

Abstract
Candida albicans frequently infects medical devices by growing as a biofilm, i.e., a community of adherent organisms entrenched in an extracellular matrix. During biofilm growth, Candida spp. acquire the ability to resist high concentrations of antifungal drugs. One recently recognized biofilm resistance mechanism involves drug sequestration by matrix β-1,3 glucan. Using a candidate gene approach, we investigated potential C. albicans β-1,3-glucan regulators, based on their homology to Saccharomyces cerevisiae, including SMI1 and protein kinase C (PKC) pathway components. We identified a role for the SMI1 in biofilm matrix glucan production and development of the associated drug resistance phenotype. This pathway appears to act through transcription factor Rlmp and glucan synthase Fks1p. The phenotypes of these mutant biofilms mimicked those of the smi1Δ/smi1Δ biofilm, and overexpression of FKS1 in the smi1Δ/smi1Δ mutant restored the biofilm resistant phenotype. However, control of this pathway is distinct from that of the upstream PKC pathway because the pkc1Δ/pkc1Δ, bck1Δ/bck1Δ, mkk2Δ/mkk2Δ, and mkc1Δ/mkc1Δ biofilms retained the resistant phenotype of the parent strain. In addition, resistance to cell-perturbing agents and gene expression data do not support a significant role for the cell wall integrity pathway during the biofilm formation. Here we show that Smi1p functions in conjunction with Rlm1p and Fks1p to produce drug-sequestering biofilm β-glucan. Our work provides new insight into how the C. albicans biofilm matrix production and drug resistance pathways intersect with the planktonic cell wall integrity pathway. This novel connection helps explain how pathogens in a multicellular biofilm community are protected from anti-infective therapy.
INTRODUCTION
Candida spp. are an increasing common cause of bloodstream infection in hospitalized patients (42). The capacity to grow in a biofilm state allows these pathogenic fungi to adhere and thrive on medical devices, such as venous catheters, urinary catheters, and dentures (12, 19). The communities of adherent cells become encased in an extracellular matrix, and the cells within the biofilm show extreme resistance to antifungal drugs and host defenses. The biofilm lifestyle allows Candida spp. to cause device-associated infections in otherwise healthy hosts (21).
Compared to planktonic, nonbiofilm cells, Candida albicans biofilm cells are up to 1,000-fold more resistant to antifungals (7, 22, 28, 44). The treatment-recalcitrant phenotype associated with the Candida biofilm state is responsible in part for the high morbidity and mortality observed for patients hospitalized with these infections. Biofilm resistance studies have shown the relevance of several mechanisms important in planktonic resistance in accounting for a portion of this biofilm phenotype (6, 28, 43). It is clear from these studies that C. albicans biofilm resistance involves contributions from a combination of mechanisms (1, 2, 8, 22, 28, 43, 47, 50). Studies exploring biofilm resistance have also investigated and identified a link between assembly of an extracellular matrix and biofilm resistance (1, 2, 8, 34, 47, 51). Our recent findings show that glucan synthesis is critical for biofilm-specific drug resistance in C. albicans (32, 34). Both production of β-glucan in the biofilm matrix and antifungal resistance to each of the four commercially available antifungal drug classes require β-1,3-glucan synthase gene FKS1/GSC1. Disruption of this process decreases drug sequestration in the matrix, rendering biofilms susceptible to antifungal agents. In the proposed resistance model, the matrix glucan covering the biofilm cells is capable of sequestering antifungal drugs and prevents them from reaching their targets. This mechanism was found to account for a large percentage of the drug resistance phenotype during biofilm growth.
Our goal is to identify genes and pathways that control biofilm matrix production and drug resistance in C. albicans. Because cell wall pathways are relatively conserved among fungi, we chose to investigate genes known to function in Saccharomyces cerevisiae glucan production (15). The protein kinase C (PKC) pathway in yeast has been shown to be important for downstream control of Fks1p and cell wall glucan production in response to stress (26). In addition, this gene network is important for cell wall integrity (26). The function of this pathway in yeast led us to ask two questions. First, is the yeast pathway conserved in C. albicans biofilms, potentially controlling β-1,3-glucan matrix and drug resistance? Second, does alteration in cell wall integrity during C. albicans biofilm growth contribute to drug resistance?
Here we show that the downstream components of the yeast PKC pathway, specifically, SMI1, RLM1, and FKS1, are important for manufacture of C. albicans cell wall and matrix β-1,3-glucan during biofilm growth. SMI1 appears to regulate glucan production through expression of FKS1 glucan synthase. Without production of biofilm β-1,3-glucan, biofilm cells exhibit increased susceptibility to antifungal drugs. Our findings suggest that the matrix production pathway intersects with cell wall integrity regulation but that this regulation is distinct from that of the PKC pathway. Because matrix production is integral for biofilm formation and drug resistance, development of drugs targeting this pathway is a potential strategy for generating antifungals effective against biofilm fungal infections.
MATERIALS AND METHODS
Media.
Strains were stored in 15% (vol/vol) glycerol stock at −80°C and maintained on yeast extract-peptone-dextrose (YPD) medium with uridine (1% yeast extract, 2% peptone, 2% dextrose, and 80 μg/ml uridine) prior to experiments. C. albicans transformants were selected on synthetic medium (2% dextrose, 6.7% yeast nitrogen base [YNB] with ammonium sulfate, and auxotrophic supplements) or on YPD plus clonNat (2% Bacto peptone, 2% dextrose, 1% yeast extract, and 400 μg/ml clonNat [Werner Bioagents]) (37). Prior to biofilm experiments C. albicans strains were grown at 30°C in YPD and biofilms were grown in RPMI 1640 buffered with morpholinepropanesulfonic acid (RPMI-MOPS) (31).
Strains and strain construction.
The C. albicans strains used in these studies are listed in Table 1. Strains SF004a (bck1Δ/bck1Δ), VIC1175 (mkc1Δ/mkc1Δ), VIC1156 (mkk2Δ/mkk2Δ), VIC1167 (pkc1Δ/pkc1Δ), BRY429 (rlm1Δ/rlm1Δ), and VIC1075 (cas5Δ/cas5Δ) were generously provided by A. Mitchell (9, 10). Their construction has been previously described (10, 36). Heterozygous mutant strain JEN118 (SMI1/smi1Δ) and the smi1Δ/smi1Δ homozygous mutant strain were constructed from the parent SN152 using disruption marker cassettes and fusion PCR, as previously described (39). For the first round of PCR, primers SMI1 F1 (CATTTCTCCGTAATATTGGT) and SMI1 F4 (GTCAGCGGCCGCATCCCTGCTCATCTACAAGACCATGACA) were paired with SMI1 F3 (CACGGCGCGCCTAGCAGCGGAAGAAAGAAAGGAAGGAAAC) and SMI1 F6 (ATCATCACCAGAAAAGATTG), respectively, to amplify the homologous sequences flanking SMI1 from template genomic DNA. Universal primers 2 (CCGCTGCTAGGCGCGCCGTGACCAGTGTGATGGATATCTGC) and 5 (GCAGGGATGCGGCCGCTGACAGCTCGGATCCACTAGTAACG) were used to amplify Candida dubliniensis HIS1 or Candida maltosa LEU2 with template pSN52 or pSN40, respectively. Flanking sequences were purified with the QIAquick PCR purification kit (Qiagen), and the markers were purified with the AxyPrep DNA gel extraction kit (Axygen Biosciences). Fusion reaction mixtures were assembled using purified templates, SMI1 F1 and F6 primers, and Extaq polymerase (Takara). Following lithium acetate transformation, correct insertion was confirmed with SMI1 Fusion US check (TATATATGCAGGCACAAGAA) and SMI1 Fusion DS check (TGAACAACCGATACAATATG) paired with HIS1 and Leu2 internal primers, as described previously (39).
Table 1.
C. albicans strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| SN152 | URA3IRO1arg4his1leu2 | 40 |
| ura3::λimm434iro1::λimm434arg4his1leu2 | ||
| JEN118 | URA3IRO1arg4his1leu2sml1::C.dHIS1 | This study |
| ura3::λimm434iro1::λimm434arg4his1leu2SMI1 | ||
| JEN136 | URA3IRO1arg4his1leu2smi1::C.dHIS1 | This study |
| ura3::λimm434iro1::λimm434arg4his1leu2smi1::C.m LEU2 | ||
| JEN188 | URA3IRO1arg4his1leu2::SMI1::C.dARG4smi1::C.d HIS1 | This study |
| ura3::λimm434iro1::λimm434arg4his1leu2smi1::C.m LEU2 | This study | |
| JEN172 | URA3IRO1arg4his1leu2smi1::C.d HIS1FKS1::pAgTEF1-NAT1-AgTEF1UTR-TDH3-SMI1 | This study |
| ura3::λimm434iro1::λimm434arg4his1leu2smi1::C.mLEU2FKS1 | ||
| BWP17 | ura3::λimm434arg4::hisGhis1::hisG | 52 |
| ura3::λimm434arg4::hisGhis1::hisG | ||
| FKS1/ | ura3::λimm434arg4::hisGhis1::hisGfks1::URA3 | 35 |
| fks1Δ | ura3::λimm434 arg4::hisGhis1::hisGFKS1 | |
| SF004a | ura3::λimm434arg4::hisGhis1::hisGbck1::Tn7-UAU1 | 9 |
| ura3::λimm434arg4::hisGhis1::hisG bck1::Tn7-URA3 | ||
| VIC1175 | ura3::λimm434arg4::hisGhis1::hisGmkc1::Tn7-UAU1 | 9 |
| ura3::λimm434arg4::hisGhis1::hisGmkc1::Tn7-URA3 | ||
| VIC1156 | ura3::λimm434arg4::hisGhis1::hisGmkk1::Tn7-UAU1 | 9 |
| ura3::λimm434arg4::hisGhis1::hisGmkk1::Tn7-URA3 | ||
| VIC1167 | ura3::λimm434arg4::hisGhis1::hisGpkc1::Tn7-UAU1 | 9 |
| ura3::λimm434arg4::hisGhis1::hisGpkc1::Tn7-URA3 | ||
| BRY429 | ura3::λimm434arg4::hisGhis1::hisGrlm1::Tn7-UAU1 | 9 |
| ura3::λimm434arg4::hisGhis1::hisGrlm1::Tn7-URA3 | ||
| VIC1075 | ura3::λimm434arg4::hisGhis1::hisGcas5::URA3 | 10 |
| ura3::λimm434arg4::hisGhis1::hisGcas5::ARG4 |
A cassette for the smi1Δ/smi1Δ+pSMI1 complementation strain was constructed by fusion PCR using plasmid pSN105. For the first round of PCR, primers C1 (GCCGAAGTCGACTATGTCATT) and C4 (GTCAGCGGCCGCATCCCTGCAAATTTCAAATACTCCAAAGTCTACAG) were paired with C3 (CACGGCGCGCCTAGCAGCGGAAAGCAGTCAAAGGGCTCTC) and C6 (GGGGATCGTTTAAACTCGAA), respectively, to amplify the homologous sequences flanking C. albicans LEU2 and the C. dubliniensis ARG4 marker using template pSN105. Primers SMI1 C2 (CCGCTGCTAGGCGCGCCGTGTTATTGAGCCACCCAAAAG) and SMI1 C5 (GCAGGGATGCGGCCGCTGACTTGGCCACAGGTGTTCAATA) were used to amplify SMI1 from genomic template DNA. Products were gel purified, and fusion reaction mixtures were assembled using purified templates, C1 and C6 primers, and Extaq polymerase (Takara). Following transformation into the smi1Δ/smi1Δ mutant, correct insertion of the cassette containing C. dubliniensis ARG4 and C. albicans SMI1, flanked by sequences homologous to C. albicans LEU2, was confirmed with check primers SMI1 end Forward (TGACGGTTTGAAAGAAGTAGAATTA) and CaLEU2downflk det R (CGAGGCCACCATTACATCTACCAG).
The smi1Δ/smi1Δ+TDH3-FKS1 strain (JEN172) was constructed using plasmids and primers as previously described (34, 37, 38). Briefly, a cassette containing a TDH3 promoter was inserted upstream of one FKS1 allele in the smi1Δ/smi1Δ mutant.
RNA isolation and real-time RT-PCR expression analysis.
RNA was collected from biofilm cells grown in 6-well plates, as described below. RNA was purified using the RNeasy Minikit (Qiagen) and quantitated using a NanoDrop spectrophotometer. TaqMan primer and probe sets designed using Primer Express (Applied Biosystems, Foster City, CA) for ACT1, FKS1, and SMI1 were as follows: for ACT1, ACT1 RT For (AGCTTTGTTCAGACCAGCTGATT), ACT1 RT Rev (GGAGTTGAAAGTGGTTTGGTCAA), and ACT1 probe (5′-6-FAM-CCAGCAGCTTCCAAACCT-3′-6-TAMSp); for FKS1, FKS1 RT For (TGTGCTGGTCCAATGTTAGGATTATGTTG), FKS1 RT Rev (TGAAACCTTCAGTGACCCACATAACAA), and FKS1 RT probe (5′-6-FAM-ACGGCAACACCATGGGCAACACCAGCA-3′-6-TAMSp); and for SMI1, SMI1 RT For (CAGGATCTGGGTCTGGTTCAAC), SMI1 RT Rev (CAATGGACTTGGAACTGGTGG), and SMI1 probe (5′-6-FAM-CCACAAATGGGAATGCTGCTTCTCCAG-3′-6-TAMSp). Cell wall damage response genes CHT2, DDR48, PHR1, and STP4 were chosen for examination of the cell wall integrity pathway, and transcript levels were measured using primers and probes as previously described (40). The QuantiTect probe reverse transcription-PCR (RT-PCR) kit (Qiagen) was used in an iQ5 PCR detection system (Bio-Rad) with the following program: 50°C for 30 min, initial denaturation at 95°C for 15 min, and then 40 cycles of 94°C for 15 s and 60°C for 1 min. Reactions were performed in triplicate. The expression of each gene relative to that of ACT1 is presented. The quantitative data analysis was completed using the 2−ΔΔCT method (23). The comparative expression method generated data as transcript fold change normalized to a constitutive reference gene transcript (ACT1) and relative to the reference strain.
In vitro biofilm model.
Biofilms were grown in 6-well or 96-well flat-bottom polystyrene plates as previously described (31, 33). The C. albicans inoculum (106 cells/ml)was prepared by growth in YPD with uridine overnight at 30°C, followed by dilution in RPMI-MOPS based on hemocytometer counts. For 6-well plates, 1 ml of culture was inoculated in each well. After a 60-min adherence period at 30°C, the nonadherent inoculum was removed and 1 ml of fresh medium (RPMI-MOPS) was applied to each well. Plates were incubated at 37°C for 48 h on an orbital shaker set at 50 rpm. Medium was removed and fresh medium was added midway through the incubation period.
In vivo C. albicans venous catheter biofilm model.
A jugular vein rat central venous catheter infection model was used for in vivo biofilm studies (5, 30). Candida strains were grown to late logarithmic phase in YPD at 30°C with orbital shaking at 200 rpm. Following a 24-h conditioning period after catheter placement, infection was achieved by intraluminal instillation of 500 μl C. albicans (106 cells/ml). After an adherence period of 6 h, the catheter volume was withdrawn and the catheter was flushed with heparinized saline. For drug treatment experiments, fluconazole (512 μg/ml) was instilled in the catheter after 24 h of biofilm growth. After a 24-h drug treatment period, the posttreatment viable burden of Candida biofilm on the catheter surface was measured by viable plate counts on Sabouraud's dextrose agar (SDA) following removal of the biofilm by sonication and vortexing. For scanning electron microscopy (SEM) studies, the catheters were removed after 24 h and placed in fixative for processing, as described below.
In vivo disseminated C. albicans infection model.
A neutropenic murine disseminated candidiasis model mimicking systemic Candida infection was used to assess strain virulence and drug susceptibility in the nonbiofilm setting (3). Mice were injected with 105 CFU/ml C. albicans via the tail vein. Animals were treated with one of three fluconazole subcutaneous regimens (3.1, 12.5, or 50 mg/kg/12 h) for 24 h; the regimens were chosen based upon treatment efficacy from previous studies (4). Total body Candida burden was estimated by measuring viable burden in kidney homogenates as previously described (3, 4).
In vitro biofilm and planktonic antifungal susceptibility testing.
A tetrazolium salt {XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt]} reduction assay was used to measure in vitro biofilm drug susceptibility (31, 33, 45). Biofilms were formed in the wells of 96-well microtiter plates, as described above. After a 6-h biofilm formation period, the biofilms were washed with phosphate-buffered saline (PBS) twice to remove nonadherent cells. Fresh RPMI-MOPS and drug dilutions were added, followed by additional periods of incubation (48 h). The antifungals studied included fluconazole at 4 to 1,000 μg/ml, amphotericin B at 0.008 to 2 μg/ml, and anidulafungin at 0.002 to 8 μg/ml. Drug treatments were reapplied after 24 h, and plates were incubated for an additional 24 h (34). Following treatment with 90 μl XTT (0.75 mg/ml) and 10 μl phenazine methosulfate (320 μg/ml) for 1 h, absorbance at 492 nm was measured using an automated plate reader. The percent reduction in biofilm growth was calculated using the reduction in absorbance compared to that of controls with no antifungal treatment. Assays were performed in triplicate, and significant differences were measured by analysis of variance (ANOVA) with pairwise comparisons using the Holm-Sidak method.
The CLSI M27 A3 broth microdilution susceptibility method was used to examine the activities of antifungal agents and biocides against planktonic C. albicans (29). The concentration ranges studied were as follows: fluconazole, 0.125 to 128 μg/ml; anidulafungin, 0.03 to 32 μg/ml; flucytosine, 0.03 to 32 μg/ml; and amphotericin B, 0.03 to 32 μg/ml. Endpoints were assessed after 24 h by visible turbidity.
Planktonic and biofilm biocide susceptibility testing.
The CLSI M27 A3 broth microdilution susceptibility method was used to examine the activities of agents associated with cell wall stress against planktonic C. albicans (29, 33). Agents with various mechanisms of action known to impact cell integrity were included (48). MICs were recorded visually and by absorbance reading at 550 nm. A 96-well XTT assay, as described above, was used for measurement of the biofilm response to stress-inducing agents. The concentration required for a 50% reduction in XTT absorbance (50% effective concentration [EC50]) was recorded as the endpoint. Assays were performed in triplicate. The following concentration ranges were tested for planktonic and biofilm studies: calcofluor white, 0.2 to 200 μg/ml; ethanol, 0.01 to 50%; and sodium dodecyl sulfate (SDS), 0.001 to 2%.
In vivo biofilm SEM.
Catheters were processed for SEM and imaged as previously described (5). After 24 h of biofilm growth, catheters were harvested and placed in fixative (4% formaldehyde and 1% glutaraldehyde in PBS) overnight. Catheter segments were then washed with PBS and treated with 1% osmium tetroxide for 30 min at ambient temperature. After a series of alcohol washes (30 to 100%), final desiccation was performed by critical-point drying. Catheter segments were mounted, gold coated, and imaged in a scanning electron microscope (JEOL JSM-6100) at 10 kV. The images were assembled using Adobe Photoshop 7.0.1.
Biofilm cell TEM.
C. albicans biofilms were grown on 6-well polystyrene plates for 48 h as described above. Cells were prepared for transmission electron microscopy (TEM) as previously described (31). Following fixation in 4% formaldehyde and 2% glutaraldehyde, cells were postfixed with 1% osmium tetroxide and 1% potassium ferricyanide, stained with 1% uranyl acetate, dehydrated in a graded series of ethanol concentrations, and embedded in Spurr's resin. Sections (70 nm) were cut, placed on copper grids, poststained with 8% uranyl acetate in 50% methanol and Reynolds' lead citrate, and analyzed by TEM (Philips CM 120). The total cell and cell wall areas of 50 reference and mutant biofilm cells were measured using NIH Image J (http://rsbweb.nih.gov/ij/). The percentages of the cell wall area, defined as the cell wall area divided by the total cellular area, were calculated. Student's t test was used to determine statistical significance of differences between strains.
Cell wall carbohydrate composition.
Biofilms growing in 6-well plates for 48 h were washed with PBS and collected for cell wall carbohydrate analysis as previously described (13, 31). Briefly, cells (5 mg dry cell weight) were washed with PBS and broken with glass beads. Isolated cell walls were alkali extracted for 60 min with 500 μl of 0.7 M NaOH at 75°C three times. The combined alkali-soluble supernatants were neutralized with 250 μl glacial acetic acid. Following neutralization, the alkali-insoluble pellet was digested with 100 U Zymolyase 20T (MP Biomedicals) at 37°C for 16 h. One half of the Zymolyase-soluble fraction was dialyzed (Slide-A-Lyzer dialysis cassette, 7,000-molecular-weight-cutoff [MWCO]; Pierce) to yield a β-1,6-glucan fraction. The β-1,3-glucan fraction was calculated as the difference between the total Zymolyase-soluble glucan and β-1,6-glucan fractions. The carbohydrate contents of each fraction were measured as hexoses by the phenol-sulfuric acid method and normalized for dry cell wall weight. ANOVA with pairwise comparisons (Holm-Sidak method) was used to determine statistical significance.
Biofilm matrix collection and matrix β-1,3-glucan measurements.
The matrix β-1,3-glucan content was measured using a Limulus lysate based assay, as previously described (31, 41). Matrix was collected from C. albicans biofilms growing in the wells of 6-well polystyrene plates for 48 h. Biofilms were dislodged using a sterile spatula, sonicated for 10 min, and centrifuged 3 times at 4,500 × g for 20 min to separate cells from soluble matrix material (27, 31). Samples were stored at −20°C, and glucan concentrations were determined using the Glucatell (1,3)-beta-d-glucan detection reagent kit (Associates of Cape Cod, MA) per the manufacturer's directions.
Accumulation of [H3]fluconazole into C. albicans biofilms.
A radiolabeled-fluconazole accumulation protocol was adapted for biofilm use as previously described (25, 34, 49). Biofilms were grown in 6-well plates, as described above, for 48 h. The biofilms were washed with sterile water twice. For stock solution preparation, radioactive [H3]fluconazole (Moravek Biochemicals; 50 μM, 0.001 mCi/ml in ethanol) was diluted 100-fold in water. The stock solution was then diluted 6-fold in RPMI-MOPS, and each biofilm well received a total of 600 μl of this medium to yield a total of 8.48 ×105 cpm of [H3]fluconazole. After incubation for 30 min at 37°C and orbital shaking at 50 rpm, unlabeled (cold) 20 μM fluconazole in RPMI-MOPS was added and biofilms were incubated for an additional 15 min. Biofilms were then washed twice with sterile water, gently dislodged with a sterile spatula, and collected as intact biofilms for scintillation counting. The biofilms were then disrupted by vortexing and sonication for 10 min to separate cells and matrix. Following centrifugation at 4,500 × g for 20 min, cells were separated from the soluble matrix material. Cells were disrupted by bead beating, and the intracellular and cell wall portions were collected by centrifugation. The fractions were then suspended in ScintiSafe 30% LSC cocktail (Fisher Scientific) and counted in a Tri-Carb 2100TR liquid scintillation analyzer (Packard). Student's t test was used to determine statistical significance of differences between strains.
RESULTS
Role of S. cerevisiae PKC pathway homologs in C. albicans biofilm formation and biofilm-associated drug resistance.
We utilized a candidate gene approach to identify regulators of C. albicans β-glucan production, with the goal of determining the control of this process during the development of biofilm drug resistance. Yeast PKC pathway homologs in C. albicans, including PKC1, BCK1, MKK2, MKC1, and SMI1, were chosen based upon demonstrated importance for control of β-1,3-glucan synthesis in S. cerevisiae. We were surprised that disruption of PKC1, BCK1, MKK2, or MKC1 did not affect biofilm antifungal drug resistance (Table 2). Each null mutant formed a biofilm in vitro that demonstrated the characteristic drug-resistant phenotype. However, we did identify C. albicans SMI1 as a regulator of biofilm-associated drug resistance. Homozygous deletion of SMI1 produced a biofilm with enhanced susceptibility to antifungal drugs (Fig. 1). While the reference strain biofilm was resistant to the highest fluconazole concentration tested (1,000 μg/ml), the homozygous smi1Δ/smi1Δ biofilm was reduced by 50% upon fluconazole treatment at concentrations of 125 and 250 μg/ml. Differences in susceptibility were observed at concentrations of as low as 7 μg/ml (data not shown). Compared to the reference strain, the smi1Δ/smi1Δ biofilm exhibited increased susceptibly to antifungals from additional drug classes (anidulafungin and amphotericin B) as well (Fig. 2). Complementation of SMI1 partially restored resistance to fluconazole. Disruption of SMI1 did not affect planktonic susceptibility to any of the antifungal agents tested using standard MIC testing methods, suggesting a biofilm-specific role for the gene products in drug resistance (Table 3).
Table 2.
Biofilm phenotypes of C. albicans cell wall integrity mutants
| Mutation | Description | Mutant phenotypea |
||
|---|---|---|---|---|
| Biofilm formationb | Matrix glucan productionc | Biofilm fluconazole resistanceb | ||
| None (reference strain) | Wild type | ++ | ++ | ++ |
| C. albicans homologs of S. cerevisiae components of the PKC pathway | ||||
| pkc1Δ/pkc1Δ | Mammalian PKC homolog | ++ | ++ | + |
| mkk2Δ/mkk2Δ | MAPKKKd | ++ | + | ++ |
| bck1Δ/bck1Δ | MAPKK | ++ | ++ | ++ |
| mkc1Δ/mkc1Δ | MAPK | ++ | ++ | + |
| smi1Δ/smi1Δ | Regulator of glucan synthesis | ++ | − | − |
| rlm1Δ/rlm1Δ | Transcription factor | ++ | − | − |
| FKS1/fks1Δ | β-1,3-Glucan synthase | ++ | − | − |
| Cell wall regulator unique to C. albicans | ||||
| cas5Δ/cas5Δ | Transcription regulator | ++ | ++ | ++ |
++, value is within 10% of reference strain level; +, value is 10 to 30% below reference strain level; −, value is less than 30% of reference strain level.
Measured using XTT reduction assay in a 96-well plate.
Measured by glucan limulus lysate assay.
MAPKKK, mitogen-activated protein kinase kinase kinase.
Fig. 1.
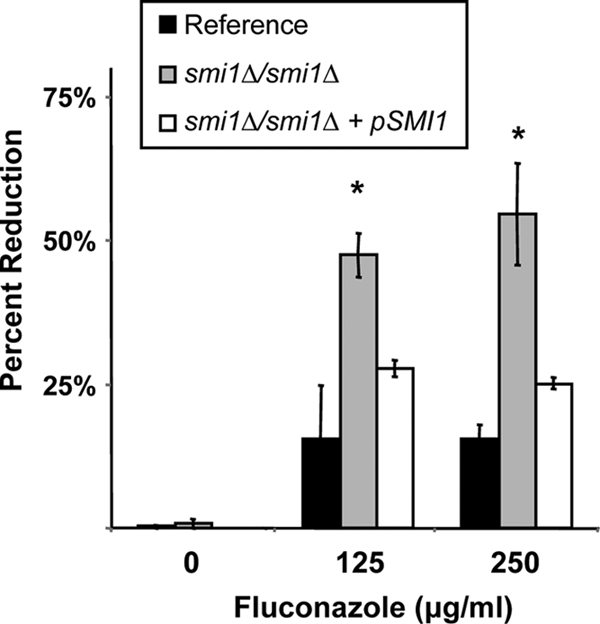
SMI1 is required for fluconazole biofilm resistance in vitro. Reference strain, homozygous mutant smi1Δ/smi1Δ, and complemented strain smi1Δ/smi1Δ+pSMI1 biofilms were treated with serial dilutions of fluconazole for 48 h, supplied as a two 24-h doses. Drug impact was determined using an XTT reduction assay. Data are expressed as percent reduction compared to untreated controls. Standard errors are shown. ANOVA with pairwise comparisons using the Holm-Sidak method was used to compare the mutant strains at each drug concentration. *, P < 0.05.
Fig. 2.

Impact of SMI1 disruption on amphotericin B and anidulafungin biofilm resistance in vitro. Reference strain and smi1Δ/smi1Δ biofilms were treated with serial dilutions of anidulafungin (A) or amphotericin B (B) for 48 h, supplied as a two 24-h doses. Drug impact was determined using an XTT reduction assay. Data are expressed as percent reduction compared to untreated controls. Standard errors are shown. Student's t test was used to compare the mutant strains at each drug concentration. *, P < 0.05.
Table 3.
Impact of SMI1 modulation on planktonic drug susceptibility
| Strain | MIC (μg/ml)a |
|||
|---|---|---|---|---|
| Fluconazole | Amphotericin B | Flucytosine | Anidulafungin | |
| Reference | 1 | 0.03 | 0.13 | 0.015 |
| smi1Δ/smi1Δ mutant | 0.5 | 0.06 | 0.06 | 0.015 |
| SMI1/smi1Δ mutant | 0.5 | 0.06 | 0.25 | 0.015 |
| smi1Δ/smi1Δ+ pSMI1 mutant | 1 | 0.06 | 0.13 | 0.015 |
MICs were determined using the CLSI method and endpoint.
Because the phenotype of the smi1Δ/smi1Δ biofilm was distinct from that of the PKC pathway mutants, we also considered the possibility of a link to CAS5, a cell wall integrity gene unique to C. albicans and required for regulation of transcription during cell wall damage (Table 2) (10). Like that of PKC pathway mutants, the cas5Δ/casΔ biofilm phenocopied the reference strain biofilm, suggesting that CAS5 is not involved in regulation of the biofilm resistance pathway.
Impact of PKC pathway disruption on biofilm matrix and cell wall β-1,3-glucan production.
We hypothesized that the enhanced antifungal drug susceptibility phenotype may be linked to decreased β-1,3-glucan production or altered assembly of the extracellular matrix during biofilm growth. To discern the role of the PKC pathway in biofilm matrix production, matrix was harvested from in vitro biofilms growing in 6-well polystyrene plates and quantified using a Limulus lysate-based assay (34). Compared to the reference strain, differences in matrix glucan were not evident for the upstream PKC pathway mutants, including the pkc1Δ/pkc1Δ, bck1Δ/bck1Δ, and mkc1Δ/mkc1Δ mutant biofilms. One exception was the mkk2Δ/mkk2Δ biofilm, which produced slightly less matrix than the reference strain (Table 2). However, matrix glucan was markedly reduced (more than 4-fold) in the smi1Δ/smi1Δ mutant biofilm (Fig. 3A) These findings suggest that Smi1p regulates biofilm glucan production and drug resistance independent of upstream PKC pathway components.
Fig. 3.

SMI1 affects biofilm cell wall and matrix glucan content. (A) SMI1 is required for matrix β-1,3-glucan production in biofilms. Matrix samples were collected from in vitro biofilms growing in 6-well polystyrene plates. β-1,3-glucan was measured using a Limulus lysate-based assay. Assays were performed in duplicate on two occasions. (B) Cell walls from reference strain, SMI1/smi1Δ, and smi1Δ/smi1Δ mutant biofilms were isolated and fractionated by alkali treatment and enzymatic digestion. ANOVA with pairwise comparisons using the Holm-Sidak method was used to compare carbohydrate of each fraction among the strains. *, P < 0.05. Assays were performed in triplicate on two occasions. Standard deviations are shown.
We used transmission electron microscopy (TEM) to examine the biofilm cell wall architecture of one of the mutant strains with enhanced antifungal susceptibility and reduced matrix glucan, the smi1Δ/smi1Δ strain. Compared to the reference strain, the cell walls of the smi1Δ/smi1Δ biofilm appeared thinner, with less electron-lucent material present (Fig. 4A). Cell wall measurements demonstrated that the total cell wall area for the mutant biofilms was approximately half that measured for the reference strain (Fig. 4B). Because the mutant biofilm cells were smaller overall than the reference strain cells based on total cellular area, we calculated the cell wall area per total cellular area. Even after correcting for the smaller cell size, the smi1Δ/smi1Δ biofilm cells contained significantly less cell wall (Fig. 4C).
Fig. 4.

Disruption of SMI1 affects biofilm cell wall ultrastructure. (A) Reference strain and smi1Δ/smi1Δ mutant biofilms were collected from 6-well polystyrene plates, fixed, processed for TEM, and imaged. Scale bars represent 0.25 μm. (B) The total cell and cell wall areas of the biofilm cells were measured using Image J. (C) The percentage of the cell wall area, defined as the cell wall area divided by the total cellular area, was calculated. Student's t test was used to determine statistical significance of differences between strains. *, P < 0.005. Standard errors are shown.
We further determined the impact of SMI1 on biofilm cell wall β-1,3-glucan production by measuring and comparing the carbohydrate contents of individual biofilm cell wall fractions. Compared to that of the reference strain, the smi1Δ/smi1Δ biofilm cell wall was composed of significantly less total glucan (Fig. 3B). Most of the observed difference was accounted for by the 40% decrease in cell wall β-1,3-glucan for the smi1Δ/smi1Δ biofilm. In addition, a smaller amount of β-1,6-glucan was detected in this mutant cell wall. Reduced biofilm cell wall glucan production was also observed for the SMI1/smi1Δ mutant. As expected for a heterozygous disruption, the extent of these differences was less than those measured for the homozygous disruption. The extent of the biochemical reduction in cell wall β-1,3-glucan for the smi1Δ/smi1Δ mutant is similar to the cell wall percent reduction measured by TEM (Fig. 4). A decrease in cell wall β-1,3-glucan is consistent with the microscopy images demonstrating a thinner electron-lucent cell wall layer.
Impact of SMI1 disruption on biofilm formation and azole susceptibility in an in vivo rat venous catheter.
We utilized a rat biofilm catheter model to examine the impact of SMI1 disruption on biofilm formation and matrix production in vivo (5). We included the in vivo model because reports have demonstrated variability in genes needed for biofilm development under in vitro and in vivo conditions (35). Following a 6-h adherence period and a 24-h growth period, catheter segments were processed for imaging by scanning electron microscopy (SEM). Both the reference strain and the smi1Δ/smi1Δ mutant were capable of adhering to the luminal catheter surface and forming heterogeneous biofilms across the surface with comparable total biofilm masses (Fig. 5). Imaging at higher magnification revealed an extracellular matrix material coating the cells, as previously described (34). However, the smi1Δ/smi1Δ biofilm appeared to produce less matrix material than the reference strain. These in vivo images correlate with the smaller amount of biofilm matrix glucan measured for the smi1Δ/smi1Δ in vitro biofilms and support a role for SMI1 in matrix glucan production and deposition.
Fig. 5.

SMI1 is not required for biofilm formation in vivo. The luminal surfaces of rat venous catheters were inoculated with the smi1Δ/smi1Δ mutant or the reference strain. Catheter segments were processed and imaged using SEM. At low magnification (×50), the reference strain and smi1Δ/smi1Δ mutant appear to form similar biofilms extending over the luminal catheter surface. At higher magnification, the smi1Δ/smi1Δ exhibits visually less matrix than the reference strain. Scale bars in the ×50, ×1,000, and ×3,000 images represent 500 μm, 20 μm, and 8 μm, respectively.
In treatment studies, the biofilm susceptible phenotype that we identified for the smi1Δ/smi1Δ mutant in vitro was corroborated in vivo. Following intraluminal fluconazole treatment (512 μg/ml), the Candida viable burden was more than 3-fold lower for the smiΔ/smi1Δ mutant strain (2.5 × 105 CFU/catheter) than for the reference strain (8.5 × 105 CFU/catheter).
Impact of SMI1 expression on uptake of [H3]fluconazole in C. albicans biofilms.
A radiolabeled-fluconazole accumulation assay was used to track fluconazole within the in vitro reference strain and mutant biofilms. Following treatment with [H3]fluconazole, less fluconazole was associated with the intact smi1Δ/smi1Δ mutant biofilm than with the reference strain biofilm (Fig. 6). Nearly all of the biofilm-associated radioactive fluconazole was localized to the biofilm matrix. Unfortunately, radioactivity levels in the intracellular component were below the level of detection (data not shown). The accumulation of drug in the biofilm matrix is consistent with sequestration of the fluconazole by the extracellular matrix. Altered sequestration in the smi1Δ/smi1Δ mutant biofilm suggests a role for this gene product in the drug sequestration process, as has been described for glucan synthase gene FKS1 (34).
Fig. 6.
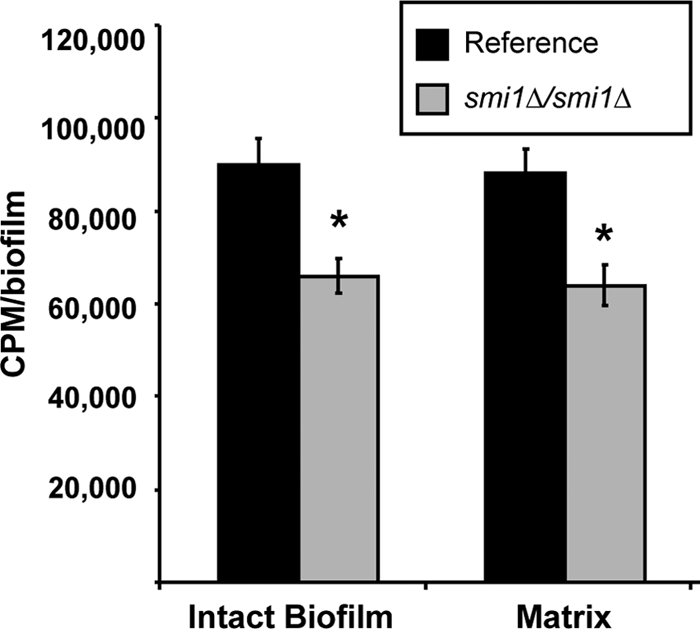
SMI1 is required for matrix sequestration of [H3]fluconazole. Intact biofilms grown from the glucan-modified strains were exposed to [H3]fluconazole, washed, and harvested. Scintillation counting was performed in triplicate to determine the fluconazole concentrations in the intact biofilms and the isolated matrix. Standard deviations are shown. Student's t test was used to determine statistical significance of differences between strains. *, P < 0.05. Standard deviations are shown.
Relationship between FKS1 and SMI1.
We considered a link between SMI1 and Fks1p, a glucan synthase required for C. albicans biofilm glucan production and drug resistance during biofilm growth (34). We examined the collection of mutants with varied SMI1 expression and measured transcript abundances of both SMI1 and FKS1 during biofilm growth by real-time RT-PCR. Heterozygous disruption of SMI1 decreased the SMI1 transcript abundance to approximately 50% of that for the reference strain (Fig. 7A). Homozygous disruption eliminated transcription entirely, while complementation partially restored SMI1 transcription. Transcript abundance of FKS1 was decreased by nearly 50% in the smi1Δ/smi1Δ mutant biofilm and by approximately 10% in the SMI1/smi1Δ mutant biofilm, consistent with a gene dose affect. To corroborate the connection between SMI1 and FKS1, we constructed a smi1Δ/smi1Δ mutant biofilm with FKS1 under the control of a TDH3 promoter and hypothesized that overexpression of FKS1 would rescue the biofilm drug susceptibility phenotype. As hypothesized, the smi1Δ/smi1Δ TDH3-FKS1 biofilm was less susceptible to fluconazole treatment than the smi1Δ/smi1Δ parent strain (Fig. 7B). These results support the hypothesis that SMI1 functions upstream of FKS1 for biofilm glucan production and drug resistance.
Fig. 7.

Modulation of SMI1 affects FKS1 expression in C. albicans biofilms. (A) RNA was isolated from reference and mutant biofilms. Real-time RT-PCR assays were used to measure transcript levels in triplicate. Mean results were normalized by ACT1 RNA measurements performed simultaneously and compared to the reference strain using the method of Livak and Schmittgen (23). Data are shown as a normalized ratio of transcript in the mutant strain divided by that in the reference strain. (B) FKS1 was placed under the control of an inserted TDH3 promoter for overexpression of FKS1 in the homozygous smi1Δ/smi1Δ mutant. Biofilms were treated with serial dilutions of fluconazole for 48 h, supplied as a two 24-h doses, and drug impact was determined using an XTT reduction assay. Data are expressed as percent reduction compared to untreated controls. Standard errors are shown. Student's t test was used to compare the mutant strains at each drug concentration. *, P < 0.05.
A potential relationship between C. albicans Smi1p and Fks1p is predicted based on S. cerevisiae Smi1/Knr4p, which modulates FKS1 expression via transcription factor Rlm1p (26). To investigate this relationship in C. albicans biofilms, we measured glucan production and drug resistance in the rlm1Δ/rlm1Δ mutant biofilm. Similar to the case for the smi1Δ/smi1Δ and FKS1/fks1Δ mutant biofilms, the rlm1Δ/rlm1Δ mutant exhibited increased susceptibility to fluconazole (Table 2). This increased susceptibility corresponded with decreased matrix glucan, consistent with disruption of drug sequestration by the matrix. Together, the data suggest that Smi1p regulates Fks1p, glucan production, and drug resistance during biofilm growth, likely through Rlm1p.
Cell wall integrity and SMI1.
We considered the possibility that disruption of SMI1 and cell wall glucan may result in global changes, rendering C. albicans more susceptible to a variety of stressors. We examined biofilm and planktonic organism susceptibility to known cell-perturbing agents (calcofluor white, ethanol, and SDS) (Table 4). With the exception of calcofluor white, the smi1Δ/smi1Δ and reference strains were similarly susceptible during biofilm and planktonic growth. As a complementary method, we examined the transcript levels of four cell wall damage response genes in reference strain and smi1Δ/smi1Δ mutant biofilms (Fig. 8) (40). In the absence of an exogenous stressor, disruption of SMI1 resulted in a minor change in each of these cell wall damage response genes that we examined. The highest expression change was approximately 1.5-fold.
Table 4.
Impact of SMI1 disruption on planktonic cell and biofilm susceptibility to biocides
| Cell type | Strain | Concn at which cells susceptible to: |
||
|---|---|---|---|---|
| Calcofluor white (μg/ml) | Ethanol (%) | SDS (%) | ||
| Planktonica | Reference | 50 | 0.8 | 0.01 |
| smi1Δ/smi1Δ mutant | 3.12 | 0.8 | 0.01 | |
| Biofilmb | Reference | 100 | 12.5 | 0.06 |
| smi1Δ/smi1Δ mutant | 25 | 12.5 | 0.03 | |
MICs were determined using the CLSI method and endpoint.
The XTT reduction assay was used to determine the drug concentration associated with a 50% reduction in optical density compared to that in the no-drug control wells (EC50).
Fig. 8.
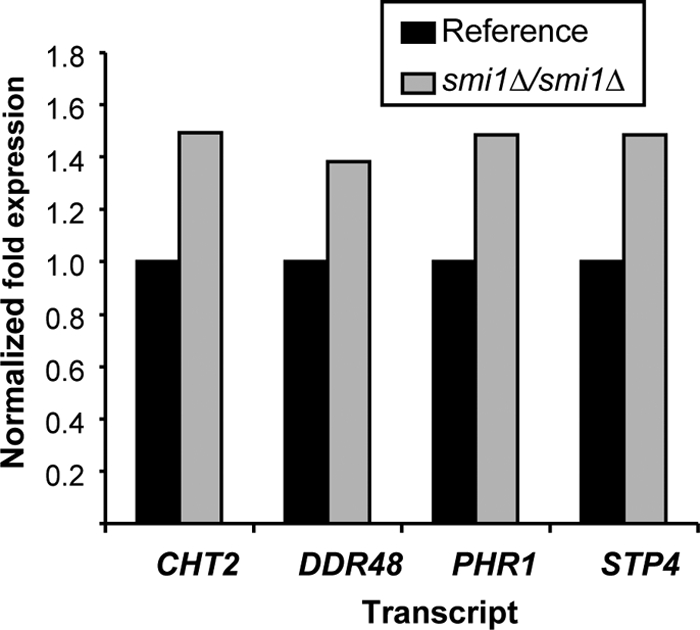
Expression of cell wall damage genes in smi1Δ/smi1Δ mutant and reference strain biofilms. RNA was isolated from reference and mutant biofilms. Real-time RT-PCR assays were used to measure transcript levels in triplicate. Mean results were normalized by ACT1 RNA measurements performed simultaneously and compared to the reference strain using the method of Livak and Schmittgen (23). Data are shown as a normalized ratio of transcript in the mutant strain divided by that in the reference strain.
Impact of SMI1 disruption on growth rate and virulence.
We measured the impact of SMI1 disruption on growth rate, filamentation, and virulence, characteristics previously demonstrated to be important for biofilm formation, in a murine disseminated candidiasis model. The smi1Δ/smi1Δ mutant and reference strain grew at similar rates under planktonic conditions (Fig. 9A). Both strains generated hyphae in response to hypha-inducing conditions, including growth in RPMI-MOPS at 37°C and on Spider medium (data not shown). Each of the SMI1-modulated strains formed biofilm in vitro in the wells of polystyrene plates, and the biofilm quantities were similar at 6 and 24 h based on XTT reduction assay (data not shown). To address the impact of SMI1 on C. albicans virulence, we used a disseminated murine neutropenic candidiasis model (3). Over the 24-h period, the viable burdens of the smi1Δ/smi1Δ mutant and reference strain increased at similar rates and to equivalent magnitudes over the study period (Fig. 9B). Together, these experiments show the disruption of SMI1 does not significantly influence growth, filamentation, or virulence in an in vivo nonbiofilm infection model.
Fig. 9.

Disruption of SMI1 does not affect growth or virulence. (A) The smi1Δ/smi1 mutant and reference strains grow at similar rates. Growth in YPD plus uridine at 30°C was estimated by measuring optical density at 550 nm. (B) The smi1Δ/smi1 mutant and reference strains are equally susceptible to fluconazole in a nonbiofilm disseminated candidiasis murine model. Kidney viable burden was measured by microbiologic plate counts and used as an estimate for total body viable burden. Standard deviations are shown.
DISCUSSION
Candida spp. infect medical devices by attaching to the surface and proliferating as a biofilm. Cells in this environment are embedded in a protective extracellular matrix and exhibit profound resistance to antifungal drugs (11, 14). Because of the lack of effective antifungal therapy against biofilm infections, the recommended treatment for Candida biofilm infections is removal of the infected device (42). Defining the pathways and mechanisms of resistance during the biofilm mode of growth is valuable for the design of innovative drug therapies targeted to treat these recalcitrant infections.
The first Candida biofilm resistance studies tested mechanisms of resistance known to be important in planktonic systems (6, 28, 43, 50). Although several of these mechanisms were found to be involved in biofilm drug resistance, much of the biofilm resistant phenotype remained an enigma. Subsequent studies have suggested that biofilm drug resistance is multifactorial, with contributions from biofilm-specific processes as well (6, 18, 20, 28, 34, 43, 47, 50). Models have demonstrated phenotypic variability among the cells in heterogeneous biofilms and have identified subsets of exquisitely resistant cells deep in the biofilm (18, 22). These studies suggest that cells throughout the biofilm may even employ different mechanisms of resistance.
Previous investigations postulating a contribution of the biofilm matrix to drug resistance measured antifungal diffusion through Candida biofilms (1, 2, 8, 47). Using variable-flow conditions to alter matrix production, the Douglas group identified a correlation between drug resistance and the extent of biofilm matrix (2, 8). Further studies with filter disk diffusion assays have also revealed a slowing of antifungal transit through Candida biofilms (1, 47). Our previous investigations linked a biofilm matrix carbohydrate, glucan, to a biofilm-specific drug resistance mechanism in C. albicans (32, 34). By producing matrix β-glucan capable of sequestering an antifungal drug, biofilm cells survive extraordinarily high drug concentrations during biofilm growth.
In the current studies, we use a candidate gene approach to explore a role for the PKC pathway in C. albicans matrix β-1,3-glucan production and biofilm resistance. This pathway has previously been described to be a positive regulator of β-1,3-glucan synthesis in S. cerevisiae, but similar studies have not been performed with C. albicans to our knowledge (17). Here we show that SMI1 and RLM1 are required for production of the characteristic drug-resistant phenotype of the biofilm lifestyle. Disruption of SMI1 and RLM1 affects the manufacture of C. albicans β-1,3 matrix and cell wall glucan during biofilm growth. The importance of these genes in S. cerevisiae β-1,3-glucan cell wall synthesis has been well characterized, but their role in C. albicans β-1,3-glucan production and biofilm resistance is a novel finding (16). Surprisingly, the upstream yeast homologs of the PKC pathway were not found to contribute to the biofilm glucan matrix resistance mechanism. Disruption of each of the four kinases in the pathway did not affect biofilm drug resistance or glucan matrix.
Consistent with previous investigations, the current studies support a role for glucan sequestration of antifungals in C. albicans biofilm drug resistance (34). Using a radioactive-fluconazole assay, we were able to track the drug accumulation and demonstrate biofilm sequestration in the matrix material. The finding that modulation of SMI1 affects the amount of matrix-sequestered drug indicates a role for the gene product in this biofilm resistance mechanism. This mechanism is specific to the biofilm mode of growth because planktonic drug resistance is not affected. The ability of biofilm matrix to act as a drug sponge, occupying the drug and preventing its activity, has been described for both bacterial and fungal biofilms (24, 34). In C. albicans biofilms, this activity is mediated, at least in part, through glucan synthase Fks1p.
These studies show that the action of Smi1p includes β-1,3-glucan matrix synthesis upstream of Fks1p. Transcription of FKS1 is modulated by SMI1 expression, and the β-1,3-glucan changes observed with disruption of SMI1 are similar to those described for FKS1/fks1Δ mutant biofilm (34). Furthermore, we show that overexpression of FKS1 in the smi1Δ/smi1Δ mutant restores the biofilm drug-resistant phenotype. In S. cerevisiae, a link between SMI1/KNR4 and FKS1 upon activation of the cell wall integrity pathway and transcription factor Rlmlp has been described (26). As predicted from S. cerevisiae, disruption of RLM1 in C. albicans produces a biofilm phenotypically similar to the SMI1 and FKS1 mutant biofilms. Together, the findings suggest that the relationship between these gene products is conserved in C. albicans.
Phenotypic studies and transcriptional profiling examining the cell wall integrity pathway in the smi1Δ/smi1Δ mutant suggest a partial link to biofilm matrix production. Disruption of SMI1 affected expression of several cell wall damage response genes, suggesting altered cell wall integrity for the mutant strain in the absence of exogenous stressors. In addition, the smi1Δ/smi1Δ mutant was significantly more susceptible to cell wall perturbation by calcofluor white. However, significant differences between the strains were not observed upon treatment of planktonic or biofilm cells with other cell stressors. We hypothesize that the differential susceptibility to calcofluor white is related to the increase in cell wall chitin in the smi1Δ/smi1Δ mutant (data not shown). Disruption of SMI1 did not affect growth at 37°C or planktonic susceptibility to additional antifungals, including amphotericin B, flucytosine, and anidulafungin. An intact cell wall integrity pathway is required for echinocandin resistance in both C. albicans and S. cerevisiae (10, 46). Therefore, similar planktonic susceptibilities to anidulafungin for the C. albicans smi1Δ/smi1Δ mutant and the reference strain do not suggest an altered cell wall integrity pathway.
Taken together, the data suggest that matrix glucan production and biofilm resistance are modulated by Smi1p and networked to the cell wall integrity pathway. However, regulation of this pathway is distinct from that of the PKC pathway. Further defining the genetic regulation of this Candida biofilm pathway may provide insight into how the organism transforms to this lifestyle and resists antifungal treatment.
ACKNOWLEDGMENTS
We thank A. Mitchell, S. Noble, and C. Nobile for strains and plasmids.
This work was supported by the National Institutes of Health (grant RO1 AI073289-01) and the Veteran Women's Health Fellowship.
Footnotes
Published ahead of print on 10 June 2011.
REFERENCES
- 1. Al-Fattani M. A., Douglas L. J. 2004. Penetration of Candida biofilms by antifungal agents. Antimicrob. Agents Chemother. 48: 3291–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Al-Fattani M. A., Douglas L. J. 2006. Biofilm matrix of Candida albicans and Candida tropicalis: chemical composition and role in drug resistance. J. Med. Microbiol. 55: 999–1008 [DOI] [PubMed] [Google Scholar]
- 3. Andes D. 2005. Use of an animal model of disseminated candidiasis in the evaluation of antifungal therapy. Methods Mol. Med. 118: 111–128 [DOI] [PubMed] [Google Scholar]
- 4. Andes D., et al. 2006. Impact of antimicrobial dosing regimen on evolution of drug resistance in vivo: fluconazole and Candida albicans. Antimicrob. Agents Chemother. 50: 2374–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andes D., et al. 2004. Development and characterization of an in vivo central venous catheter Candida albicans biofilm model. Infect. Immun. 72: 6023–6031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baillie G. S., Douglas L. J. 1998. Effect of growth rate on resistance of Candida albicans biofilms to antifungal agents. Antimicrob. Agents Chemother. 42: 1900–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baillie G. S., Douglas L. J. 1999. Candida biofilms and their susceptibility to antifungal agents. Methods Enzymol. 310: 644–656 [DOI] [PubMed] [Google Scholar]
- 8. Baillie G. S., Douglas L. J. 2000. Matrix polymers of Candida biofilms and their possible role in biofilm resistance to antifungal agents. J. Antimicrob. Chemother. 46: 397–403 [DOI] [PubMed] [Google Scholar]
- 9. Blankenship J. R., Fanning S., Hamaker J. J., Mitchell A. P. 2010. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 6: e1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruno V. M., et al. 2006. Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog. 2: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandra J., et al. 2001. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J. Bacteriol. 183: 5385–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Costerton J. W., Stewart P. S., Greenberg E. P. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284: 1318–1322 [DOI] [PubMed] [Google Scholar]
- 13. Dijkgraaf G. J., Brown J. L., Bussey H. 1996. The KNH1 gene of Saccharomyces cerevisiae is a functional homolog of KRE9. Yeast 12: 683–692 [DOI] [PubMed] [Google Scholar]
- 14. Douglas L. J. 2002. Medical importance of biofilms in Candida infections. Rev. Iberoam. Micol. 19: 139–143 [PubMed] [Google Scholar]
- 15. Fuchs B. B., Mylonakis E. 2009. Our paths might cross: the role of the fungal cell wall integrity pathway in stress response and cross talk with other stress response pathways. Eukaryot. Cell 8: 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong Z., et al. 1994. Cloning and characterization of KNR4, a yeast gene involved in (1,3)-beta-glucan synthesis. Mol. Cell. Biol. 14: 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hong Z., Mann P., Shaw K. J., Didomenico B. 1994. Analysis of beta-glucans and chitin in a Saccharomyces cerevisiae cell wall mutant using high-performance liquid chromatography. Yeast 10: 1083–1092 [DOI] [PubMed] [Google Scholar]
- 18. Khot P. D., Suci P. A., Miller R. L., Nelson R. D., Tyler B. J. 2006. A small subpopulation of blastospores in Candida albicans biofilms exhibit resistance to amphotericin B associated with differential regulation of ergosterol and beta-1,6-glucan pathway genes. Antimicrob. Agents Chemother. 50: 3708–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kojic E. M., Darouiche R. O. 2004. Candida infections of medical devices. Clin. Microbiol. Rev. 17: 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumamoto C. A. 2005. A contact-activated kinase signals Candida albicans invasive growth and biofilm development. Proc. Natl. Acad. Sci. U. S. A. 102: 5576–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumamoto C. A., Vinces M. D. 2005. Alternative Candida albicans lifestyles: growth on surfaces. Annu. Rev. Microbiol. 59: 113–133 [DOI] [PubMed] [Google Scholar]
- 22. LaFleur M. D., Kumamoto C. A., Lewis K. 2006. Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob. Agents Chemother. 50: 3839–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 24. Mah T. F., et al. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426: 306–310 [DOI] [PubMed] [Google Scholar]
- 25. Mansfield B. E., et al. 2010. Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 6: e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin-Yken H., Dagkessamanskaia A., Basmaji F., Lagorce A., Francois J. 2003. The interaction of Slt2 MAP kinase with Knr4 is necessary for signalling through the cell wall integrity pathway in Saccharomyces cerevisiae. Mol. Microbiol. 49: 23–35 [DOI] [PubMed] [Google Scholar]
- 27. McCourtie J., Douglas L. J. 1985. Extracellular polymer of Candida albicans: isolation, analysis and role in adhesion. J. Gen. Microbiol. 131: 495–503 [DOI] [PubMed] [Google Scholar]
- 28. Mukherjee P. K., Chandra J., Kuhn D. M., Ghannoum M. A. 2003. Mechanism of fluconazole resistance in Candida albicans biofilms: phase-specific role of efflux pumps and membrane sterols. Infect. Immun. 71: 4333–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. NCCLS 2002. Reference method for broth dilution antifungal susceptibility testing. Document M27-A2, 2nd ed. National Committee for Clinical Laboratory Standards, Wayne, PA [Google Scholar]
- 30. Nett J., Lincoln L., Marchillo K., Andes D. 2007. Beta-1,3 glucan as a test for central venous catheter biofilm infection. J. Infect. Dis. 195: 1705–1712 [DOI] [PubMed] [Google Scholar]
- 31. Nett J., et al. 2007. Putative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrob. Agents Chemother. 51: 510–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nett J. E., Crawford K., Marchillo K., Andes D. R. 2010. Role of Fks1p and matrix glucan in Candida albicans biofilm resistance to an echinocandin, pyrimidine, and polyene. Antimicrob. Agents Chemother. 54: 3505–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nett J. E., Guite K. M., Ringeisen A., Holoyda K. A., Andes D. R. 2008. Reduced biocide susceptibility in Candida albicans biofilms. Antimicrob. Agents Chemother. 52: 3411–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nett J. E., Sanchez H., Cain M. T., Andes D. R. 2010. Genetic basis of Candida biofilm resistance due to drug-sequestering matrix glucan. J. Infect. Dis. 202: 171–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nobile C. J., et al. 2006. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog. 2: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nobile C. J., Mitchell A. P. 2009. Large-scale gene disruption using the UAU1 cassette. Methods Mol. Biol. 499: 175–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nobile C. J., et al. 2008. Complementary adhesin function in C. albicans biofilm formation. Curr. Biol. 18: 1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nobile C. J., et al. 2008. Candida albicans transcription factor Rim101 mediates pathogenic interactions through cell wall functions. Cell. Microbiol. 10: 2180–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Noble S. M., Johnson A. D. 2005. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell 4: 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Norice C. T., Smith F. J., Jr., Solis N., Filler S. G., Mitchell A. P. 2007. Requirement for Candida albicans Sun41 in biofilm formation and virulence. Eukaryot. Cell 6: 2046–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Odabasi Z., et al. 2004. Beta-d-glucan as a diagnostic adjunct for invasive fungal infections: validation, cutoff development, and performance in patients with acute myelogenous leukemia and myelodysplastic syndrome. Clin. Infect. Dis. 39: 199–205 [DOI] [PubMed] [Google Scholar]
- 42. Pappas P. G., et al. 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48: 503–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramage G., Bachmann S., Patterson T. F., Wickes B. L., Lopez-Ribot J. L. 2002. Investigation of multidrug efflux pumps in relation to fluconazole resistance in Candida albicans biofilms. J. Antimicrob. Chemother. 49: 973–980 [DOI] [PubMed] [Google Scholar]
- 44. Ramage G., Lopez-Ribot J. L. 2005. Techniques for antifungal susceptibility testing of Candida albicans biofilms. Methods Mol. Med. 118: 71–79 [DOI] [PubMed] [Google Scholar]
- 45. Ramage G., Vande Walle K., Wickes B. L., Lopez-Ribot J. L. 2001. Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms. Antimicrob. Agents Chemother. 45: 2475–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reinoso-Martin C., Schuller C., Schuetzer-Muehlbauer M., Kuchler K. 2003. The yeast protein kinase C cell integrity pathway mediates tolerance to the antifungal drug caspofungin through activation of Slt2p mitogen-activated protein kinase signaling. Eukaryot. Cell 2: 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samaranayake Y. H., Ye J., Yau J. Y., Cheung B. P., Samaranayake L. P. 2005. In vitro method to study antifungal perfusion in Candida biofilms. J. Clin. Microbiol. 43: 818–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sampaio P., et al. 2009. Increased number of glutamine repeats in the C-terminal of Candida albicans Rlm1p enhances the resistance to stress agents. Antonie Van Leeuwenhoek 96: 395–404 [DOI] [PubMed] [Google Scholar]
- 49. Sanglard D., et al. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39: 2378–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uppuluri P., Nett J., Heitman J., Andes D. 2008. Synergistic effect of calcineurin inhibitors and fluconazole against Candida albicans biofilms. Antimicrob. Agents Chemother. 52: 1127–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vediyappan G., Rossignol T., d'Enfert C. 2010. Interaction of Candida albicans biofilms with antifungals: transcriptional response and binding of antifungals to beta-glucans. Antimicrob. Agents Chemother. 54: 2096–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson R. B., Davis D., Mitchell A. P. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181: 1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]