

Abstract
Rifampin, a potent antibacterial agent, is one of the main drugs used in the treatment of mycobacterial infections. Hepatotoxicity is a well-documented adverse event. The aim of this study was to investigate the effect of rifampin on the production of inflammatory mediators in human epithelial HepG2 liver cells in the absence or presence of proinflammatory cytokines. Incubation of HepG2 cells with a cytokine mix plus rifampin was associated with a significant dose-dependent increase in the production of nitric oxide compared to incubation with the cytokine mix alone (P < 0.05) as well as with an increase in inducible nitric oxide synthase protein and mRNA expression. Rifampin significantly increased the secretion of interleukin 8 (IL-8) in both untreated cells (P < 0.001) and cytokine-treated cells (P < 0.006). An array screening assay revealed that rifampin stimulated the production of IL-1β and gamma interferon-induced protein-10 (IP-10) in untreated cells and increased the secretion of RANTES in cytokine-treated cells. Together, these results indicate that rifampin may exert proinflammatory effects on liver cells.
INTRODUCTION
Rifampin is one of the main drugs used in the treatment of mycobacterial infections and also exerts potent antibacterial activity against staphylococci, legionellae, Haemophilus spp., and other bacteria. Rifampin has long been considered an immunosuppressive drug. However, despite the many studies describing its immunosuppressive activities (15, 21, 25, 27, 32, 35), physicians have been puzzled by the lack of clinically relevant immunosuppressive adverse effects, even during prolonged treatment and in patients with AIDS (10, 12). Additionally, rifampin activates the nuclear pregnane X receptor (PXR) of the cytochrome P-450 enzymes that metabolize drugs in the liver (23, 30). Rifampin accumulates in the liver, and one of its well-documented adverse effects in the treatment of tuberculosis is hepatotoxicity (3, 15, 29).
Nitric oxide (NO) is a major immune mediator and an important part of the host defense against Mycobacterium tuberculosis (4, 18). It acts as an intercellular and intracellular messenger, regulating expression of many genes and the activity of a wide range of proteins and signal transduction pathways. NO modulates the production of several cytokines and chemokines, among them gamma interferon (IFN-γ)-induced protein-10 (IP-10) (CXCL-1), regulated on activation, normal T-cell expressed and secreted protein (RANTES), monocyte chemotactic protein-1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1), tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), macrophage inflammatory protein-1 (MIP-1), and IL-8, in different cell types by different mechanisms (8, 17, 20, 22). The production of NO is controlled by constitutive and inducible isoforms of nitric oxide synthase (NOS), each associated with a different mode of regulation and a different amount of NO produced. High levels are generated in response to bacterial components or a combination of proinflammatory cytokines, such as IL-1β, TNF-α, and IFN-γ (13, 14). Although elevated levels of NO have beneficial antimicrobial effects, they have been implicated in the pathogenesis of some inflammatory diseases (4, 13). NO is produced in the liver by constitutive and inducible NOS (iNOS) and is associated with many cellular and biochemical functions. The constitutive level of NO is important for hepatic microcirculation and endothelial integrity, whereas the high level of NO generated by iNOS can be both protective and harmful, depending on the kind of insult. Thus, in models of sepsis, hepatitis, and liver regeneration, NO was found to protect against apoptotic cell death. By contrast, in warm ischemia reperfusion and hemorrhagic-shock models, it enhanced the hepatic oxidative injury (5, 9, 19).
Interleukin 8 (IL-8) is a potent chemokine that functions as a leukocyte chemoattractant in immune and inflammatory responses. It is induced by bacterial and viral products and proinflammatory cytokines. IL-8 is involved in the immune response to M. tuberculosis (2, 24) and has been linked to the pathology of chronic lung and liver diseases (1, 2, 6, 11).
Our previous studies have shown that rifampin augments expression of inducible NOS (iNOS) and NO in human alveolar epithelial cells (31, 33). Given the important role of rifampin in the liver, we sought to determine its effect on NO production and on expression of inflammatory mediators, particularly IL-8, in human hepatic cells.
MATERIALS AND METHODS
Reagents.
The cell culture medium and its supplements were obtained from Biological Industries (Beit HaEmek, Israel), and recombinant human IL-1β, IFN-γ, and TNF-α were obtained from ProSpect-Tant TechnoGene Ltd. (Rehovot, Israel). The NOS inhibitor NG-nitro-l-arginine methyl ester (L-NAME) and rifampin were obtained from Sigma Chemical (St. Louis, MO). A stock solution of rifampin (50 g/liter) was prepared using dimethyl sulfoxide (DMSO; Sigma). The final concentration of DMSO in culture medium was 0.1% (vol/vol) in all experiments.
Cell culture.
Human hepatic epithelial HepG2 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and l-glutamine (2 mM) and with penicillin (100 U/ml), streptomycin (100 mg/liter), and nystatin (12.5 U/ml) (PSN) at 37°C in a humidified incubator in a 5% CO2 atmosphere. The cells were incubated in serum- and antibiotic-free medium for 24 h before stimulation and were then exposed to a cytokine mix (TNF-α, IL-1β, and IFN-γ [100 μg/liter each]) with or without rifampin. Control, untreated cells were incubated with DMSO (0.1%). Cell culture viability was evaluated by the use of a neutral red uptake viability assay. No differences in viability among differently treated cells were found.
NO measurement.
Nitrite (NO2) concentration was measured as an indicator of NO production by the spectrophotometry method based on the Griess reaction (31).
Western blot analysis.
Cytosolic extract preparation and Western blot analysis of iNOS were performed as described previously (31) with rabbit anti-human iNOS antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
Cytokine array.
One milliliter of culture medium was centrifuged to remove cell debris, and the supernatants were analyzed using a Proteome Profiler Array Human Cytokine Panel A kit (R&D system) according to the manufacturer's instructions.
IL-8 and IL-1β determinations.
The amounts of IL-8 and IL-1β were determined using an enzyme-linked immunoassay kit from R&D Systems (Minneapolis, MN) according to the manufacturer's instructions.
Real-time PCR for mRNA determinations.
RNA was isolated and transcribed to cDNA with a TaqMan cells-to-CT kit (Ambion Inc.—The RNA Company, Austin, TX). The cDNA samples were examined for iNOS and IL-8 using TaqMan Assay-on-Demand primers and a TaqMan Gene Expression mix with an ABI 7000 real-time PCR system and a Prism 7000 sequence detector (Applied Biosystems, Foster City, CA). The results determined for iNOS mRNA were normalized to human β-actin mRNA and, for IL-8, to human RPLP0 ribosomal protein mRNA.
Statistical analysis.
The results are presented as means ± standard errors of the means. Unpaired t test was used to compare the results among treatments. Significance was defined a priori as a P value of <0.05.
RESULTS
Effect of rifampin on NO production.
Incubation of HepG2 cells with the cytokine mix induced time-dependent production of NO. The addition of rifampin to the cytokine mix significantly increased NO release in a concentration-dependent manner at 24 h and 48 h after stimulation (Fig. 1a).
Fig. 1.

Rifampin (Rif.) effect on NO production in HepG2 cells stimulated with cytokine mix. (a) NO levels in cell supernatant at 24 and 48 h after stimulation. At 24 h, for bars a and b, P < 0.05 for mix versus mix + rifampin at 10 mg/liter and for mix versus mix + rifampin at 25 mg/liter, and for bar c, P = 0.008 for mix versus mix + rifampin at 50 mg/liter; at 48 h, for bar d, P = 0.02 for mix versus mix + rifampin at 25 mg/liter, and for bar e, P = 0.009 for mix versus mix + rifampin at 50 mg/liter (n = 3). Data represent the results of one of three similar experiments. (b) iNOS protein expression at 24 h after stimulation. Western blot analysis showed a rifampin dose-dependent increase in iNOS expression compared to β-actin results. A blot representative of one of two similar experiments is shown. (c) Densitometric analysis of the blot presented in panel b. (d) INOS mRNA expression 19 h after stimulation. The values represent the change compared with the results for the control cells. Data represent the results of one of three similar experiments (P = 0.004 for mix versus mix + rifampin at 50 mg/liter) (n = 3).
For protein analysis, total cell protein was extracted 24 h after stimulation and subjected to Western blotting. Unstimulated cells and cells treated with rifampin alone did not express iNOS protein. Incubation with the cytokine mix induced iNOS protein expression, and the addition of rifampin increased iNOS expression in a dose dependent manner (Fig. 1b and c). Quantitative real-time PCR showed that rifampin acts at the transcriptional level: 19 h after stimulation, iNOS mRNA was undetectable in cells incubated with medium only (control). Incubation with cytokines induced a 120- ± 12-fold increase in iNOS mRNA transcription, and the addition of rifampin (10 and 50 mg/liter) further increased transcription 158- ± 22-fold and 308- ± 19-fold, respectively (P < 0.003 for cytokine versus cytokine with rifampin at 50 mg/liter). Treatment with rifampin alone (50 mg/liter) was associated with very low levels of iNOS mRNA (Fig. 1d).
Effect of rifampin on inflammatory mediator production.
To gain more information on the effect of rifampin on inflammatory processes, we screened 35 inflammatory mediators by cytokine array assays. Cells were incubated with medium (control), the cytokine mix alone, the cytokine mix with rifampin (50 mg/liter), or rifampin alone. The supernatants were collected after 24 h and analyzed for cytokine levels. The untreated HepG2 cells spontaneously secreted 3 cytokines: migration inhibition factor (MIF), soluble intercellular adhesion molecule (sICAM), and Serpin E1. Rifampin alone induced the production of 3 cytokines: IL-1β, IL-8, and IP-10 (CXCL-10). Incubation of the cells with the cytokine mix alone induced the production of granulocyte colony-stimulating factor (G-CSF), growth-regulated oncogene alpha (GROa; CXCL-11), IP-10, IFN-inducible T-cell α chemoattractant (I-TAC), sICAM, Serpin E1, and IL-8; the addition of rifampin increased the concentration of IL-8 and RANTES over the levels reached with the cytokine mix alone (Fig. 2a). The effect of rifampin alone on IL-1β production was further demonstrated by the results of a quantitative enzyme-linked immunoassay (Fig. 2b).
Fig. 2.

Effect of rifampin on the production of inflammatory mediators. (a) HepG2 cells were treated with rifampin (50 mg/liter) alone, the cytokine mix, or the cytokine mix with rifampin (50 mg/liter). The culture medium was collected 24 h after stimulation and analyzed using a human cytokine array. The strongly upregulated genes are highlighted by rectangles. PC, positive control. A blot representative of one of two similar experiments is shown. (b) IL-1b levels in cells supernatant 48 h after the addition of rifampin, determined using an enzyme-linked immunoassay (n = 4).
Effect of rifampin on IL-8 production.
Prompted by the array results showing that rifampin alone strongly induced IL-8 production and further enhanced the level of IL-8 stimulated by the cytokine mix, we focused on the effect of rifampin on IL-8 production. Cells were incubated with the cytokine mix alone, rifampin alone, or the cytokine mix plus different doses of rifampin. IL-8 levels in the cells supernatant were measured 48 h after stimulation with a quantitative enzyme-linked immunoassay. The results confirmed the array screening test data and also demonstrated that the HepG2 cells spontaneously secreted IL-8. Incubation with the cytokine mix alone increased the amount of IL-8 in the cell supernatant from 2.5 ± 0.2 μg/liter (untreated cells) to 8.3 ± 0.4 μg/liter (P < 0.001), and the addition of rifampin at doses of 10 mg/liter and 50 mg/liter significantly increased the IL-8 level to 29.9 ± 0.5 μg/liter and 19.6 ± 2.1 μg/liter, respectively (P < 0.006). Interestingly, rifampin at 10 mg/liter was more effective than rifampin at 50 mg/liter. As demonstrated in the array screening, rifampin alone (50 mg/liter) markedly increased the spontaneous release of IL-8 (P < 0.001) (Fig. 3 a).
Fig. 3.
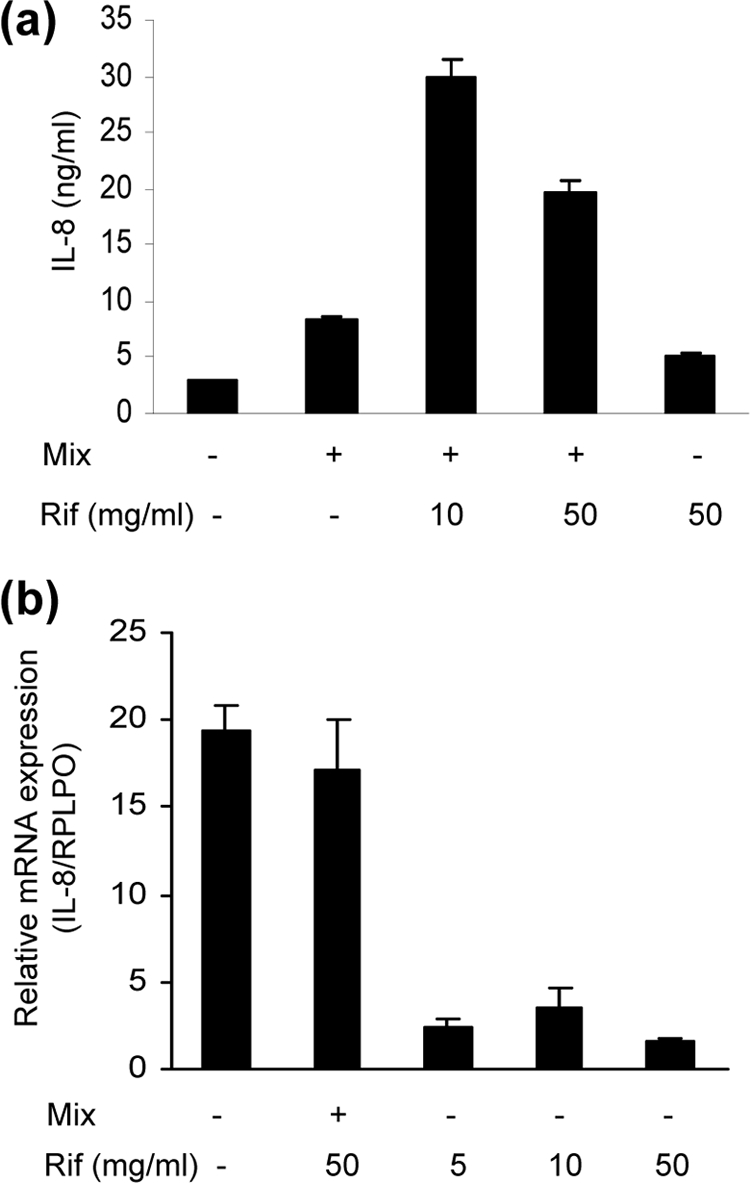
Effect of rifampin (Rif) on IL-8 production in HepG2 cells. (a) IL-8 levels in cell supernatants 48 h after stimulation with cytokine mix in the presence and absence of rifampin. P < 0.001 for control versus mix and versus rifampin at 50 mg/liter; P < 0.006 for mix versus mix + rifampin at 10 mg/liter and for mix versus mix + rifampin at 50 mg/liter) (n = 5 for each treatment). Data represent the results of one of three similar experiments. (b) IL-8 mRNA expression at 19 h after the addition of cytokine with or without rifampin and with different doses of rifampin. The values represent changes compared to the results determined for the control cells. Data are presented as the means ± standard errors of the means (n = 3) of the results of 1 of 3 similar experiments.
Analysis of RNA extracted from the cells 19 h after stimulation revealed that incubation with cytokines increased expression of IL-8 mRNA by 20-fold and that the addition of rifampin (50 mg/liter or 10 mg/liter) had no significant effect. However, rifampin alone at doses of 5 to 50 mg/liter increased the level of IL-8 mRNA compared to control (untreated) cell results in a dose-dependent manner. Interestingly, rifampin at a dose of 10 mg/liter was the most effective treatment (Fig. 3b).
NO modulation of rifampin-induced IL-8 production.
NO has been reported to modulate IL-8 expression in several models (7, 8, 17, 20, 22). In view of our finding of rifampin augmentation of NO production in HepG2 cells, we investigated the potential involvement of NO in the rifampin-induced increase in IL-8 production. Cells were incubated with rifampin in the presence or absence of the iNOS inhibitor L-NAME (20 mM) for 24 h. L-NAME at this concentration completely inhibited cytokine-induced NO production. The addition of L-NAME to rifampin significantly augmented the concentration of IL-8 in the cell supernatants, as follows: from 539 ± 20 ng/liter to 1,006 ± 10 ng/liter in cells treated with rifampin at 10 mg/liter (P < 0.006) and from 454 ± 75 to 2621 ± 600 ng/liter in cells treated with rifampin at 50 mg/liter (P < 0.007). RNA analysis showed that incubation of the cells with combined rifampin and L-NAME treatment for 19 h increased expression of IL-8 mRNA from 3.0 ± 0.3 to 4.4 ± 0.2 ng/liter in cells treated with rifampin at 10 mg/liter and from 2.8 ± 0.25 to 7.8 ± 1.6 ng/liter in cells treated with rifampin at 50 mg/liter (P < 0.03) (Fig. 4a and b).
Fig. 4.
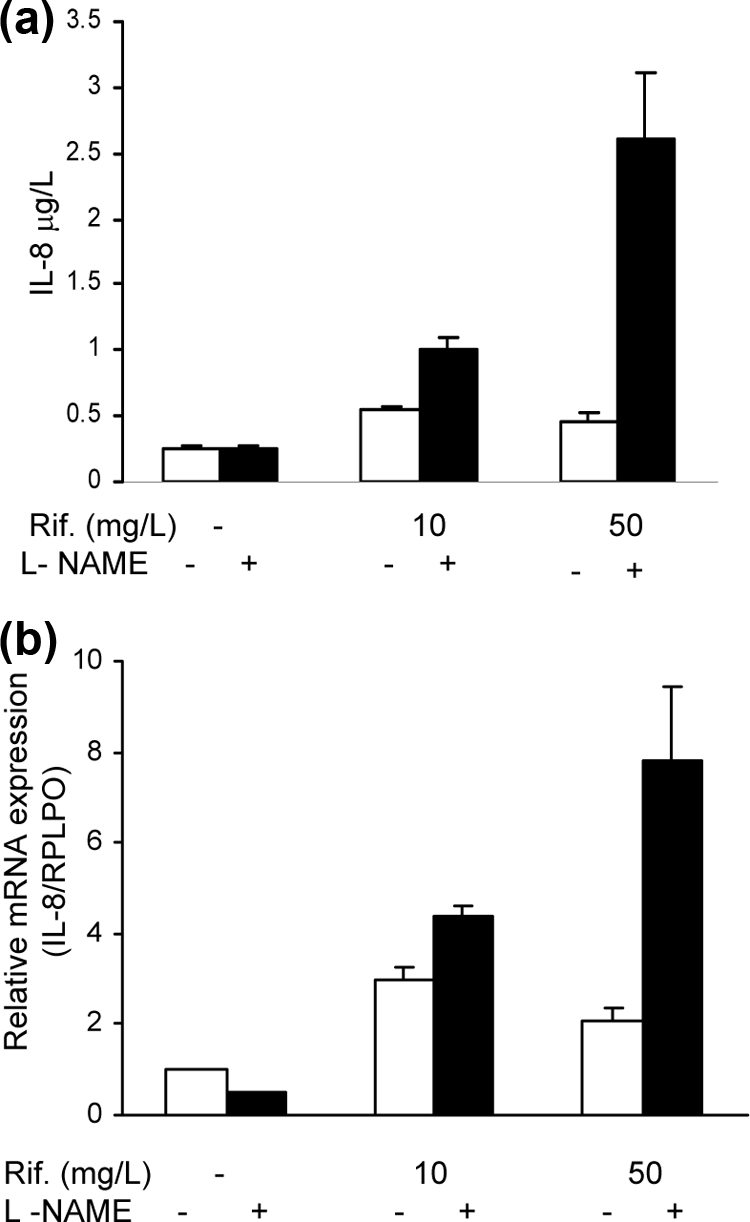
Effect of L-NAME on rifampin stimulation of IL-8 production. (a) HepG2 cells were treated with rifampin in the presence or absence of L-NAME (NOS inhibitor) (20 μM). IL-8 levels were determined 24 h after stimulation. P < 0.007 for rifampin at 50 mg/liter versus rifampin at 50 mg/liter + L-NAME; P < 0.006 for rifampin at 10 mg/liter versus rifampin at 10 mg/liter + L-NAME) (n = 4). Data represent the results of one of three similar experiments. (b) IL-8 mRNA expression 19 h after the addition of rifampin with or without L-NAME (20 μM). The values represent changes compared with the results determined for the control untreated cells. Data are presented as the means ± standard errors of the means. P < 0.04 for rifampin at 10 mg/liter versus rifampin at 10 mg/liter + L-Name and for rifampin at 50 mg/liter versus rifampin at 50 mg/liter + L-NAME) (n = 3).
DISCUSSION
This report demonstrates that rifampin increases the cytokine-induced production of NO and IL-8 and the constitutive release of IL-8 and IL-1β in human hepatic epithelial HepG2 cells. In cytokine array screening assays, incubation of the cells with rifampin alone was associated with increased production of IP-10 and increased levels of cytokine-stimulated RANTES, both of which are proinflammatory. Interestingly, like IL-8, both IP-10 and RANTES belong to the family of chemotactic cytokines (chemokines) that recruit leukocytes to inflammatory sites.
Rifampin increased the production of NO in the HepG2 cells by increasing iNOS mRNA transcription and iNOS protein expression. We previously noted similar findings for A549 and H-27 cells (31, 33). Furthermore, although rifampin upregulated the constitutive release of IL-8 at the transcriptional level, its addition to the cytokine mix had no significant effect on cytokine-induced IL-8 mRNA expression. Previous studies have reported that IL-8 production is both transcriptionally and posttranscriptionally controlled (11). Bérubé et al. (2) observed that, in human bronchial epithelial cells activated by TLR pathways, the p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase 1 (ERK1)-ERK2 pathways regulated IL-8 production posttranscriptionally, whereas in HeLa cells stimulated by IL-1β, the role of the p38 MAPK pathways was confined to transcription only. They concluded that different agonists may activate distinct pathways of regulation. Our results further demonstrated the complex regulation of IL-8 production.
Interestingly, the strongest augmentation of IL-8 levels occurred with the addition of rifampin at 10 mg/liter, and higher concentrations were less efficient.
Concomitant incubation of the cells with rifampin and the NOS inhibitor L-NAME increased the concentration of IL-8 and its mRNA expression, suggesting that NO inhibits rifampin-induced IL-8 production. In previous studies, NO either increased or decreased IL-8 production, depending on the experimental model. Using NOS inhibitors and NO donors, Gilchrist and Befus (7) found that NO inhibited IL-8 production and mRNA expression in phorbol myristate acetate (PMA)-stimulated human mast HMC1 cells. Similarly, Marion et al. (20) reported that the inhibition of NO production was associated with a significant increase in IL-8 production and IL-8 mRNA expression in the human intestinal epithelial HCT-8 cell line. By contrast, Mühl et al. (22) reported that NO augmented IL-8 release from monocystic U937 cells, and Ma et al. (17) reported that NO posttranscriptionally upregulated IL-8 expression in a human monocytic THP-1 cell line. Apparently, IL-8 induction is an intricate process that is differently regulated in different cells by different inducers (2). In our model, the inhibitory effect of NO on IL-8 production explains why the concentration of IL-8 induced by rifampin at 10 mg/liter was higher than that induced by rifampin at 50 mg/liter; indeed, in the presence of L-NAME, the amounts of IL-8 were interchanged. Although we did not find NO in the supernatant of cells treated with rifampin alone, there was a low level of iNOS mRNA, indicating that undetectable levels of NO may have been present. Also, we cannot rule out the possibility of the presence of undetectable amounts of NO produced by endothelial NOS. It should be noted that, in the study by Toell et al. (28), a PXR binding site was found to be present in the promoter of human iNOS; thus, induction of NO by rifampin most probably occurs in various other cell types in addition to that reported by us (32, 34).
Both NO and IL-8 exert proinflammatory effects in the liver. Cumulative evidence suggests that, in accordance with the amount of NO and the kind of insult, the increase in the level of NO in the liver may be either protective or harmful (5, 9, 16). It is generally accepted that prolonged or massive production of NO can lead to hepatic inflammation and tumor development. Although the mechanism of rifampin toxicity in the liver is unknown (29), its administration to rats together with isoniazid led to hepatotoxicity accompanied by a significant increase in total liver nitrite, and the administration of drugs that significantly ameliorated the levels of serum hepatic marker enzymes also decreased liver NO levels (26). IL-8 has been implicated in neutrophil-mediated tissue injury and organ failure, conditions of reperfusion injury and septic shock, and chronic diseases of the liver (1, 2, 11). It remains unclear, however, whether an increase in IL-8 production contributes to the hepatic damage induced by rifampin. One study reported lymphocytic infiltration in a patient who died of rifampin hepatotoxicity (29). Although HepG2 cells are widely used as a reliable in vitro model for the study of drug-induced hepatotoxicity (23), further investigation is required to determine whether rifampin also augments NO and IL-8 levels in vivo. If rifampin-related liver toxicity is caused, at least partly, by inflammatory processes and increased NO levels, the role of anti-inflammatory agents in reducing the toxicity should be explored, and coadministration of rifampin with other drugs that stimulate NO should be avoided.
The ability of rifampin to enhance NO production may have implications that are even more extensive. Since NO regulates expression of many genes, it can actually modulate the production of many mediators in the immune system. It is possible, therefore, that some of the reported immunomodulatory effects of rifampin stem, at least partly, from its ability to increase NO.
It is noteworthy that, in the present study, the effect of rifampin was achieved in pharmacologically relevant concentrations, since the peak concentration of rifampin in the serum following the usual 600-mg dose is 10 μg/ml, whereas in other tissue and fluids, rifampin may reach higher levels (25, 34).
In conclusion, this report demonstrated that rifampin can induce inflammatory mediators by itself and enhance cytokine-induced production of NO and IL-8 in a liver epithelial cell line. The elevation of NO production induced by rifampin may further affect the induction of other immunomodulators, which may explain the extensive impact of rifampin in the immune system.
Footnotes
Published ahead of print on 19 September 2011.
REFERENCES
- 1. Bautista A. P. 2002. Neutrophilic infiltration in alcoholic hepatitis. Alcohol 27: 17–21 [DOI] [PubMed] [Google Scholar]
- 2. Bérubé J., Bourdon C., Yao Y., Rousseau S. 2009. Distinct intracellular signaling pathways control the synthesis of IL-8 and RANTES in TLR1/TLR2, TLR3 or NOD1 activated human airway epithelial cells. Cell Signalling 21: 448–456 [DOI] [PubMed] [Google Scholar]
- 3. Blanchard J. S. 1998. The ying and yang of rifampicin. Nat. Med. 4: 14–15 [DOI] [PubMed] [Google Scholar]
- 4. Bogdan C. 2001. Nitric oxide and the immune response. Nat. Immunol. 2: 907–916 [DOI] [PubMed] [Google Scholar]
- 5. Chen T., Zamora R., Zuckerbraun B., Billiar T. R. 2003. Role of nitric oxide in liver injury. Curr. Mol. Med. 3: 519–526 [DOI] [PubMed] [Google Scholar]
- 6. Dong W., et al. 1998. Cytokine expression in hepatocytes: role of oxidant stress. J. Interferon Cytokine Res. 18: 629–638 [DOI] [PubMed] [Google Scholar]
- 7. Gilchrist M., Befus A. D. 2007. Interferon-γ regulates chemine expression and release in the human mast cell line HMC1: role of nitric oxide. Immunology 123: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giustizieri M. L., Albanesi C., Scarponi C., De Pita O., Girolomoni F. 2002. Nitric oxide donors suppress chemokine production by keratinocytes in vitro and in vivo. Am. J. Pathol. 161: 1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harbrecht B. G., Billiar T. R. 1995. The role of nitric oxide in Kupffer cell-hepatocyte interactions. Shock 3: 79–87 [PubMed] [Google Scholar]
- 10. Herr A. S., Wochnik G. M., Rosenhagen M. C., Holsboer F., Rein T. 2000. Rifampicin is not an activator of glucocorticoid receptor. Mol. Pharmacol. 57: 732–737 [DOI] [PubMed] [Google Scholar]
- 11. Hoffmann E., Dittrich-Breiholz O., Holtmann H., Kracht M. 2002. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 72: 847–855 [PubMed] [Google Scholar]
- 12. Jaffuel D., et al. 1999. Rifampicin is not an activator of the glucocorticoid receptor in A549 human alveolar cells. Mol. Pharmacol. 55: 841–846 [PubMed] [Google Scholar]
- 13. Kleinert H., Schwarz P. M., Forstermann U. U. 2003. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 384: 1343–1364 [DOI] [PubMed] [Google Scholar]
- 14. Kwon S., George S. C. 1999. Synergistic cytokine-induced nitric oxide production in human alveolar epithelial cells. Nitric Oxide 3: 348–357 [DOI] [PubMed] [Google Scholar]
- 15. Labro M.-T. 2005. Anti-inflammatory activity of ansamycins. Expert Rev. Anti Infect. Ther. 3: 91–103 [DOI] [PubMed] [Google Scholar]
- 16. Li J., Billiar T. R. 1999. Nitric oxide. IV. Determinants of nitric oxide protection and toxicity in liver. Am. J. Physiol. 276: G1069–G1073 [DOI] [PubMed] [Google Scholar]
- 17. Ma P., et al. 2004. Nitric oxide post-transcriptionally up-regulates LPS-induced IL-8 expression through p38 MAPK activation. J. Leukoc. Biol. 76: 278–287 [DOI] [PubMed] [Google Scholar]
- 18. MacMicking J. D., et al. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94: 5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Majano P. L., et al. 2004. N-acetyl-cysteine modulates inducible nitric oxide syntase gene expression in human hepatocytes. J. Hepatol. 40: 632–637 [DOI] [PubMed] [Google Scholar]
- 20. Marion R., et al. 2005. L-arginine modulates CXC chemokines in the human intestinal epithelial cell line HCT-98 by the NO pathway. Biochimie 87: 1048–1055 [DOI] [PubMed] [Google Scholar]
- 21. Mlambo G, Sigola L. B. 2003. Rifampicin and dexamethasone have similar effects on macrophage phagocytosis of zymosan, but differ in their effect on nitrite and TNF-α production. Int. Immunopharmacol. 3: 513–522 [DOI] [PubMed] [Google Scholar]
- 22. Mühl H., et al. 2000. Nitric oxide augments release of chemokines from monocytic U937 cells: modulation by anti-inflammatory pathways. Free Radic. Biol. Med. 29: 969–980 [DOI] [PubMed] [Google Scholar]
- 23. Neuman M. G., et al. 1999. Inducers of cytochrome P450 2E1 enhance methotrexate-induced hepatocytotoxicity. Clin. Biochem. 32: 519–536 [DOI] [PubMed] [Google Scholar]
- 24. O'Kane C. M., Boyle J. J., Horncastle D. E., Elkington P. T., Friedland J. S. 2007. Monocyte-dependent fibroblast CXCL8 secretion occurs in tuberculosis and limits survival of mycobacteria within macrophages. J. Immunol. 178: 3767–3776 [DOI] [PubMed] [Google Scholar]
- 25. Pahlevan A. A., Wright D. J., Bradley L., Smith C., Foxwell B. M. 2002. Potential of rifamides to inhibit TNF-induced NF-kappaB activation. J. Antimicrob. Chemother. 49: 531–534 [DOI] [PubMed] [Google Scholar]
- 26. Saad E. I., El-Gowilly S. M., Sherhaa M. O., Bistawroos A. E. 2010. Role of oxidative stress and nitric oxide in the protective effects of alpha-lipoic acid and aminoguanidine against isoniazid-rifampicin-induced hepatotoxicity in rats. Food Chem. Toxicol. 48: 1869–1875 [DOI] [PubMed] [Google Scholar]
- 27. Tentori L., et al. 1998. Rifampin increases cytokine-induced expression of the CD1b molecule in human peripheral blood monocytes. Antimicrob. Agents Chemother. 42: 550–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toell A., Kroncke K.-D., Kleinert H., Carlberg C. 2002. Orphan nuclear receptor binding site in the human inducible nitric oxide synthase promoter mediates responsiveness to steroid and xenobiotic ligands. J. Cell Biochem. 85: 72–82 [PubMed] [Google Scholar]
- 29. Tostmann A., et al. 2008. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J. Gastroenterol. Hepatol. 23: 192–202 [DOI] [PubMed] [Google Scholar]
- 30. Yasuda K., et al. 2008. A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab. Dispos. 36: 1689–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuhas Y., Berent E., Ovadia H., Azoulay I., Ashkenazi S. 2006. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob. Agents Chemother. 50: 396–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yuhas Y., Azoulay-Alfaguter I., Berent E., Ashkenazi S. 2007. Rifampin inhibits prostaglandin E2 production and arachidonic acid release in human alveolar epithelial cells. Antimicrob. Agents Chemother. 51: 4225–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuhas Y., Berent E., Cohen R., Ashkenazi S. 2009. Roles of NF-κB activation and peroxisome proliferator-activated receptor gamma inhibition in the effect of rifampin on inducible nitric oxide synthase transcription in human lung epithelial cells. Antimicrob. Agents Chemother. 53: 1539–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ziglam H. M., Baldwin D. R., Daniels I., Andrews J. M., Finch R. G. 2002. Rifampin concentrations in bronchial mucosa, epithelial lining fluid, alveolar macrophages and serum following a single 600 mg oral dose in patients undergoing fibre-optic bronchoscopy. J. Antimicrob. Chemother. 50: 1011–1015 [DOI] [PubMed] [Google Scholar]
- 35. Ziglam H. M., Daniels I., Finch R. G. 2004. Immunomodulating activity of rifampicin. J. Chemother. 16: 357–361 [DOI] [PubMed] [Google Scholar]