

Abstract
We determined the crystal structure of an inactive Ser70Gly mutant of CTX-M-9 in complex with the bulky penicillin piperacillin at precovalent and posthydrolytic stages in the catalytic process. The structures obtained at high resolution were compared with the corresponding structures for the small penicillin benzylpenicillin and the bulky cephalosporin cefotaxime. The findings highlight the key role of the configuration of the carbon adjacent to the acylamino group of the side chain of β-lactams in the precovalent recognition of substrates.
INTRODUCTION
The production of β-lactamases is the predominant cause of resistance to β-lactam antibiotics in Gram-negative bacteria. These enzymes are divided on the basis of amino acid sequences into four classes designated A to D, with class A β-lactamases being the most prevalent in Enterobacteriaceae (1).
The oximino cephalosporins such as cefotaxime have been used since the 1980s to overcome these resistance mechanisms because of their relative stability to serine β-lactamases and their broad antibacterial spectrum. These β-lactams typically contain a bulky group such as a 2-(2-aminothiazole-4-yl)-2-oximino substituent at position C-7 of the cephalosporin nucleus, which prevents hydrolysis by serine β-lactamases. However, the intensive use of these molecules was quickly followed by the emergence of extended-spectrum β-lactamases (ESBLs), which can accommodate and hydrolyze such large β-lactams. The CTX-M enzymes of the class A family, first described in 1989, have spread worldwide since the late 1990s and are now the most prevalent extended-spectrum β-lactamases (2, 3, 5, 6, 25, 29).
The hydrolysis process catalyzed by these enzymes is based on an acid-base catalytic mechanism, as for the other class A enzymes. It comprises three major steps (8, 9, 14, 19): the formation of a precovalent encounter complex with the substrate; the nucleophilic attack on the β-lactam ring by the active serine residue resulting in an acyl-enzyme; and the nucleophilic attack on this covalent complex by the catalytic water molecule, which produces a transient noncovalent product-enzyme complex and subsequently results in the release of both the product and the active enzyme.
Several studies have looked at the crystal structure of CTX-M enzymes in complexes with β-lactams or transition-state analogs (8, 10, 11, 26). Surprisingly, CTX-M-9 efficiently binds and hydrolyzes the large oximino cephalosporin cefotaxime despite its small active site. Recently, the structures of benzylpenicillin and cefotaxime CTX-M-9 Michaelis complexes revealed that the accommodation of the bulky cephalosporin cefotaxime can be accommodated by a widening of the catalytic pocket, which is not observed for the small molecule benzylpenicillin (10). In this work, we determined the crystal structure of an inactive Ser70Gly mutant of CTX-M-9 in complex with the hydrolyzed and nonhydrolyzed forms of piperacillin to investigate the recognition of this bulky penicillin by CTX-M-type ESBLs. The structure shows that the size of the substrate is not the exclusive determinant in the widening of the catalytic pocket.
MATERIALS AND METHODS
Enzyme expression and purification.
The replacement of the active serine (Ser70) by an inactive Gly residue was performed in CTX-M-9 by site-directed mutagenesis using a CTX-M-9-producing pET-9a-based expression vector (11). DNA sequence was checked by gene sequencing. The mutated protein, designated CTX-M-9-S70G, was expressed in Escherichia coli BL21(DE3) (Novagene) as previously described (7). Briefly, E. coli was inoculated in 2× YT medium (Qbiogene, Irvine, CA) supplemented with 30 μg/ml kanamycin for aerobic growth at 37°C up to an A600 of 0.8. Expression was induced for 36 h at 30°C with 0.2 mM isopropyl-β-d-thiogalactopyranoside (Sigma Chemical Co., St. Louis, MO). Bacteria were harvested with 20 mM morpholineethanesulfonic acid (MES)-NaOH (pH 6.0). The cell pellet was disrupted by one cycle of freezing and thawing, followed by sonication. After centrifugation (10,000 × g for 10 min and 18,000 × g for 60 min at 4°C), the clarified supernatant was loaded onto an ion-exchange CM-Fast Flow column (100 ml; Amersham Pharmacia Biotech, Orsay, France) equilibrated with 20 mM MES-NaOH (pH 6.0). Proteins were eluted with a linear NaCl gradient (0 to 150 mM). The β-lactamase-containing elution peak was loaded on a gel filtration Superose 12 column (25 ml; Amersham Pharmacia Biotech) and eluted with a 5 mM Tris-HCl (pH 7.0) and 50 mM NaCl buffer. The protein was concentrated by ultrafiltration to 10 mg/ml for crystallization. The protein concentration was estimated by the Bio-Rad protein assay (Bio-Rad, Richmond, CA). Homogeneity was estimated to be more than 98% by SDS-PAGE.
Crystallization.
Crystals of CTX-M-9-S70G were obtained by vapor diffusion in hanging drops, using microseeding techniques with CTX-M-9 crystals. To a solution of 10 mg/ml protein in 5 mM Tris-HCl (pH 7.0) and 50 mM NaCl was added an equal volume of 1.2 M potassium phosphate buffer (pH 8.2) as previously described (10). Crystals appeared within 24 to 48 h after equilibration at 20°C and were soaked overnight at 20°C with hydrolyzed and native β-lactams at a concentration of 50 mM in 1.2 M potassium phosphate buffer (pH 8.2). Piperacillin was purchased from Dakota Pharm. Hydrolyzed piperacillin was obtained by 24 h of incubation with the wild-type CTX-M-9 at 30°C. Crystals were cryoprotected with 30% sucrose in 1.2 M potassium phosphate (pH 8.2) and flash frozen in liquid nitrogen.
Data collection and structure determination.
Data were collected on beam line ID23-1 of the European Synchrotron Radiation Facility at Grenoble, France. Reflections were indexed, integrated, and scaled using the HKL software package (21). The space group of crystals was P21, with two molecules in the asymmetric unit. Phases were calculated by molecular replacement with the MolRep program (28) of the CCP4 package (24), using the apoenzyme structure of CTX-M-9 (9) with water molecules and ions removed. Marvin version 5.3.8 was used for drawing compound structures (ChemAxon). The structures were refined with program Refmac5 (27) of the CCP4 package and Coot (12). Cross-validation was used throughout, and 5% of the data were used for the Rfree calculation. The stereochemical quality of the models was monitored with the Procheck program (15), and the figures were generated by Chimera (22). Between 91.3 and 91.9% of residues, excluding proline and glycine, were in the favored region, and 7.4 to 8.3% of residues were in the allowed region of the Ramachandran plot (17).
Molecular modeling.
The starting structure for simulations was taken from the ultrahigh-X-ray structure of CTX-M-9 (9). The GROMACS package and the geometric and charge parameters of the OPLSAA force field were used to carry out all energy minimizations and molecular dynamics simulations (16). Topology and RESP charges of hydrolyzed piperacillin were obtained from ab initio calculations using GAMESS (HF/6-31G* theory level) with RED-II software. Steered molecular dynamics (SMD) simulation was performed as previously described (10).
Protein structure accession numbers.
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 3HRE, 3Q07, and 3Q1F.
RESULTS AND DISCUSSION
Overall crystal structures.
The residue Ser70 of CTX-M-9 is responsible for the nucleophilic attack against the carbonyl carbon of β-lactams, which forms a covalent bond with the substrate resulting in the acyl-enzyme. Ser70 side chain can also bind the carboxylate function resulting from β-lactam hydrolysis. A Ser70Gly mutant of CTX-M-9 was therefore constructed to capture intact and hydrolyzed forms of piperacillin in the binding site as noncovalent adducts. As expected, no hydrolytic activity of this mutant against piperacillin was detected by using a computerized microacidimetric method (7).
The structures were determined at high resolution (Table 1). Two monomers were observed in the crystallographic asymmetric unit. The final Rwork and Rfree varied from 14.4 to 15.4% for the structure in complex with the intact form of piperacillin and from 17.5 to 18.4% for the hydrolyzed form, respectively. Electronic density maps confirmed the presence of the Ser70Gly substitution and of the catalytic water molecule. This water molecule was hydrogen bonded to the side chain of Glu166 and Asp170 (2.5 to 2.9 Å) as observed in CTX-M-9 apoenzyme (2P74). Electronic density maps also revealed the presence of ligands in each of the two active sites of the crystallographic asymmetric unit. Densities compatible with the dioxopiperazine cycle identified the piperacillin molecules. Analysis of the structure of the crystal soaked with intact piperacillin identified the intact and hydrolyzed forms of piperacillin in the active site. However, the intact form, which harbors a four-membered β-lactam, was the predominant adduct (80% occupancy). Analysis of the structure of the crystal soaked with hydrolyzed piperacillin identified only the hydrolyzed molecule in the active site. The Cα atom root mean square deviation (RMSD) calculated from the overlay of CTX-M-9-S70G apoenzyme structure with the corresponding complex structures varied from 0.08 to 0.18 Å, showing that the insertion of piperacillin in the catalytic pocket did not modify the overall structure of the enzyme. In addition, the hydrogen bonds between the key residues of the binding site were similar to those previously observed in CTX-M enzymes (Table 2).
Table 1.
Data collection and refinement statisticsa
| Parameter | CTX-M-9 S70G | CTX-M-9 S70G/piperacillin | CTX-M-9 S70G/hydrolyzed piperacillin |
|---|---|---|---|
| Space group | P21 | P21 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 45.07, 106.29, 47.54 | 45.13, 106.34, 47.61 | 45.10, 106.61, 47.54 |
| α, β, γ (°) | 90.00, 102.17, 90.00 | 90.00, 101.90, 90.00 | 90.00, 101.92, 90.00 |
| Rmerge | 0.047 (0.180) | 0.048 (0.950) | 0.064 (0.334) |
| I/σI | 22.4 (5.4) | 14.7 (5.9) | 16.6 (3.5) |
| Completeness (%) | 96.2 (91) | 94.5 (90.1) | 99.8 (98.4) |
| Redundancy | 4.9 (4.8) | 5.0 | 4.9 |
| Resolution (Å) | 42.6–1.5 | 19.47–1.50 | 19.20–1.50 |
| No. of reflections | 74,760 | 62,848 | 66,447 |
| Rwork/Rfree | 0.156/0.184 | 0.145/0.175 | 0.144/0.176 |
| RMSD | |||
| Bond length (Å) | 0.008 | 0.010 | 0.011 |
| Bond angle (°) | 1.536 | 1.623 | 1.287 |
Values in parentheses are for the highest-resolution shell. One crystal was used for each data set.
Table 2.
Key interactions in the complex structures with β-lactams
| Type of distance | Complex | Distance (Å) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CTX-M-9 S70G |
CTX-M-9 S70G/ benzylpenicillin |
CTX-M-9 S70G/piperacillin |
CTX-M-9 S70G/ hydrolyzed benzylpenicillin |
CTX-M-9 S70G/ hydrolyzed piperacillin |
|||||||
| Chain A | Chain B | Chain A | Chain B | Chain A | Chain B | Chain A | Chain B | Chain A | Chain B | ||
| Distance between key residues of the binding sitea | H2O cat-Glu166 (Oε1) | 2.57 | 2.50 | 2.62 | 2.67 | 2.54 | 2.50 | 2.62 | 2.65 | 2.50 | 2.47 |
| H2O cat-Asn170 (Oδ1) | 2.69 | 2.74 | 2.83 | 2.94 | 2.80 | 2.89 | 2.80 | 2.78 | 2.79 | 2.89 | |
| Lys73 (Nζ)-Asn132 (Oδ1) | 2.70 | 2.69 | 2.68 | 2.73 | 2.69 | 2.67–2.76 | 2.65–2.75 | 2.68 | 2.68 | 2.68–2.78 | |
| Lys73(Nζ)-Ser130 (Oγ) | 2.89 | 2.84 | 2.90 | 2.88 | 2.86 | 2.93 | 2.92–3.05 | 2.84 | 2.88 | 2.92 | |
| Ser130 (Oγ)-Lys234 (Nζ) | 2.84 | 2.82 | 2.86 | 2.88 | 2.77 | 2.82 | 2.83 | 2.82 | 2.81 | 2.86 | |
| Glu166 (Oε2)-Asn170 (Nδ2) | 2.74 | 2.76 | 2.74 | 2.78 | 2.79 | 2.81 | 2.73 | 2.81 | 2.81 | 2.83 | |
| Pro167 (O)-Asn170 (N) | 3.07 | 3.17 | 3.04 | 3.09 | 3.16 | 3.26 | 3.12 | 3.20 | 3.20 | 3.27 | |
| Pro167 (O)-Asn170 (Nδ2) | 3.23 | 3.07 | 3.02 | 2.94 | 3.03 | 3.00 | 3.00 | 2.99 | 3.09 | 3.03 | |
| Asn170 (O)-Asp240 (N) | 3.05 | 3.05 | 3.15 | 3.11 | 3.22 | 3.29 | 3.15 | 3.14 | 3.20 | 3.28 | |
| Distance between key residues and amide function of β-lactams | N-12 (β-lactam)–Ser237 (O) | NAb | NA | 2.85 | 2.89 | 2.95 | 2.97 | 2.84 | 2.83 | 2.95 | 3.00 |
| O-14 (β-lactam)–Asn104 (Nδ2) | NA | NA | 3.02 | 3.10 | 3.03 | 2.99 | 3.08 | 3.12 | 3.07 | 3.00 | |
| O-14 (β-lactam)–Asn132 (Nδ2) | NA | NA | 2.90 | 2.93 | 2.91 | 2.90 | 3.01 | 3.01 | 2.90 | 2.91 | |
| Distance between key residues and conserved C-3 carboxylate function of β-lactams | O-11 (β-lactam)–Ser130 (Oγ) | NA | NA | 2.48c | 2.52 | 3.03 | 2.88 | 4.35 | 4.22 | 5.45 | 5.55 |
| O-11 (β-lactam)–Thr235 (Oδ) | NA | NA | 2.56 | 2.62 | 2.70 | 2.92 | 2.61 | 2.65 | 4.82 | 5.01 | |
| O-11 (β-lactam)–Lys234 (Nζ) | NA | NA | 3.03 | 3.07 | 3.65 | 3.74 | 4.53 | 4.44 | 6.74 | 6.89 | |
| O-10 (β-lactam)–Ser237 (Oγ) | NA | NA | 4.36 | 4.00 | 2.79 | 2.62 | 4.51 | 4.51 | 3.03 | 3.03 | |
| Distance between key residues and carbonyl function of the β-lactam ring or the carboxylate resulting from hydrolysis | O-8 (β-lactam)–Gly70 (N) | NA | NA | 2.83 | 2.83 | 2.82 | 2.88 | 2.77 | 2.76 | 2.83 | 2.84 |
| O-8 (β-lactam)–Ser237 (N) | NA | NA | 2.83 | 2.85 | 2.88 | 2.95 | 2.82 | 2.76 | 2.86 | 2.86 | |
| O-8′ (β-lactam)–Lys73 (Nζ) | NA | NA | NA | NA | NA | NA | 2.66 | 2.83 | 2.66 | 2.46 | |
| O-8′ (β-lactam)–Ser130 (Oγ) | NA | NA | NA | NA | NA | NA | 2.49 | 2.43 | 3.00 | 2.98 | |
Two distances in monomers A and B. In cases with two distances shown within one monomer, multiple conformations are present.
NA, not applicable.
Boldface indicates a significant difference in enzyme-ligand distances between complexes.
Binding of piperacillin into the CTX-M-9-S70G active site.
The electrostatic interactions, previously identified as critical for hydrolytic activity (8, 13, 14, 18, 26), were observed between piperacillin and key residues of the catalytic pocket (Fig. 1A). The oxygen atom of the β-lactam ring was located in an oxyanion hole formed by Ser70N and Ser237N. C-3 carboxylate function interacted with conserved residues Ser130 and Thr235 of the active site. C-6 amide function interacted with the residues at positions 104, 132, and 237. The other atoms of the C-6 substituent did not establish additional electrostatic interactions with the CTX-M-9-S70G protein. The dioxopiperazine substituent of piperacillin adopted a positioning similar to that of the cefotaxime methoximino group. The latter bulky group can be accommodated only if the catalytic pocket is opened and the conserved hydrogen bond between the residues N170 and D240 is broken (10). The binding of piperacillin did not induce such conformational change, although the dioxopiperazine substituent is bigger than the methoximino group (volume, 265 versus 153 Å3). This paradox can be explained by the configuration of the α-carbon of the carbonyl of the C-6/C-7 side chain harboring these two substituents. The imino sp2 of the α-carbon of the side chain of cefotaxime imposes a planar configuration on the carbons harboring the aminothiazol methoximino groups, which are difficult to accommodate and open the CTX-M-9 binding site. The sp3 hybridization of the α-carbon of the side chain of piperacillin confers a tetrahedral configuration to the C bearing the benzyl dioxopiperazine group, in which the H atom is oriented toward the β3 strand and allows the accommodation of the substituents without notable constraint (Fig. 2). The steric hindrance of the C-6/7 substituent of β-lactams is reported as an important factor in the resistance to β-lactamases (11, 18, 23). Our crystallographic data reveal that the presence of a bulky side chain is not sufficient to explain a restricted recognition of substrate by the class A enzymes; topographic constraints must also be imposed on substituents of the side chain. In oximino cephalosporins, this constraint is supplied by the imino group, in which the α-carbon adjacent to the acylamino group adopts a planar hybridization (sp2), which puts the substituents in steric hindrance with the β3 strand and Ω loop (Fig. 2). In contrast, the accommodation of the C-6 substituent of piperacillin occurs without notable constraint. Accordingly, the effect linked to the modifications of the C-6 substituent in penicillins is slight when considering the Km values (25 and 20 μM for benzylpenicillin and piperacillin, respectively [2]).
Fig. 1.

Key polar interactions observed with crystallographic structures between CTX-M-9-S70G and piperacillin (A), benzylpenicillin (B), cefotaxime (C), hydrolyzed piperacillin (D), and hydrolyzed benzylpenicillin (E). Carbon atoms are in green for the protein or in yellow for the adduct; oxygen atoms are red, and nitrogen atoms are blue. The α-carbon of the carbonyl of the side chain of β-lactams is magenta.
Fig. 2.
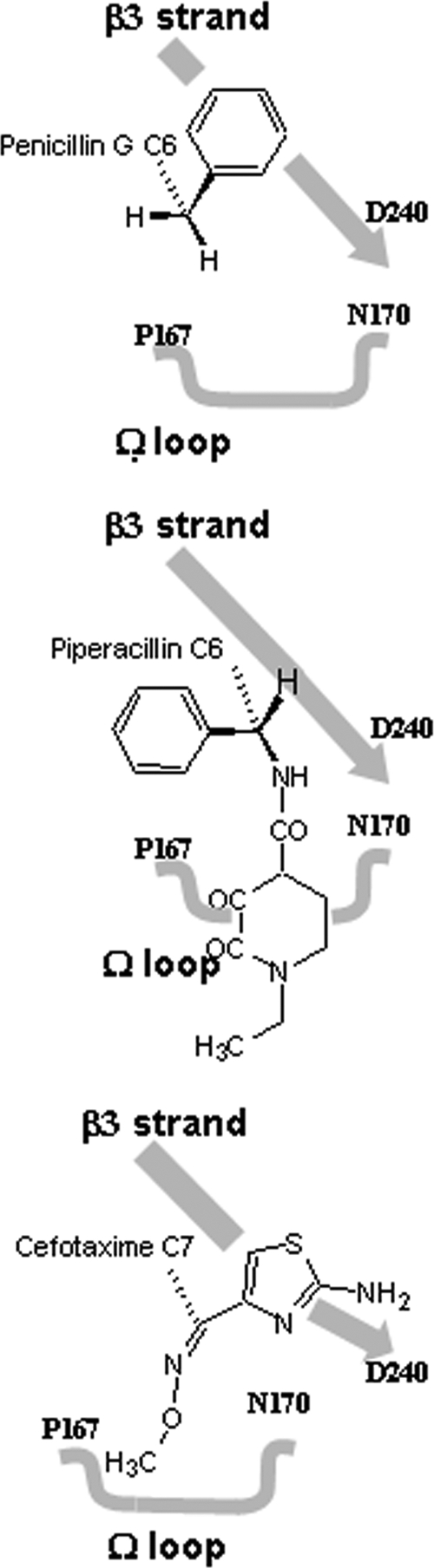
Schematic representation of the α-carbon of the carbonyl of the C-6/C-7 side chain of benzylpenicillin, cefotaxime, and piperacillin, in relation to the β3 strand and Ω loop of CTX-M-9. The α-carbon of cefotaxime adopts planar (sp2) hybridization; the α-carbon of penicillins adopts a tetrahedral (sp3) hybridization.
However, the positioning of the C-6 side chain of the piperacillin toward the β3 strand, to avoid steric hindrance between the dioxopiperazine cycle and the residue Pro167 of the Ω loop, induces an 0.7-Å shift in the opposite direction of its thiazolidine ring toward Asn132, in comparison with benzylpenicillin. Consequently, the interactions of the C-3 carboxylate functions of piperacillin and benzylpenicillin were different. Piperacillin C-3 carboxylate was at a hydrogen bond distance of Ser237Oγ (2.6 to 2.8 Å), unlike benzylpenicillin C-3 carboxylate (4.0 to 4.4 Å). Interestingly, no hydrogen bond was established between the C-3 carboxylate function of piperacillin and lysine 234, in contrast to penicillin and cefotaxime (3.7 versus 3.0 and 3.2 Å, respectively; Fig. 1A and 1B). Lysine 234 is conserved among the class A β-lactamases; it belongs to the KTG motif. This cationic residue binds the C-3 carboxylate common to most β-lactamase substrates and is important for the stabilization of the transition state in acylation, much more so than for the complementarity between the enzyme and the substrate ground states, which is responsible for the initial binding (4). The protonated lysine of the KTG motif is also important in penicillin-binding proteins, in which it aids in the proper positioning of the substrate during acyl-enzyme complex formation (30). The absence of a bond between the lysine 234 and the C-3 carboxylate of piperacillin can affect the acylation and may be therefore involved in the decrease of the kcat value observed for piperacillin in comparison to those measured for penicillin and cefotaxime (110 s−1 versus 295 s−1 and 450 s−1, respectively) (2).
Product release from CTX-M binding site.
In the structure in complex with the hydrolysis product of piperacillin, the C-7 carbonyl function of the β-lactam ring was replaced by a carboxylate function, which interacted with active-site residues (Fig. 1D) as observed in the complex structure of CTX-M-9-S70G with hydrolyzed benzylpenicillin (Fig. 1E). Because of electrostatic repulsion between this carboxylate function and the C-3 carboxylate function of the thiazolidine ring, the thiazolidine ring of both hydrolyzed penicillins rotated ∼60°. It disrupted interactions with residues Ser130 (5.5 to 5.6 Å) and Thr235 (4.8 to 5.0 Å) for piperacillin, but the hydrogen bond with the Oδ atom of Ser237 (2.9 Å) was conserved. To complete the crystallographic data, the release of hydrolyzed piperacillin was investigated by steered molecular dynamics (SMD) simulations. The thiazolidine ring rotated 180° over the β3 strand, and the interactions between the C-3 carboxylate group of the thiazolidine ring and the electropositive region of the CTX-M binding site (Ser130, Lys234, Thr235) were disrupted. During this rotation, the C-3 carboxylate group interacted with Ser237 until the hydrolyzed piperacillin exited from the active site. Piperacillin and cefotaxime are therefore expelled from the active site of CTX-M-9 by a closely related mechanism (10). However, the differences in the structures of the penicillin and cephalosporin products may be involved in the higher turnover of CTX-M against cephalosporins than against penicillins (kcat = 3,000, 450, 295, and 110 s−1, respectively, for cephalothin, cefotaxime, benzylpenicillin, and piperacillin [2]). Indeed, the carbon adjacent to the carboxylate function of cephalosporins, such as cefotaxime, adopts a planar hybridization sp2 and that of penicillins adopts a tetrahedral hybridization sp3. Consequently, the orientation of cephalosporin carboxylate function is in the plane of the 6-membered ring. Because of this, the rotation of the ring observed with penicillin-derived products may create a steric clash between carboxylate of cephalosporins and the Ser237 side chain. This steric conflict, absent in penicillins, can help an efficient autoexpulsion of cephalosporin-derived product.
In conclusion, we determined the crystal structure of CTX-M-9-S70G in complex with intact and hydrolyzed forms of piperacillin. The findings revealed the determinants of the interaction between the CTX-M binding site and piperacillin at precovalent and posthydrolytic steps of the catalytic process. From these data, we show that the configuration of the α-carbon of the carbonyl of the side chain of β-lactams is crucial for their accommodation and that the absence of a bond between Lys234 and the C-3 carboxylate function of piperacillin could explain, in part, a lower catalytic activity of the CTX-M enzymes against this penicillin.
ACKNOWLEDGMENTS
This work was supported, in part, by a grant from INRA and Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche (Paris, France).
We thank Rolande Perroux and Marlene Jan for technical assistance. Provision of beam time at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) is gratefully acknowledged.
Footnotes
Published ahead of print on 19 September 2011.
REFERENCES
- 1. Ambler R. P., et al. 1991. A standard numbering scheme for the class A beta-lactamases. Biochem. J. 276:269–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonnet R. 2004. Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob. Agents Chemother. 48:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradford P. A. 2001. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 14:933–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brannigan J., et al. 1991. The mutation Lys234His yields a class A beta-lactamase with a novel pH-dependence. Biochem. J. 278:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantón R., et al. 2008. Prevalence and spread of extended-spectrum beta-lactamase-producing Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 14(Suppl. 1):144–153 [DOI] [PubMed] [Google Scholar]
- 6. Cantón R., Coque T. M. 2006. The CTX-M beta-lactamase pandemic. Curr. Opin. Microbiol. 9:466–475 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y., Delmas J., Sirot J., Shoichet B., Bonnet R. 2005. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 348:349–362 [DOI] [PubMed] [Google Scholar]
- 8. Chen Y., Shoichet B., Bonnet R. 2005. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J. Am. Chem. Soc. 127:5423–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y., Bonnet R., Shoichet B. K. 2007. The acylation mechanism of CTX-M β-lactamase at 0.88 Å resolution. J. Am. Chem. Soc. 129:5378–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delmas J., et al. 2010. Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J. Mol. Biol. 400:108–120 [DOI] [PubMed] [Google Scholar]
- 11. Delmas J., et al. 2008. Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J. Mol. Biol. 375:192–201 [DOI] [PubMed] [Google Scholar]
- 12. Emsley P., Cowtan K. 2004. Coot: model-building tolls for molecular graphics. Acta Cyrstallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 13. Gazouli M., Tzelepi E., Sidorenko S. V., Tzouvelekis L. S. 1998. Sequence of the gene encoding a plasmid-mediated cefotaxime-hydrolyzing class A beta-lactamase (CTX-M-4): involvement of serine 237 in cephalosporin hydrolysis. Antimicrob. Agents Chemother. 42:1259–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ibuka A. S., et al. 2003. Crystal structure of extended-spectrum β-lactamase Toho-1 insights into the molecular mechanism for catalytic reaction and substrate specificity expansion. Biochemistry 42:10634–10643 [DOI] [PubMed] [Google Scholar]
- 15. Laskowski R. A., Moss D. S., Thornton J. M. 1993. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231:1049–1067 [DOI] [PubMed] [Google Scholar]
- 16. Lindahl E., Hess B., van der Spoel D. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis, J. Mol. Model. 7:306–317 [Google Scholar]
- 17. Lovell S. C., et al. 2003. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50:437–450 [DOI] [PubMed] [Google Scholar]
- 18. Massova I., Mobashery S. 1996. Molecular bases for interactions between beta-lactam antibiotics and beta-lactamases. Acc. Chem. Res. 30:162–168 [Google Scholar]
- 19. Matagne A., Lamotte-Brasseur J., Frere J. M. 1998. Catalytic properties of class A beta-lactamases: efficiency and diversity. Biochem. J. 330:581–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reference deleted.
- 21. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 276:307–326 [DOI] [PubMed] [Google Scholar]
- 22. Pettersen E. F., et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 23. Philippon A., Arlet G., Jacoby G. A. 2002. Plasmid-determined AmpC-type beta-lactamases. Antimicrob. Agents Chemother. 46:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Potterton E., McNicholas S., Krissinel E., Cowtan K., Noble M. 2002. The CCP4 molecular-graphics project. Acta Crystallogr. Sect. D 58:1955–1957 [DOI] [PubMed] [Google Scholar]
- 25. Rossolini G. M., D'Andrea M. M., Mugnaioli C. 2008. The spread of CTX-M-type extended-spectrum beta-lactamases. Clin. Microbiol. Infect. 14(Suppl. 1):33–41 [DOI] [PubMed] [Google Scholar]
- 26. Shimamura T., et al. 2002. Acyl-intermediate structures of the extended-spectrum class A β-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J. Biol. Chem. 277:46601–46608 [DOI] [PubMed] [Google Scholar]
- 27. Steiner R. A., Lebedev A. A., Murshudov G. N. 2003. Fisher's information in maximum-likelihood macromolecular crystallographic refinement. Acta Crystallogr. D Biol. Crystallogr. 59:2114–2124 [DOI] [PubMed] [Google Scholar]
- 28. Vagin A., Teplyakov A. 2000. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56:1622–1624 [DOI] [PubMed] [Google Scholar]
- 29. Walther-Rasmussen J., Høiby N. 2004. Cefotaximases (CTX-M-ases), an expanding family of extended-spectrum beta-lactamases. Can. J. Microbiol. 50:137–165 [DOI] [PubMed] [Google Scholar]
- 30. Zhang W., Shi Q., Meroueh O., Vakulenko S. B., Mobashery S. 2007. Catalytic mechanism of penicillin-binding protein 5 of Escherichia coli. Biochemistry 46:10113–10121 [DOI] [PubMed] [Google Scholar]