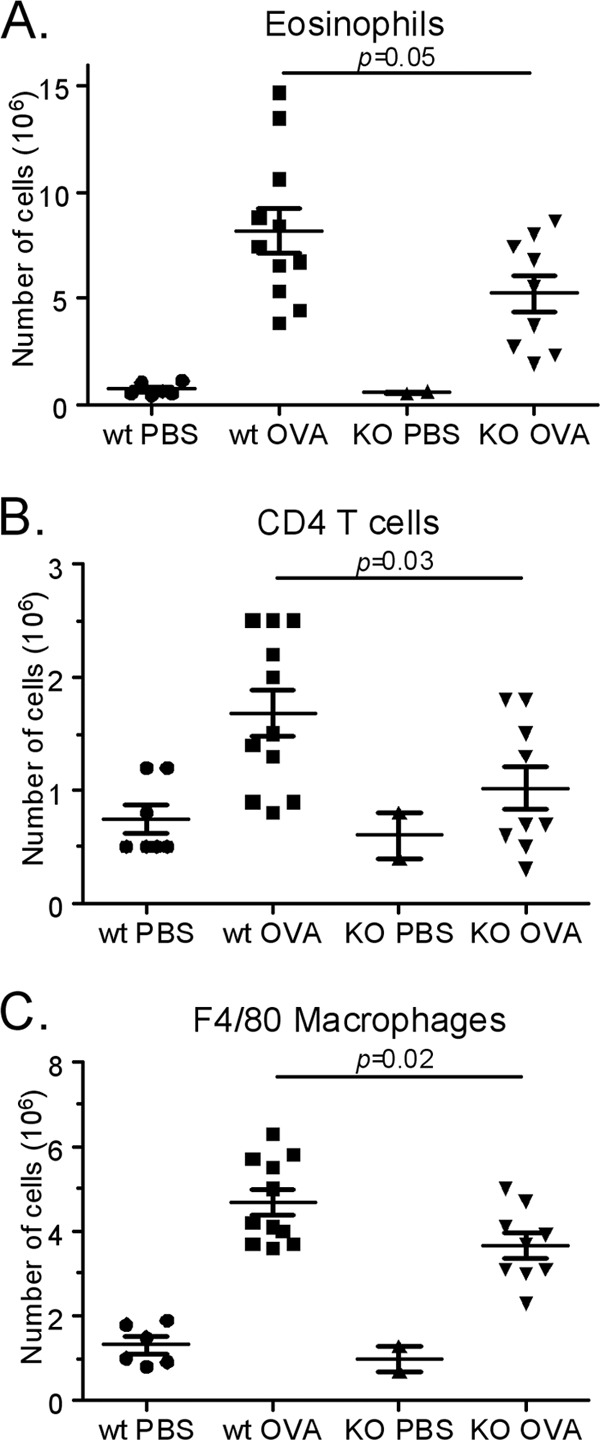
Significant reduction of inflammatory cells in challenged CCR8 knockout mice. The numbers of eosinophils (CCR3 in combination with scatter profile) (A), T cells (CD3 in combination with CD4) (B), and monocytes/macrophages (F4/80 in combination with scatter profile) (C) were all significantly reduced in CCR8 knockout animals compared to wild-type (wt) animals. Results are from one representative out of two experiments showing a similar inhibition of cellular influx. Each symbol represents one animal.
Expression in human COPD lung tissue.
With the observation that macrophages express CCR8 in murine inflamed lung tissue, we evaluated the expression of CCR8 in lung tissue from patients with COPD, a disease characterized by an elevated number of macrophages. It is clear that in human lung tissue follicular lymphocytes and endothelial cells are highly CCR8 positive (Fig. 3A and B). Importantly, the vast majority of the macrophages in the lung tissue expressed high levels of CCR8 (Fig. 3A and B). These observations are in accordance with the mouse expression pattern and strengthen the connection of CCR8 to the monocytic cell lineage during inflammation. On a cellular level, a homogenous expression of CCR8 was seen in almost all of the macrophages derived from GOLD stage IV COPD patients, which was confirmed by both immunocytochemistry (ICC) and flow cytometric analyses (Fig. 3C, D, and F). The number of CCR8-expressing macrophages from healthy controls and the level of CCR8 expression per cell were markedly lower than those for macrophages from COPD patients (Fig. 3E and F). Taken together, the data clearly show that macrophages in human lung tissue express CCR8 and that COPD lung tissue macrophages express enhanced levels of CCR8.
Fig. 3.

Expression of CCR8 in human COPD lung tissue. (A and B) CCR8 is expressed on endothelial cells, lymphocytes, and alveolar macrophages in human COPD lung tissue. (C and D) Enriched macrophages from human GOLD stage IV COPD lung explant tissue express significant levels of CCR8 (C), whereas the isotype control does not show any staining (D). (E) Enriched macrophages from healthy human lung explants tissue express only low levels of CCR8. (F) The number of CCR8 positive cells was quantified from five patient samples in a flow cytometric analysis.
Human M2 macrophages express high levels of CCR8.
In order to evaluate the function of CCR8 on macrophages, human peripheral blood-derived monocytes were differentiated into M1 (by GM-CSF) or M2 (by M-CSF) macrophage subtypes in vitro. Freshly isolated peripheral blood monocytes expressed negligible levels of CCR8 (Fig. 4). M-CSF-derived macrophages had significantly upregulated surface CCR8, with peak expression on day 2 (40-fold enhanced). GM-CSF-derived macrophages also had enhanced CCR8 expression, but to a significantly lower degree than M-CSF-derived macrophages (Fig. 4). At all time points evaluated, M-CSF-derived and GM-CSF-derived macrophages expressed significantly elevated levels of surface CCR8 compared to monocytes (P values of <0.001 and <0.05, respectively).
Fig. 4.

Kinetic analysis of CCR8 expression on M-CSF- or GM-CSF-derived human macrophages. Expression is shown as geometric mean fluorescence intensity (MFI). The standard deviation was routinely less than 20%. Values are means from three independent donors. Data are from one representative out of three experiments.
Thus, CCR8 is induced on both M1 and M2 macrophages. Due to the maximum induction of CCR8 on day 2, all functional analysis was conducted at this time point. With the significantly higher CCR8 expression on M-CSF-derived cells, a more in-depth analysis was performed on these cells than on the GM-CSF-derived cells.
Adhesion molecules on human macrophages are modulated by CCL1 and LPS.
To evaluate whether CCR8 signaling modulates expression of adhesion molecules and activation markers, the M-CSF- and GM-CSF-derived macrophages were evaluated by flow cytometric analysis after challenge with CCL1. These markers all play important roles in the rolling, local tissue adhesion and accumulation of inflammatory cells.
CCL1 treatment of M-CSF-derived macrophages significantly upregulated CD18, whereas CD49b was significantly downregulated compared to that in unchallenged M-CSF-derived macrophages (Fig. 5B and D; Table 1). All markers, with the exception of CD29, were significantly modulated by LPS (Fig. 5C; Table 1). Interestingly, although neither CCL1 nor LPS independently modulated CD29 expression significantly, the combination of CCL1 and LPS did significantly downregulate CD29 expression (Fig. 5C; Table 1). The expression of CD18 was normalized to levels noted in unchallenged M-CSF-derived macrophages when the cells were cotreated with LPS and CCL1 (Fig. 5B; Table 1). CCL1 did not induce a significant effect by itself and was not significantly modulated by the LPS response in GM-CSF-derived macrophages (Fig. 5A to F; Table 1).
Fig. 5.

Expression of integrin molecules on human macrophages after treatment with 1 nM CCL1 or 10 ng/ml LPS. (A to F) Expression of CD11b (A), CD18 (B), CD29 (C), CD49b (D), CD49d (E), and CD49f (F) in M-CSF- or GM-CSF-derived macrophages. Expression is shown as geometric mean fluorescence intensity. (G) Results from one representative experiment to show the gating strategy. Viability was monitored in all samples and was routinely >95% (data not shown). The standard deviation was routinely less than 20%. Values are means from three independent donors. Data are from one representative out of three experiments.
Table 1.
Statistical change of expression of integrin molecules on human macrophages after treatment with 1 nM CCL1 and/or 10 ng/ml LPS
| Molecule | Comparison | Significance of differencea for macrophages derived with: |
|
|---|---|---|---|
| M-CSF | GM-CSF | ||
| CD11b | Medium vs LPS | * | NS |
| Medium vs CCL1 | NS | NS | |
| Medium vs LPS + CCL1 | * | NS | |
| LPS vs CCL1 | ** | NS | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | ** | NS | |
| CD18 | Medium vs LPS | NS | NS |
| Medium vs CCL1 | ** | NS | |
| Medium vs LPS + CCL1 | NS | NS | |
| LPS vs CCL1 | *** | NS | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | ** | NS | |
| CD29 | Medium vs LPS | NS | NS |
| Medium vs CCL1 | NS | NS | |
| Medium vs LPS + CCL1 | * | NS | |
| LPS vs CCL1 | NS | NS | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | ** | NS | |
| CD49b | Medium vs LPS | *** | NS |
| Medium vs CCL1 | ** | NS | |
| Medium vs LPS + CCL1 | *** | NS | |
| LPS vs CCL1 | * | NS | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | ** | NS | |
| CD49d | Medium vs LPS | *** | * |
| Medium vs CCL1 | NS | NS | |
| Medium vs LPS + CCL1 | *** | NS | |
| LPS vs CCL1 | *** | * | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | *** | * | |
| CD49f | Medium vs LPS | *** | NS |
| Medium vs CCL1 | NS | NS | |
| Medium vs LPS + CCL1 | ** | NS | |
| LPS vs CCL1 | ** | NS | |
| LPS vs LPS + CCL1 | NS | NS | |
| CCL1 vs LPS + CCL1 | * | NS | |
The significance of differences between groups was calculated using one-way ANOVA with Bonferroni's multiple-comparison test, using GraphPad Prism. Statistical power is described as follows: NS, P > 0.05; *, P ≤ 0.05 to P ≥ 0.01; **, P ≤ 0.01 to P ≥ 0.001; and ***, P < 0.001.
Taken together, the data demonstrate that CCL1 affects expression of adhesion molecules differently in M-CSF- and GM-CSF-derived macrophages. This suggests that CCR8 signaling may potentially affect macrophage functionality differently in the two subsets.
Chemotaxis, superoxide release, and cytokine release are modulated by interactions of CCL1 and TLR4.
To further evaluate the potential role of CCL1/CCR8 signaling in macrophages, chemotaxis, superoxide generation, and production of COPD-associated cytokines in the presence of LPS were evaluated. Fresh blood monocytes isolated directly from peripheral blood did not migrate in response to CCL1 (data not shown). M-CSF-derived macrophages responded to CCL1 with a classical bell-shaped chemotaxis response, with maximum migration at 30 nM (Fig. 6A). Most interestingly, the presence of LPS during the migration completely inhibited the CCL1-induced migration of M-CSF-derived macrophages (Fig. 6A). CCL1 inhibited spontaneous migration of GM-CSF-derived macrophages, which was even further augmented by addition of LPS (Fig. 6B).
Fig. 6.

Macrophage functions are modulated by CCL1 and 10 ng/ml LPS. (A and B) Migration of M-CSF (A)- or GM-CSF (B)-derived macrophages toward increasing concentrations of CCL1 in the presence or absence of LPS. Negative values indicate less migration than for unstimulated cells (i.e., medium control). (C) Inhibition of superoxide generation compared to that with medium alone (i.e., spontaneous superoxide release) in M-CSF- and GM-CSF-derived macrophages. (D) Synergistic agonistic activity on superoxide generation between 10 ng/ml LPS and CCL1 in M-CSF-derived macrophages. The standard deviation was routinely less than 20%. Values are means from three independent donors. Data are from one representative out of three experiments.
CCL1 challenge of M-CSF-derived cells inhibited spontaneous superoxide release in M-CSF-derived cells in a dose-dependent manner, with a 50% effective concentration (EC50) of 4 nM. A weak inhibition was also observed in the GM-CSF-derived macrophages (Fig. 6C). LPS treatment alone also inhibited spontaneous release of superoxide in both macrophage subpopulations (Fig. 6D). In sharp contrast, CCL1 induced superoxide release in the presence of LPS in a dose-dependent manner (Fig. 6D). Importantly, this phenomenon was observed at concentrations of CCL1 which showed no effect on their own (Fig. 6C and D).
CCL1 induced significant production of TNF-α and IL-6 in M-CSF-derived macrophages, while no induction of IL-1β was detected (Fig. 7A, C, and E). As expected, LPS induced highly significant levels of all three cytokines in the macrophages (Fig. 7A, C, and E). Most interestingly, CCL1 effectively downregulated, in a dose-dependent manner, the LPS-induced production of all three cytokines. The most potent inhibitory effect of CCL1 on LPS-mediated cytokine production was noted for TNF-α production, with an EC50 of 2 nM, followed by IL-1β and IL-6 (EC50 of 19 and 71 nM, respectively).
Fig. 7.

Production of inflammatory cytokines is modulated by 10 nM CCL1 and 10 ng/ml LPS. Production of IL-1β (A), TNF-α (C), and IL-6 (E) in M-CSF-derived macrophages and inhibition of LPS-induced IL-1β (B), TNF-α (D), and IL-6 (F) by increasing concentrations of CCL1 are shown. Values are means from three independent donors. Data are from one representative out of three experiments.
Taken together, the results show that CCL1 either positively or negatively modulates effector functions in human macrophages. The data suggest that the CCL1 and TLR4 pathways may influence each other's activities in human macrophages.
DISCUSSION
In this report, we describe impaired monocyte infiltration in a murine airway inflammation model in CCR8 KO mice. In addition, we demonstrate CCR8 expression in human COPD lung tissue macrophages. Importantly, we show a novel role for CCR8 on human macrophages that may have implications in innate immunity responses to pathogen elimination during inflammatory conditions.
The relevance of CCR8 in inflammatory disease progression has not yet been firmly established. With the recognition that asthma comprises a range of heterogeneous phenotypes that differ in presentation, etiology, and pathophysiology (2), it is important to note that in our hands expression of both CCL1 and CCR8 is spatially and temporally regulated during the inflammatory response. The demonstration of a significant upregulation of CCL1 in close proximity to activated endothelial cells early during the inflammatory response suggests that these cells may contribute to creating a gradient of CCL1 from the lung tissue to the circulation which will attract inflammatory cells. Previous studies by Haque et al. have demonstrated, in a human umbilical vein endothelial cell (HUVEC) model of atherosclerosis, that endothelial cells stimulated with apolipoprotein(a) can be induced to express CCL1 (16). In the atherosclerosis model, the endothelial CCL1 production results in attraction of peripheral monocytes and macrophages. In our OVA model of airway inflammation, we also detected enhanced numbers of monocytes/macrophages in the lung tissue. The activated endothelial cells also upregulated CCR8 at later time points. This indicates that the endothelial cells may both contribute to the CCL1 gradient and respond to CCL1 intrinsically in an autocrine manner. Indeed, HUVECs and human aortic smooth muscle cells have been shown to respond to CCL1 with chemotaxis in a pro-matrix metalloproteinase 2 (pro-MMP2)-dependent manner (15). This autocrine loop creates a microenvironment highly responsive to and dependent on the interaction of CCL1 and CCR8. The significant expression observed in the inflamed lungs in our study suggests that CCR8 may have a role on structural cells not previously recognized during airway inflammatory responses, which should be further evaluated.
The biological importance of CCR8 in airway inflammation, e.g., asthma, is not firmly established (4–6, 12, 13). Recently, Gonzalo et al. proposed an explanation for the contradictory results generated in the CCR8 knockout animals by demonstrating a key role for mast cells as the CCL1 producer (12). In addition to the murine data, the importance of CCR8 in human diseases is demonstrated firmly by a clear demonstration of a correlation between the number of CCR8+ T cells in asthma lung tissue and a decline in FEV1 (28, 29). Previously, most of the focus on the role for CCR8 in airway inflammatory models has been to establish a link to Th2 cells. Our data show for the first time a clear relationship between CCR8 and the monocytic lineage in murine lung tissue. Importantly, our data demonstrate that in addition to a reduction of the number of T cells and eosinophils, there was a significant reduction of monocytes/macrophages in the lung tissue in OVA-challenged CCR8 KO animals. Monocytes/macrophages from explanted human COPD lung were also demonstrated to express significant levels of CCR8. Our novel observation in COPD patients is in alignment with observations on multiple sclerosis (MS) lesions from patients who previously has been demonstrated to express CCR8 and correlated with demyelization activity in the brain and in foam cells in arthrosclerosis plaque (16, 34). In preclinical models of peritoneal adhesion, macrophages were described to produce CCL1 and respond in an autocrine fashion through CCR8 (17). These cells regulate expression of several adhesion molecules in the integrin family, allowing formation of aggregates of cells that over time result in fibrous tissue (17). In context of this observation, it is interesting that adhesion molecule expression on in vitro-generated human M2 macrophages (M-CSF derived) is more sensitive to CCL1 than that on M1 macrophages (GM-CSF derived). Kang et al. have previously demonstrated that prolonged exposure to LPS downregulates expression of certain VLA components, while brief transient exposure upregulates expression (18). Our observations suggest that M2 macrophages may be more involved in conditions such as peritoneal adhesion surgery complications and that CCL1 may play a contributing role.
The observation that CCL1 negatively regulates superoxide release from macrophages in a dose-dependent manner is intriguing. Matsukawa et al. elegantly demonstrated that CCR8 negatively impacts the host defense during septic peritonitis (23). In their murine experiments, LPS induced superoxide production, which was augmented in CCR8 knockout animals. In contrast, we failed to induce superoxide in our human M-CSF-derived macrophages with LPS. In fact, a significant downregulation of spontaneous superoxide was observed. However, in combination with CCL1, LPS induces significant reactive oxygen species (ROS) generation. This is in agreement with a previous study that suggested a requirement of “priming/activation” of the macrophage to induce generation of significant amounts of ROS (7). The effect of CCL1 together with LPS suggests a potential cross talk of the CCR8 pathway with the TLR4 pathway during bacterial infections at the site of inflammation. Given that exacerbation of severe asthma and COPD often is driven by bacterial infections, the observation that CCL1 positively influences ROS generation is of great importance since it may positively affect antimicrobial responses (26, 27, 36). On the other hand, LPS released during subchronic bacterial infections, which are often observed in these patients, may together with CCR8 activation on the macrophages account for the excessive release of ROS in these patients, thereby contributing to the lung tissue damage and accelerated inflammation (1).
The discrepancy between the murine macrophages (23) and our human macrophages is intriguing. Further attention is warranted, and responsiveness to LPS, CCR8 sequence homology between species, and the influence of natural antagonists to CCR8 should be evaluated.
Failure to mount appropriate innate/adoptive immune responses to invading bacteria may result in systemic responses leading to an overwhelming cytokine/chemokine storm commonly known as the systemic inflammatory response syndrome (SIRS) (14). LPS induces production of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β though TLR4 interactions in both murine and human macrophages (8). The common view is that signaling though Gi proteins on macrophages favors a proinflammatory cytokine response (8). However, inhibition of signaling though certain Gαi subunits in murine peritoneal macrophages has actually been shown to augment LPS-induced cytokine production (8). Since Gi proteins do not directly couple to nonheptahelical receptors such as TLR4, the interaction most likely takes place through trans-activation of other receptors in lipid rafts (19, 20, 31, 35). Our data demonstrating that CCL1 by itself generates cytokine release support the hypothesis that Gi-coupled receptors cause a proinflammatory response in macrophages. It is intriguing that the presence of both TLR4 and CCR8 signaling in human macrophages reduces the TLR4-induced cytokine production in a dose-dependent fashion. The results further strengthen the conclusion that CCR8 signaling may influence TLR4 pathways activated by bacterial components. Our human macrophage data showing a negative impact on cytokine production by CCL1 treatment support the observation in the CCR8 knockout mouse that the absence of CCR8 signaling further enhances LPS-induced cytokine production (23).
Taken together, our data demonstrate that human COPD lung macrophages express CCR8. The absence of CCR8 negatively influences not only T cells and eosinophils but also the number of lung monocytes/macrophages in an airway inflammation model. Our study suggests a novel functional role for CCR8 in human macrophages, with impacts on adhesion, chemotaxis, ROS generation, and cytokine production. The observation that human macrophages respond to LPS differently in the presence of CCR8 could be of great importance for several diseases and indicate a potential role for the host defense and should be further evaluated. This novel information on the role of CCR8 on macrophages suggests that future therapeutic approaches to target CCR8 need to be carefully evaluated in context of bacterial load.
ACKNOWLEDGMENTS
We thank Katarina Walles and Karin Hellmann for excellent technical assistance and Elisabet Wieslander, John D. Taylor, and Harbans Lal for help with study design and valuable discussion.
Footnotes
Published ahead of print on 5 October 2011.
REFERENCES
- 1. Bowler R. P., Barnes P. J., Crapo J. D. 2004. The role of oxidative stress in chronic obstructive pulmonary disease. COPD 1:255–277 [DOI] [PubMed] [Google Scholar]
- 2. Burke W., Fesinmeyer M., Reed K., Hampson L., Carlsten C. 2003. Family history as a predictor of asthma risk. Am. J. Prev. Med. 24:160–169 [DOI] [PubMed] [Google Scholar]
- 3. Cantor J., Haskins K. 2007. Recruitment and activation of macrophages by pathogenic CD4 T cells in type 1 diabetes: evidence for involvement of CCR8 and CCL1. J. Immunol. 179:5760–5767 [DOI] [PubMed] [Google Scholar]
- 4. Chensue S. W., et al. 2001. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J. Exp. Med. 193:573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiu B. C., Chensue S. W. 2002. Chemokine responses in schistosomal antigen-elicited granuloma formation. Parasite Immunol. 24:285–294 [DOI] [PubMed] [Google Scholar]
- 6. Chung C. D., et al. 2003. CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J. Immunol. 170:581–587 [DOI] [PubMed] [Google Scholar]
- 7. Cunnick J., Kaur P., Cho Y., Groffen J., Heisterkamp N. 2006. Use of bone marrow-derived macrophages to model murine innate immune responses. J. Immunol. Methods 311:96–105 [DOI] [PubMed] [Google Scholar]
- 8. Fan H., et al. 2007. Differential regulation of lipopolysaccharide and Gram-positive bacteria induced cytokine and chemokine production in macrophages by Galpha(i) proteins. Immunology 122:116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fife B. T., Paniagua M. C., Lukacs N. W., Kunkel S. L., Karpus W. J. 2001. Selective CC chemokine receptor expression by central nervous system-infiltrating encephalitogenic T cells during experimental autoimmune encephalomyelitis. J. Neurosci. Res. 66:705–714 [DOI] [PubMed] [Google Scholar]
- 10. Fleetwood A. J., Dinh H., Cook A. D., Hertzog P. J., Hamilton J. A. 2009. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 86:411–421 [DOI] [PubMed] [Google Scholar]
- 11. Gombert M., et al. 2005. CCL1-CCR8 interactions: an axis mediating the recruitment of T cells and Langerhans-type dendritic cells to sites of atopic skin inflammation. J. Immunol. 174:5082–5091 [DOI] [PubMed] [Google Scholar]
- 12. Gonzalo J. A., et al. 2007. Coordinated involvement of mast cells and T cells in allergic mucosal inflammation: critical role of the CC chemokine ligand 1:CCR8 axis. J. Immunol. 179:1740–1750 [DOI] [PubMed] [Google Scholar]
- 13. Goya I., et al. 2003. Absence of CCR8 does not impair the response to ovalbumin-induced allergic airway disease. J. Immunol. 170:2138–2146 [DOI] [PubMed] [Google Scholar]
- 14. Hack C. E., Aarden L. A., Thijs L. G. 1997. Role of cytokines in sepsis. Adv. Immunol. 66:101–195 [DOI] [PubMed] [Google Scholar]
- 15. Haque N. S., Fallon J. T., Pan J. J., Taubman M. B., Harpel P. C. 2004. Chemokine receptor-8 (CCR8) mediates human vascular smooth muscle cell chemotaxis and metalloproteinase-2 secretion. Blood 103:1296–1304 [DOI] [PubMed] [Google Scholar]
- 16. Haque N. S., et al. 2000. CC chemokine I-309 is the principal monocyte chemoattractant induced by apolipoprotein(a) in human vascular endothelial cells. Circulation 102:786–792 [DOI] [PubMed] [Google Scholar]
- 17. Hoshino A., et al. 2007. Inhibition of CCL1-CCR8 interaction prevents aggregation of macrophages and development of peritoneal adhesions. J. Immunol. 178:5296–5304 [DOI] [PubMed] [Google Scholar]
- 18. Kang Y. H., Lee C. H., Brummel S. E., Newball H. H., Forrester J. 1995. Effects of endotoxin on expression of VLA integrins by human bronchoalveolar lavage macrophages. J. Leukoc. Biol. 57:624–634 [DOI] [PubMed] [Google Scholar]
- 19. Ling K., et al. 1999. Five-transmembrane domains appear sufficient for a G protein-coupled receptor: functional five-transmembrane domain chemokine receptors. Proc. Natl. Acad. Sci. U. S. A. 96:7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luttrell L. M., Daaka Y., Lefkowitz R. J. 1999. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr. Opin. Cell Biol. 11:177–183 [DOI] [PubMed] [Google Scholar]
- 21. Mantovani A. 1999. The chemokine system: redundancy for robust outputs. Immunol. Today 20:254–257 [DOI] [PubMed] [Google Scholar]
- 22. Martinon F., Mayor A., Tschopp J. 2009. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27:229–265 [DOI] [PubMed] [Google Scholar]
- 23. Matsukawa A., et al. 2006. Absence of CC chemokine receptor 8 enhances innate immunity during septic peritonitis. FASEB J. 20:302–304 [DOI] [PubMed] [Google Scholar]
- 24. Montes-Vizuet R., et al. 2006. CC chemokine ligand 1 is released into the airways of atopic asthmatics. Eur. Respir. J. 28:59–67 [DOI] [PubMed] [Google Scholar]
- 25. Murphy P. M., et al. 2000. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52:145–176 [PubMed] [Google Scholar]
- 26. Murray H. W., Cohn Z. A. 1980. Macrophage oxygen-dependent antimicrobial activity. III. Enhanced oxidative metabolism as an expression of macrophage activation. J. Exp. Med. 152:1596–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murray H. W., Juangbhanich C. W., Nathan C. F., Cohn Z. A. 1979. Macrophage oxygen-dependent antimicrobial activity. II. The role of oxygen intermediates. J. Exp. Med. 150:950–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mutalithas K., et al. 2010. Expression of CCR8 is increased in asthma. Clin. Exp. Allergy 40:1175–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Panina-Bordignon P., et al. 2001. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J. Clin. Invest. 107:1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roos R. S., et al. 1997. Identification of CCR8, the receptor for the human CC chemokine I-309. J. Biol. Chem. 272:17251–17254 [DOI] [PubMed] [Google Scholar]
- 31. Saito N., et al. 2002. Possible involvement of C-C chemokines in functional augmentation of adhesion molecules in asthmatic patients. Lung 180:251–263 [DOI] [PubMed] [Google Scholar]
- 32. Smit J. J., Lukacs N. W. 2006. A closer look at chemokines and their role in asthmatic responses. Eur. J. Pharmacol. 533:277–288 [DOI] [PubMed] [Google Scholar]
- 33. Thelen M. 2001. Dancing to the tune of chemokines. Nat. Immunol. 2:129–134 [DOI] [PubMed] [Google Scholar]
- 34. Trebst C., et al. 2003. CC chemokine receptor 8 in the central nervous system is associated with phagocytic macrophages. Am. J. Pathol. 162:427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Triantafilou K., Triantafilou M., Dedrick R. L. 2001. A CD14-independent LPS receptor cluster. Nat. Immunol. 2:338–345 [DOI] [PubMed] [Google Scholar]
- 36. Wilson C. B., Tsai V., Remington J. S. 1980. Failure to trigger the oxidative metabolic burst by normal macrophages: possible mechanism for survival of intracellular pathogens. J. Exp. Med. 151:328–346 [DOI] [PMC free article] [PubMed] [Google Scholar]