

Abstract
In Escherichia coli, low-molecular-mass penicillin-binding proteins (LMM PBPs) are important for correct cell morphogenesis. These enzymes display dd-carboxypeptidase and/or dd-endopeptidase activities associated with maturation and remodeling of peptidoglycan (PG). AmpH has been classified as an AmpH-type class C LMM PBP, a group closely related to AmpC β-lactamases. AmpH has been associated with PG recycling, although its enzymatic activity remained uncharacterized until now. Construction and purification of His-tagged AmpH from E. coli permitted a detailed study of its enzymatic properties. The N-terminal export signal of AmpH is processed, but the protein remains membrane associated. The PBP nature of AmpH was demonstrated by its ability to bind the β-lactams Bocillin FL (a fluorescent penicillin) and cefmetazole. In vitro assays with AmpH and specific muropeptides demonstrated that AmpH is a bifunctional dd–endopeptidase and dd-carboxypeptidase. Indeed, the enzyme cleaved the cross-linked dimers tetrapentapeptide (D45) and tetratetrapeptide (D44) with efficiencies (kcat/Km) of 1,200 M−1 s−1 and 670 M−1 s−1, respectively, and removed the terminal d-alanine from muropeptides with a C-terminal d-Ala-d-Ala dipeptide. Both dd-peptidase activities were inhibited by 40 μM cefmetazole. AmpH also displayed a weak β-lactamase activity for nitrocefin of 1.4 × 10−3 nmol/μg protein/min, 1/1,000 the rate obtained for AmpC under the same conditions. AmpH was also active on purified sacculi, exhibiting the bifunctional character that was seen with pure muropeptides. The wide substrate spectrum of the dd-peptidase activities associated with AmpH supports a role for this protein in PG remodeling or recycling.
INTRODUCTION
Bacterial peptidoglycan (PG) is an essential and specific structural component of the cell wall which is critical for preserving cell integrity and providing a defined cell shape (29). Escherichia coli PG assembly (murein synthesis) requires the polymerization of glycan strands composed of alternating N-acetylglucosamine (NAcGlc) and N-acetylmuramic acid (NAcMur) residues and subsequent cross-linking by short peptides (28). Penicillin-binding proteins (PBPs) are a family of enzymes of common evolutionary origin responsible for the polymerization and cross-linking of PG. PBPs share the ability to bind to β-lactam antibiotics that are substrate analogues of PG constituents, the natural substrates of PBPs in vivo (6). The PBPs have been organized into three classes based on sequence similarities (9). Class A high-molecular-mass PBPs (HMM PBPs) synthesize nascent glycan chains and cross-link them (transglycosylation and transpeptidation), but class B HMM PBPs catalyze cross-linking reactions only between stem peptides (transpeptidation). Peptidoglycan remodeling, and possibly some aspects of synthesis, is mediated by class C LMM PBPs. Class C PBPs display two predominant catalytic activities in vivo: dd-carboxypeptidase activity, which removes the terminal d-alanine from muropeptides with C-terminal d-Ala-d-Ala dipeptides, and dd-endopeptidase activity, which hydrolyzes peptide bridges linking adjacent glycan strands (23). LMM PBPs are monofunctional or bifunctional dd-peptidases (12, 17, 16), but up to now it has not been clear what their roles are and which of these activities are predominant in vivo.
The ampH gene of E. coli codes for AmpH, a class C LMM PBPs of the AmpH type (23), and is included in the cluster of orthologous genes COG1680 (26), a family of genes whose products include AmpC-type β-lactamases and dd-carboxypeptidases. Although closely related to AmpC and other class C β-lactamases, AmpH did not show β-lactamase activity in a previous study (11). The phenotypes of certain (multiple) mutants suggest that although it is dispensable under laboratory conditions, AmpH might be relevant for PG metabolism and morphogenesis (11).
In this study, we wanted to define the enzymatic activities of E. coli AmpH on a broad range of purified muropeptides as well as on intact, purified sacculi. According to our results, AmpH is a bifunctional dd-endopeptidase-dd-carboxypeptidase which accepts a wide variety of muropeptides as substrates for both activities. Additionally, we have shown that AmpH appears to be processed when exported to the periplasm but remains membrane associated. The possible significance of these findings is discussed.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and enzymes.
Escherichia coli DH5α (F− lacZΔM15 recA1) and E. coli BL21(DE3) (F− ompT hsdSB[rB− mB−] gal dcm [DE3]) were used as cloning hosts, and DV900 (CS-109 Δ[ponB dacA dacB dacC dacD pbpG ampH ampC pbp4b]) (27) was used for peptidoglycan and muropeptide purification. DV900(DE3) was constructed by using a λDE3 lysogenization kit (Novagen, Merck KGaA, Darmstadt, Germany), following the manufacturer's recommendations, and was used to overexpress and purify the AmpH-ENd2 protein. Bacterial cultures were grown in one of the following media (1): Luria-Bertani (LB) medium, Super Optimal broth with catabolite repression (SOC) medium, or minimal M9 medium supplemented with 1 mM MgSO4 and 0.2% (wt/vol) Casamino Acids. Ampicillin (Amp) (100 μg/ml) and kanamycin (Kn) (30 μg/ml) were added as required.
Plasmid pGEM-T Easy vector (Promega, Madison, WI) carrying E. coli ampH (pGEM-H) was from our laboratory collection, and pET-28b(+) Knr (Novagen) was purchased from Merck Chemicals Ltd. (Nottingham, United Kingdom). Restriction enzymes were from Fermentas, Life Sciences (Madrid, Spain), and T4 DNA ligase and Pfu DNA polymerase were from Biotools B&M Labs, S.A. (Madrid, Spain). All DNA manipulations were performed using standard methods, and DNA samples were purified using a Promega Wizard Plus SV miniprep (Promega, Madison, WI) DNA purification kit. PCR DNA products were cleaned using Promega Wizard SV gel and a PCR cleanup system (Promega, Madison, WI).
Chemical reagents.
All chemicals were of analytical grade (Merck, Darmstadt, Germany). Imidazole, sodium dodecyl sulfate (SDS), and N-lauroylsarcosine sodium salt, ≥94% (Sarkosyl), were from Sigma-Aldrich (Saint Louis, MO), high-pressure liquid chromatography (HPLC)-grade methanol was obtained from Scharlau S.L. (Sentmenat, Spain), and ultrapure water (for the preparation of HPLC eluants) was generated on a Millipore Super-Q water purification system. Nitrocefin was from Oxoid (Cambridge, United Kingdom). Protein assays were performed with an RC DC protein assay (Bio-Rad, Hercules, CA).
Cloning, overexpression, and purification of E. coli AmpH.
The E. coli ampH gene (10), previously inserted forward (opposite to p-lac) into pGEM-T Easy (pGEM-H), was amplified by PCR, using the primers (Sigma-Aldrich, Saint Louis, MO) NcI-H (5′-CCATGGGCTTGAAACGTAGTCTGCT-3′), NdI-H (5′-GCCATATGGCTTTGAAACGTAGTCTGCT-3′), and RI-H (5′-TCGAATTCGAGGACGCGGGGATAACCA-3′). The resulting fragments from PCR (a 1.28-kbp NdeI-EcoRI fragment with primers NdI-H and RI-H and a 1,221-kbp NcoI-EcoRI fragment with primers NcI-H and RI-H) were purified, digested with NdeI and EcoRI and with NcoI and EcoRI, and cloned into plasmid pET28b, following the manufacturer's instructions. Vectors carrying ampH were used to transform E. coli DH5α and, after verification by DNA sequencing, were transformed into E. coli BL21(DE3), in which the induction assay was done. After transformation, competent E. coli cells were recovered in SOC medium with vigorous shaking for 1 h at 37°C; cells were then plated on LB agar plates supplemented with kanamycin (Kn) (30 μg/ml) and incubated at 37°C overnight. The recombinant proteins carried His6 tags either at both termini (AmpH-ENd2) or at the C terminus (AmpH-ENc1).
For overexpression, E. coli BL21(DE3)/p28H-ENc1 (producing AmpH-ENc1) and BL21(DE3)/p28H-ENd2 (producing AmpH-ENd2) were grown in a 30-liter fermentor (UD-30 B; Braun, Germany) in minimal M9 medium supplemented with 30 μg/ml Kn at 37°C for 1 to 2 h with vigorous agitation (220 rpm) until an optical density at 600 nm (OD600) of 0.3 was reached. Induction of protein expression was achieved by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubation for a further 2 h at 37°C. Cells were then harvested and frozen at −70°C. To purify the expressed proteins, a portion of the cell paste (5 g) was thawed and suspended in 30 ml of saline phosphate buffer (PBS; 150 mM NaCl, 2.5 mM KCl, 8 mM Na2HPO4·12H2O, and 1.45 mM NaH2PO4), pH 8.0. Cells were disrupted by two passes through a French press (American Instrument Co., Urbana, Ill.) at 20,000 lb/in2, and the lysate was centrifuged (257,000 × g, 15 min, 4°C). The resulting pellet was suspended to a final volume of 10 ml in PBS (pH 8.0) with 2% Sarkosyl and stirred in a rotary wheel for 4 h at room temperature. Insoluble material was removed by ultracentrifugation (345,000 × g, 15 min, 20°C). The supernatant (10 ml) was dialyzed three times, for 1 h each, against 1 liter of PBS–0.2% Triton X-100, pH 8.0 (solubilization buffer), at room temperature. The dialyzed supernatant was mixed with 2 M imidazole (20 mM final concentration) and with 1 ml (2 mg of protein/ml of resin) of high-density nickel 6BCL-QHNi-25 resin (ABT, Tampa, FL) that had been equilibrated with PBS–0.2% Triton X–100-20 mM imidazole, pH 8.0 (rinse buffer). The protein solution and resin were mixed in a rotary wheel for 4 h at room temperature. Unbound proteins were removed from the resin washing with 3 ml of rinse buffer three times. Proteins bound to the nickel-resin were eluted stepwise with 3 ml each of 125 mM, 250 mM, and 500 mM imidazole in solubilization buffer. Each 3-ml elution fraction was dialyzed threefold against 1 liter of solubilization buffer at room temperature for 1 h each, and then fractions were aliquoted and stored at −20°C.
Fractions containing AmpH-ENd2 or AmpH-ENc1 were analyzed by polyacrylamide gel electrophoresis (PAGE) on a NuPAGE system with Novex 10% bis-Tris gels in MOPS (morpholinepropanesulfonic acid)-SDS running buffer and SeeBlue Plus2 prestained standard molecular markers (Invitrogen, Carlsbad, CA). The gels were stained with Coomassie blue G-250, and the protein was electroblotted onto Immobilon-P transfer membranes (Millipore Corporation, Bedford, MA) by means of a Criterion blotter (Bio-Rad) by a modification of the method described previously (22). The membrane was immersed in blocking buffer (Tris-buffered saline–0.05% Tween 20 [TBS-T]) containing 3% (wt/vol) nonfat dry milk for 1 h. Immobilized proteins were probed with anti-His-tagged rabbit IgG (SC-803; Santa Cruz Biotechnology, Heidelberg, Germany), diluted 3,000-fold in TBS-T, as the primary antibody. The blot was washed three times for 5 min in TBS-T and exposed to a second antibody, goat anti-rabbit IgG–horseradish peroxidase conjugate, diluted 3,000-fold in TBS-T (GAR-HRPO 170-651; Bio-Rad, Hercules, CA), for 1 h. After a washing in TBS-T, protein was identified by luminol visualization by adding a premixed solution of 5 μl luminol [200× stock solution containing 88.6 mg luminol (A8511; Sigma-Aldrich, Saint Louis, MO) in 1 ml dimethyl sulfoxide] and 4.3 μl luciferin [218× stock solution: 10 mg d-luciferin (411400; Roche Diagnostics, Mannheim, Germany) in 2.1 ml Tris (100 mM), pH 7.8 to 8.0] in 1 ml Tris (100 mM), pH 7.8 to 8.0, to 1 ml of an H2O2 (Merck, Darmstadt, Germany) 15% solution in Tris, 100 mM, pH 7.8 to 8.0. The blot was placed between two squares of plastic wrap in contact with 1 ml of luminol solution and laid over an X-ray film for a few minutes. The peptide sequence was characterized by MALDI-TOF analysis, and the N-terminal amino acid sequence was determined with standard Edman chemistry.
In order to exclude any possible contamination of the purified protein with any putative activity from other PBPs or β-lactamases, the ENd2 protein was also overproduced and purified, by the procedure described above, from the strain DV900(DE3)-p28H-ENd2, which is defective for all known LMM PBPs.
Separation of soluble and envelope fractions from spheroplasts.
Spheroplasts were prepared by a protocol based on the procedure of Birdsell and Cota-Robles (3) from 25 ml of cell culture at an OD550 of 1.0. Cultures were harvested and cell pellets resuspended in 1 ml of 30 mM Tris-HCl (pH 8.0)–20% sucrose with 10 μl of EDTA, 0.5 M (pH 8.0), lysozyme was added to cell suspensions at a final concentration of 250 μg/ml, and the suspensions were kept on ice for 10 to 20 min. After addition of CaCl2 (20 mM final concentration) and NaCl (100 mM final concentration), samples were centrifuged at 40,000 × g for 15 min at 4°C. The supernatants constituted the periplasmic fraction and were stored. The pellets were resuspended in low-osmotic-strength buffer (0.5 ml of 30 mM Tris-HCl [pH 8.0], 5 mM MgCl2) with DNase (1 μg/ml) to favor disruption of spheroplasts and centrifuged at 40,000 × g for 30 min at 4°C. Supernatant (cytoplasmic fraction) was recovered and stored at 4°C, and pellets (membrane fraction) were resuspended in 50 μl of 30 mM Tris-HCl (pH 8.0) and 1 mM EDTA. Aliquots of periplasmic (15 μl), cytoplasmic (5 μl), and membrane (1 μl) fractions were analyzed by SDS-PAGE in 10% acrylamide gels, and AmpH-ENd2 and AmpH-ENc1 were identified by Western blot immunodetection. Aliquots of the membrane fraction were washed with KCl, NaCl, and LiCl at final concentrations of 0.5 and 1 M to attempt dissociation of His-tagged AmpH forms from cell membranes.
β-Lactamase assay.
β-Lactamase activity was assayed using the chromogenic substrate nitrocefin as described by O'Callaghan et al. (19). Nitrocefin stock solution (500 μg/ml) was made in PBS, pH 8.0. Samples with 125 μl (25 μg, 4 μM final concentration) of the purified AmpH-ENc1 or AmpH-ENd2 fraction or 20 μl of a pure stock solution of AmpC (0.1 μg, 1.7 × 10−2 μM final concentration) were added to 25 μl of nitrocefin stock solution (160 μM final concentration) in a final volume of 150 μl of PBS, pH 8.0, and incubated at 37°C; samples were taken every 5 min for 1 h. Parallel samples without enzyme were used as blanks for each sample. The change in absorbance at 482 nm over time was measured on a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA).
β-Lactam binding assays.
The in vitro assays for PBP activity were based on modifications of the procedures described by Spratt and Pardee (24) and Koyasu et al. (14). Membranes of E. coli BL21/p28H-ENd2 and E. coli CS-109 were prepared as described before. Membranes from E. coli CS109 were used as a standard for the molecular weights of E. coli PBPs. Protein concentration was determined with a D-C protein assay kit (Bio-Rad, Hercules, CA) and adjusted to 5 mg/ml in PBS, pH 8.0. Purified AmpH-ENc1 (9.75 μg/15 μl) was preincubated with 4 μl of cefmetazole (20-μg/ml final concentration) at 37°C for 5 min, and then 3 μl of Bocillin FL (Invitrogen, Carlsbad, CA) was added (10 μM final concentration) in a final volume of 40 μl, and the mixtures were incubated for 30 min at 37°C. SDS sample buffer (10 μl) was added, and samples were boiled for 10 min. Insoluble materials were removed by centrifugation (Eppendorf centrifuge at maximum speed for 10 min at 20°C). Proteins in the sample were separated by SDS-PAGE in 8% acrylamide gels and detected directly on the gels on a Thyphon 9410 variable-mode imager (General Electric) at 588 nm, with a 520BP40 emission filter.
Preparation of peptidoglycan.
Peptidoglycan was prepared from cultures of E. coli DV900 (a mutant with deletions of eight PBPs and AmpC and depleted of dd-carboxypeptidase activity) grown in LB medium at 37°C with aeration. The cells from a 1-liter culture were harvested by centrifugation for 15 min at 4,300 × g at room temperature, resuspended in 20 ml of culture medium, and slowly mixed with an equal volume of 8% (wt/vol) boiling SDS with vigorous stirring. The suspension was boiled for 4 h and left overnight with moderate stirring at room temperature. Sacculi were concentrated by centrifugation for 15 min at 265,000 × g. The pellet was washed with water until no SDS was detected by the method of Hayashi (10). The last pellet of the washing procedure was suspended in 10 ml of 10 mM Tris-HCl (pH 7.2) and digested first with 100 μg/ml α-amylase (EC 3.2.1.1; Sigma-Aldrich, Saint Louis, MO) for 1 h at 37°C and then with 100 μg/ml preactivated pronase E (EC 3.4.24.4; Merck, Darmstadt, Germany) at 60°C for 90 min. The enzymes were inactivated by boiling for 20 min in 1% (final concentration) SDS. The cell walls were collected by centrifugation as described above and washed three times with water. The peptidoglycan was stored in water at 4°C.
Preparation and separation of muropeptides.
Peptidoglycan was digested in 50 mM phosphate buffer (pH 4.9) with Cellosyl (Hoechst AG, Frankfurt, Germany) 100 μg/ml final concentration at 37°C overnight. The enzyme reaction was stopped by boiling the sample for 2 min in a water bath and centrifuged (Eppendorf centrifuge at maximum speed for10 min) to remove insoluble debris. The supernatant was mixed with 1/3 volume of 0.5 M sodium borate buffer (pH 9.0) and reduced with excess sodium borohydride (NaBH4) for 30 min at room temperature. The pH was tested with pH indicator strips (Acilit, Merck) and adjusted to 3 with orthophosphoric acid. All samples were filtered (Millex-GV filters; 0.22-μm pore size, 2.5-mm diameter; Millipore, Cork, Ireland) and stored at −20°C.
Separation of the reduced muropeptides by HPLC (325 system; Kontron Instruments) was performed essentially by the method of Glauner et al. (7, 8). The eluted muropeptides were monitored by measuring absorbance at 204 nm (Jasco UV-1570 spectrophotometer). When required, the individual peaks were collected, vacuum dried, and stored at −20°C.
Quantification of muropeptides.
Individual muropeptides were quantified from their integrated areas using samples of known concentration as standards. Concentration of the standard muropeptides was determined as described by Work (30).
Enzymatic assay for determination of dd-carboxypeptidase activity in vitro.
Purified His-tagged AmpH forms were assayed using the tripeptide Nα,Nε-diacetyl-Lys-d-Ala-d-Ala (Sigma-Aldrich, Saint Louis, MO) as the substrate using a modification of the method of Frère et al. (5) as described by Vega and Ayala (27).
Reaction mixtures containing 10 μl of Nα,Nε-diacetyl-Lys-d-Ala-d-Ala (8.3 mM final concentration), 3 μl of 10× buffer (Tris-HCl buffer, 300 mM, pH 7.5), and 17 μl each of purified His-tagged AmpH forms (1 μg, 0.8 μM final concentration) were incubated at 37°C for 60 min. At this time, 5 μl of O-dianisidine (10 mg/ml; Sigma-Aldrich, Saint Louis, MO) (in methanol) and 70 μl of enzyme/coenzyme mix (flavin adenine dinucleotide [FAD], peroxidase, and d-amino acid oxidase) were added to each sample. After a further 5 min at 37°C, 400 μl of 50% (vol/vol) methanol-water was added, and samples were incubated for another 2 min. The absorbance of each sample at 460 nm was measured immediately. Control reaction mixtures containing only enzyme and controls for natural degradation of the tripeptide without enzyme were made for each sample. Standard samples with known amounts of d-alanine and unknown samples were tested in triplicate.
HPLC assay of E. coli AmpH dd- peptidase activities.
All enzymatic reactions were analyzed in triplicate. The dd-carboxypeptidase activity was assayed by monitoring the appearance of the monomeric reaction product disaccharide tetrapeptide, NAcGlc-NAcMur-L-Ala-d-Glu-DAP-d-Ala (M4) (where DAP is diaminopimelic acid), in mixtures containing PBS buffer (pH 7.3), enzyme (17 μg; final concentration, 2.05 μM), and various concentrations (6.3 × 10−3 to 9.5 × 10−2 mM) of monomer disaccharide pentapeptide, NAcGlc-NAcMur-L-Ala-d-Glu-DAP-d-Ala-d-Ala (M5), as a substrate in a final volume of 200 μl. Reaction mixtures were incubated at 37°C for 240 min. The dd-endopeptidase activity was determined with the dimeric compounds bis-disaccharide tetrapentapeptide (D45) and disaccharide tetratetrapeptide (D44), as well as their 1→6 anhydromuramic acid-containing derivatives D45N and D44N, respectively, as substrates. Sample mixtures consisted of 200 μl PBS (pH 7.3) and 0.4 μM enzyme (3.4 μg) at various concentrations of substrates: for D45 and D44, 4 × 10−3 to 8 × 10−2 mM, and for D45N and D44N, 1 × 10−3 to 2 × 10−2 mM. Reaction mixtures were incubated at 37°C for 15 min (for D45) or 30 min (for D44, D44N, and D45N).
To study the activity of His-tagged AmpH on macromolecular PG, reaction mixtures containing 160 μg of PG and 24.5 μg (2.17 μM) of AmpH-ENc1 in 270 μl PBS (pH 7.3) were incubated overnight at 37°C. All enzymatic reactions were terminated by boiling the samples for 2 min. Once cooled, samples were digested with muramidase (100 μg/ml, final concentration) overnight and further processed for HPLC analysis as described above. Enzyme activities were estimated from the variation in the abundance of presumed substrate and product muropeptides relative to a control sample in the HPLC analyses of digested sacculi.
Analysis of kinetic data.
The dependence of the reaction rate on concentration of substrates for the dd-peptidase activities considered here was examined under the conditions described above for concentrations in the range of 10−3 to 10−1 mM. Apparent Km and Vmax values were obtained from double-reciprocal Lineweaver-Burk plots of the data. kcat was determined as Vmax/[E0], where [E0] = nmol of protein/ml (His-tagged AmpH) (18). Graphical and statistical analyses were performed using Microsoft Excel (Microsoft Inc., Redmond, WA).
RESULTS
Cloning, overexpression, and identification of AmpH.
Upon induction with 1 mM IPTG, the E. coli BL21 derivatives containing the plasmids p28H-ENd2 (producing AmpH-ENd2, containing His6 tags at both termini) and p28H-ENc1 (producing AmpH-ENc1, containing a His6 tag at the C terminus) overexpressed a single protein that migrated to a position around 45 kDa as determined by SDS-PAGE (Fig. 1, lanes 2 and 4) in accordance with the molecular masses of 46.39 kDa and 44.22 kDa expected from the known DNA sequences of the fusion proteins, respectively. Furthermore, the DNA sequences of the 1.28-kb EcoRI-NdeI (AmpH-ENd2) and 1.22-kb EcoRI-NcoI (AmpH-ENc1) fragments were identical to the sequence of E. coli ampH deposited in GenBank (accession number AAC73479). MALDI-TOF analyses of purified His-tagged AmpH-ENd2 and AmpH-ENc1 peptide sequences determined that both clones contain a major fragment of a 41.86-kDa protein with a pI value of 9.33 calculated by the Mascot program. This molecular mass is compatible with the mobility of the protein band in SDS-PAGE (Fig. 2) but is smaller than the 46.393-kDa and 44.215-kDa theoretical masses calculated from the nucleotide sequences of AmpH-ENd2 and AmpH-ENc1, respectively. However, that mass correlated precisely with the band observed by Western blotting of the purified proteins extracted from the membrane of the overproducing strain (Fig. 2), suggesting that both proteins, having a cleavable sequence, are actually cleaved at that site. Liquid chromatography-tandem mass spectrometry confirmed the mass loss of a 23-amino-acid peptide (MGLKRSLLFSAVLCAASLTSVHA) at the N terminus of AmpH-ENc1. Although the final products for both fusion proteins were identical, we recovered mainly the mature form (His labeled at the C terminus) for ENc1 protein but most often got two bands for ENd2, a major component corresponding to the mature form (labeled at the C terminus) and a second band with the expected size of the precursor (His labeled at both termini). We speculate that presence of the His6 tag at the N terminus might slow maturation of the protein. Mapping of the C terminus showed that it remained intact in both proteins. Although both His-tagged forms of AmpH seem to be cleaved at the amino terminus, the mature protein remains membrane associated (see below). However, inspection of the amino acid sequence did not reveal potential hydrophobic membrane-anchoring sequences other than the N-terminal signal peptide.
Fig. 1.
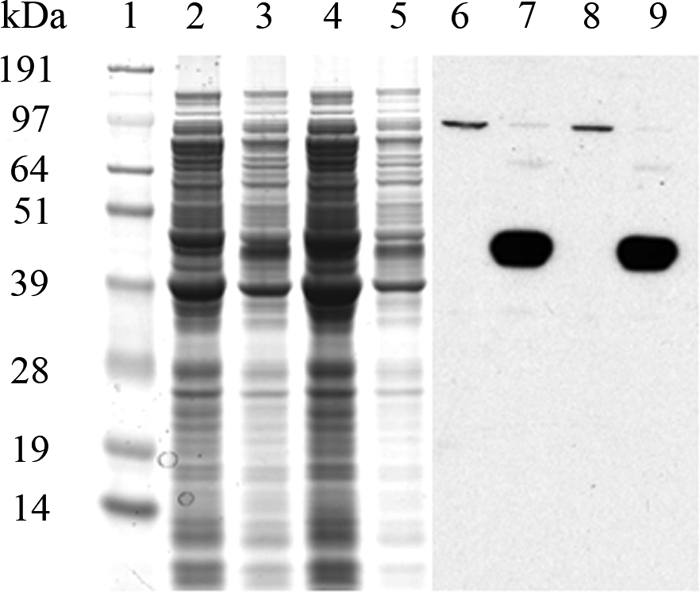
SDS-PAGE analysis of overexpressed His-tagged AmpH from E. coli BL21/p28H. Cells were grown in minimal M9 medium and induced with 1 mM isopropyl-β-d-thiogalactopyranoside as described in Materials and Methods. Lane 1, prestained protein molecular weight standards (SeeBluePlus2); lanes 2, 3, 4, and 5, Coomassie blue staining of cell extracts from 50 μl of culture from noninduced (lane 2) and induced (lane 3) BL21/p28H-ENd2 and noninduced (lane 4) and induced (lane 5) BL21/p28H-ENc1; lanes 6, 7, 8, and 9, Western blot analysis using anti-His tag antibodies to detect the corresponding noninduced (lanes 6 and 8) and induced (lanes 7 and 9) samples, performed simultaneously.
Fig. 2.

Purified His-tagged AmpH from E. coli BL21/p28H-ENd2. Cells overproducing AmpH-ENd2 were analyzed by Coomassie-stained SDS-PAGE (A) and Western blotting (B) as described in Materials and Methods. Lane 1, prestained protein molecular weight standards (SeeBlue Plus2); lanes 2 and 8, total membrane fraction treated with 2% Sarkosyl; lanes 3 and 9, soluble fraction; lanes 4 and 10, flowthrough from the nickel affinity column; lanes 5 and 11, first eluted fraction with 125 mM imidazole; lanes 6 and 12, second eluted fraction with 250 mM imidazole; lanes 7 and 13, third eluted fraction with 500 mM imidazole. Molecular masses are on the right.
Membrane localization of AmpH.
Exponentially growing cultures of E. coli BL21/p28H clones overexpressing AmpH-ENd2 and AmpH-ENc1 were subjected to cell fractionation to separate periplasmic, cytoplasmic, and membrane fractions and determine His-tagged AmpH location. As shown in Fig. 3, Western blot analysis of the fractions indicated that His-tagged AmpH-ENd2 derivatives were mostly associated with the membrane fractions but were essentially absent in both the periplasmic and the cytoplasmic fractions. Furthermore, AmpH derivatives could not be extracted when membrane fractions were washed with NaCl, KCl, or LiCl at high concentrations (0.5 M to 1 M) (data not shown), and only in the presence of strong detergents (1% SDS or 2% sodium sarcosylate) could AmpH be removed from the membranes (Fig. 4). The same results were obtained for AmpH-ENc1 (data no shown).
Fig. 3.
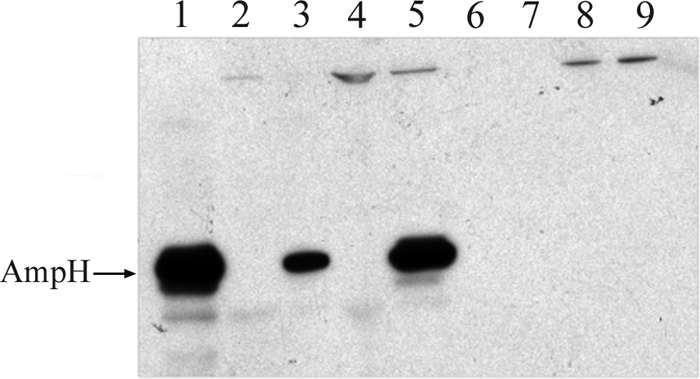
Western blot analysis of His-tagged AmpH in cellular fractions from overexpressed E. coli spheroplasts. Membrane, cytoplasm, and periplasm fractions from E. coli BL21/p28H-ENd2 induced by isopropyl-β-d-thiogalactopyranoside were prepared as described in Materials and Methods. Lane 1, purified AmpH-ENd2 extract (lane 1); lanes 2 and 3, noninduced (lane 2) and induced (lane 3) total spheroplasts; lanes 4 and 5, noninduced (lane 4) and induced (lane 5) membrane fractions; lanes 6 and 7, noninduced (lane 6) and induced (lane 7) periplasmic fractions; lanes 8 and 9, noninduced (lane 8) and induced (lane 9) cytoplasmic fractions.
Fig. 4.
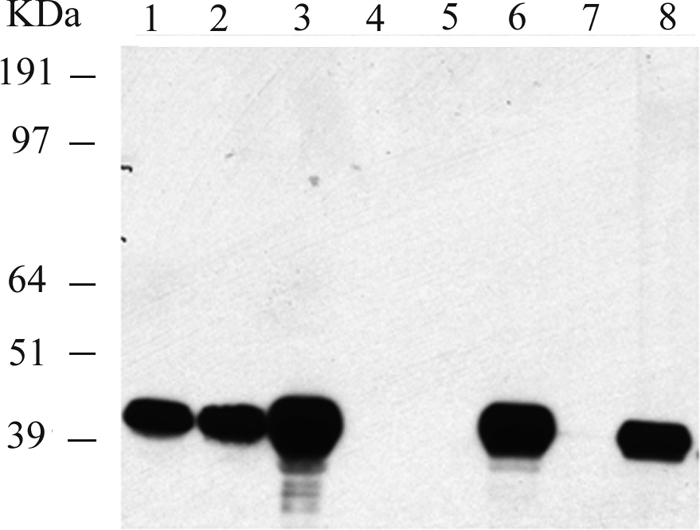
Western blot analysis of His-tagged AmpH in isolated and detergent-extracted membranes from E. coli BL21/p28H-ENd2. Cells overexpressing AmpH-ENd2 were disrupted with a French press. The resulting cell membrane pellet fraction was treated with 1% SDS and 2% Sarkosyl followed by ultracentrifugation to recover membrane pellet and soluble fraction. Lane 1, whole cells overexpressing AmpH-ENd2; lane 2, total cells disrupted with a French press; lane 3, resulting membrane cell pellet; lane 4, supernatant fraction; lanes 5 and 6, membrane pellet and soluble fraction after 1% SDS treatment; lanes 7 and 8, membrane pellet and soluble fraction after 2% Sarkosyl treatment. Positions of the protein standards are shown on the left.
β-Lactam-binding capacity of purified AmpH.
Membrane extracts from induced E. coli BL21/p28H-ENc1 cells with 1 mM IPTG and its corresponding purified AmpH-ENc1 form were used in a binding assay with fluorescent antibiotic. The four major PBPs of E. coli (PBP1a/b, PBP2, PBP3, and PBP5) were easily detected by SDS-PAGE, as well as a new, intense band matching the molecular weight calculated for the AmpH derivatives (Fig. 5). Cefmetazole is a β-lactam with high affinity for the LMM PBPs of E. coli. To confirm the β-lactam binding ability of AmpH-ENc1, cefmetazole was used as a competitor for Bocillin FL in competition assays. Indeed, preincubation of His-tagged AmpH-containing samples with 20 μg/ml of cefmetazole abolished binding to Bocillin FL (Fig. 5). The two bands corresponding to AmpH-ENd2, and overlapping PBP5 in lane 1 of Fig. 5, are most probably due to partial cleavage of the signal peptide, producing both precursor and mature forms.
Fig. 5.
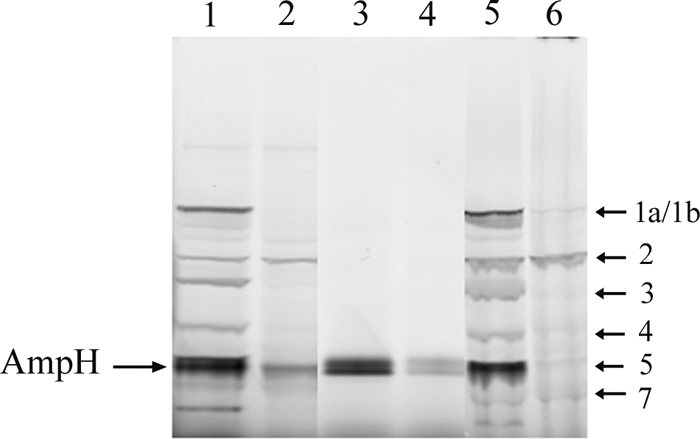
SDS-PAGE analysis of Bocillin FL binding of membrane-bound and purified His-tagged AmpH. Bocillin FL binding assays were done with 35 μg of membrane extracts (lanes 1 and 2) and with 0.65 μg of purified AmpH-ENd2 (lanes 3 and 4) obtained from induced E. coli BL21/p28H-ENd2 cultures. Membranes from E. coli CS109 (lanes 5 and 6) were used as standards for PBP molecular weights. For lanes 2, 4, and 6, reaction mixtures were preincubated with 20 μg/ml cefmetazole. The major and minor bands of the PBP profile are indicated on the right.
Enzymatic activity of AmpH.
Because AmpH is closely related to the class C β-lactamases, we tested purified His-tagged AmpH forms for β-lactamase activity, with purified AmpC as a reference. AmpH displayed a clearly positive, although reduced, level of β-lactamase activity in assays using the chromogenic β-lactam nitrocefin as the substrate (1.4 × 10−3 nmol/μg protein/min), about 1/1,000 the rate for AmpC (8.7 × 10−1 nmol/μg protein/min) under identical conditions.
The potential dd-peptidase activities of His-tagged AmpH derivatives were studied by monitoring the effect of the protein on a series of purified muropeptides. Initial assays indicated that AmpH derivatives exhibited both dd-carboxypeptidase activity, as they were able to convert M5 and M5N into M4 and M4N, respectively, and dd-endopeptidase activity, because they cleaved the dd-peptide bridge in cross-linked muropeptides, releasing the monomeric subunits (Fig. 6). Once the activities were confirmed, a more detailed analysis was performed with AmpH-ENc1 and a number of substrates to define both the specificity of each reaction and the basic kinetic parameters. In all instances, the enzyme activity followed saturation kinetics and could be fitted to double-reciprocal Lineweaver-Burk plots to determine the apparent Km, Vmax, and kcat by nonlinear regression.
Fig. 6.

HPLC analysis of His-tagged-AmpH dd-peptidase activities on muropeptides. (A) The dd-endopeptidase activity of AmpH-ENc1 (0.28 μM) was assayed by monitoring the appearance of the monomeric compounds M4 (NAcGlc-NAcMur-L-Ala-d-Glu-DAP-d-Ala) and M5 (NAcGlc-NAcMur-L-Ala-d-Glu-DAP-d-Ala-d-Ala) in mixtures containing dimer D45 as the substrate. (B) The dd-carboxypeptidase activity of AmpH-ENc1 (1.38 μM) was assayed by monitoring the appearance of M4 in mixtures containing M5 as the substrate. Reactions were performed in the presence of cefmetazole (CF) 40 μM (a and d) or without CF (b and e) as described in Materials and Methods. Control samples with muropeptides without enzyme (c and f) were incubated simultaneously under the same conditions.
dd-Carboxypeptidase activity was measured with the natural substrate M5 and the synthetic tripeptide Nα,Nε-diacetyl-Lys-d-Ala-d-Ala, often used as an alternative dd-carboxypeptidase substrate (27, 13). AmpH-dd-carboxypeptidase had a low turnover number and a low specificity constant for M5 (Table 1). The activity of His-AmpH on the synthetic tripeptide was (6 ± 0.8) × 10−3 nmol of d-Ala/μg protein/min, which was comparable to the rates obtained using the complete muropeptide M5 as the substrate ([4.98 ± 0.48] × 10−3 nmol/μg protein/min), as shown in Table 1.
Table 1.
Kinetic of E. coli His-tagged AmpH dd-peptidase activitiesa
| Enzymatic activity | Substrate | Km (μM) | Vmax [nmol min−1 μg protein−1] | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|---|
| dd-Carboxypeptidase | M5 | 225 ± 35 | (4.98 ± 0.48) × 10−3 | 3.4 × 10−3 | 15 |
| Tripeptide | ND | (6 ± 0.8) × 10−3 | ND | ND | |
| dd-Endopeptidase | D45 | 102 ± 5 | (174 ± 15.6) × 10−3 | 1.2 × 10−1 | 1.2 × 103 |
| D45N | 28.7 ± 7.5 | (27.6 ± 9) × 10−3 | 1.9 × 10−2 | 6.6 × 102 | |
| D44 | 134 ± 25 | (162 ± 15.6) × 10−3 | 9 × 10−2 | 6.7 × 102 | |
| D44N | 31.6 ± 1.5 | (23.4 ± 3) × 10−3 | 1.6 × 10−2 | 5 × 102 |
dd-Carboxypeptidase activity in vitro on tripeptide (Nα,Nε-diacetyl-Lys-d-Ala-d-Ala) as the substrate was measured as described in Materials and Methods. All kinetic constants were calculated using data obtained with 2.05 μM purified His-tagged AmpH extract with various amounts (6.3 × 10−3 to 9.5 × 10−2 mM) of monomer dissacharide pentapeptide (M5). All kinetic constants of dd-endopeptidase activity were calculated using data obtained with 0.4 μM purified His-tagged AmpH extract and various amounts (4 × 10−3 to 8 × 10−2 mM) of dimers dissacharide tetrapentapeptide (D45) or dissacharide tetratetrapeptide (D44) and various amounts of the analogous (1→6)anhydro compounds D45N and D44N (1 × 10−3 to 2 × 10−2 mM) as described in Materials and Methods. The enzymatic reactions were analyzed by HPLC assay as described in Materials and Methods. All kinetic constants must be considered apparent values because of the impossibility of calculating initial enzyme velocities by HPLC. Values are means ± standard deviations. ND, not determined.
To gain further insight into the properties of His-tagged AmpH dd-endopeptidase activity, the cross-linked dimers D44 and D45 and their anhydro derivatives D44N and D45N were assayed as substrates to define the basic kinetic parameters (Table 1). The kinetics of E. coli AmpH-ENc1 dd-endopeptidase activity showed higher efficiencies for dimers D45 and D44 than for D45N and D44N, respectively. Interestingly, the presence or absence of d-Ala at position 5 in the acceptor stem peptide had little or no influence on endopeptidase activity, but the presence of anhydromuramic acid strongly affected the reaction. Cross-linked trimers and tetramers were also substrates for both E. coli AmpH-ENc1 and AmpH-ENd2 (data not shown), but detailed data could not be gathered because of the difficulty of obtaining appropriate amounts of these muropeptides. Both dd-peptidase activities were inhibited by 40 μM cefmetazole (20 μg/ml) with isolated muropeptides as substrates (Fig. 6), in accordance with the results of penicillin binding experiments reported above.
In order to exclude any contamination from other LMM PBPs, we cloned, expressed, and purified the ENd2 protein using a strain lacking all LMM PBPs and β-lactamases [DV900(DE3)]. Analysis of the three enzymatic activities (dd-endopeptidase, dd-carboxypeptidase, and β-lactamase) on this preparation produced the same or equivalent results ([2.94 ± 0.40] × 10−3 nmol/μg protein/min on M5, [134 ± 40] × 10−3 nmol/μg protein/min on D45, and 2.6 × 10−3 nmol/μg protein/min with nitrocefin) as first found for the protein purified from the BL21(DE3) strain, supporting the multifunctional character of AmpH.
Activity of AmpH on macromolecular peptidoglycan.
Peptidoglycan hydrolases may or may not accept sacculi as substrates. Those that do not are generally associated with peptidoglycan turnover or recycling rather than biosynthesis. Therefore, we assayed the ability of AmpH-ENc1 to accept sacculi as substrates for both activities. As sacculi of wild-type E. coli are essentially free of pentapeptides, sacculi from DV900, a multiple dd-carboxypeptidase mutant which accumulates high proportions of d-Ala-d-Ala-containing muropeptides, were used to check AmpH dd-carboxypeptidase activity. HPLC analyses showed that AmpH-ENc1 displayed both dd-peptidase activities on macromolecular PG (Table 2; Fig. 7). The proportion of monomeric pentapeptide (peak M5), dimeric compounds (peaks D45, D44, and D45N), and trimeric compounds (peaks T445 and T445N) fell drastically when PG from E. coli DV900 was incubated in the presence of His-tagged AmpH. In addition, the relative abundance of essentially all muropeptides cross-linked by dd-peptide bridges also fell upon AmpH digestion of sacculi from both strains, indicating a broad substrate specificity of the dd-endopeptidase activity and a high efficiency on macromolecular peptidoglycan. These results indicate that His-tagged AmpH exhibits both dd-endopeptidase and dd-carboxypeptidase activities on sacculi.
Table 2.
His-tagged AmpH activity on sacculi purified from E. coli strainsa
| Muropeptide | Muropeptide composition (%) |
|||
|---|---|---|---|---|
| CS109 | CS109/AmpHb | DV900 | DV900/AmpHb | |
| M3 | 19 | 18.8 | ||
| M3G | 1.3 | 2.8 | ||
| M4 | 29.2 | 43.5 | 6.34 | 46.62 |
| M3L | 4.2 | 4.8 | ||
| D34D | 5.1 | 4.9 | ||
| D43 | 10.4 | 6.2 | ||
| M5 | 37.73 | 21.27 | ||
| D44 | 19.5 | 10.7 | 6.53 | 3.08 |
| D45 | 40 | 17.58 | ||
| M4N | 0.51 | 6.05 | ||
| T444 | 2.1 | 1.6 | ||
| D43L | 8.5 | 5.8 | ||
| T445 | 7.1 | 4.05 | ||
| M5N | 0.98 | 2.04 | ||
| D44N | 0.7 | 0.9 | ||
| D45N | 0.28 | 0.13 | ||
| T445N | 0.49 | 0.16 | ||
| Total | 100 | 100 | 100 | 100 |
Relative molar abundance of muropeptides was calculated from the areas of the corresponding peaks as described previously (8). Muropeptides are abbreviated according to the following notation: the first letter indicates monomer (M), cross-linked dimer (D), or cross-linked trimer (T); the numbers indicate the length of stem peptides, where 3 stands for l-Ala-d-Glu-meso-DAP, 4 stands for l-Ala-d-Glu-meso-DAP-d-Ala, and 5 stands for l-Ala-d-Glu-meso-DAP-d-Ala-d-Ala; the last letter indicates a muropeptide cross-linked through an (ld)-DAP-DAP peptide bridge (D), a Braun's lipoprotein anchoring muropeptide (L), or a muropeptide with a (1→6)anhydromuramic acid residue (N).
AmpH dd-peptidase activities were measured in reaction mixtures with enzyme and peptidoglycan as described in Materials and Methods.
Fig. 7.

HPLC analysis of His-tagged AmpH dd-peptidase activities on macromolecular peptidoglycan. The changes in absorbance (Abs) at 204 nm of each muropeptide reaction product are displayed. (A) PG substrate from E. coli DV900 incubated with purified His-tagged AmpH enzyme as described in Materials and Methods; (B) PG substrate incubated under the same conditions without added enzyme. dd-Peptidase activity is showed by decrease of dimeric and trimeric compounds, i.e., tetrapentapeptide (D45), tetratetrapeptide (D44), tetratetrapentapeptide (T445), and analogous (1→6)anhydro-D45N and -T445N compounds. dd-Carboxypeptidase activity is displayed as a decrease of monomeric pentapeptide (M5). Both dd-endopeptidase and dd-carboxypeptidase activities display an increase of monomeric tetrapeptide (M4) and the analogous (1→6)-anhydromuramic acid-containing derivatives M4N and M5N.
DISCUSSION
A general understanding of PG structure and metabolism, and specifically the bacteriolytic effect of β-lactam antibiotics, depends on detailed knowledge of the activity of enzymes involved in murein biochemistry. β-Lactam antibiotics exert their action through inhibition of HMM PBPs responsible for the polymerization of PG. The LMM PBPs are also inhibited by β-lactams, but since they are not essential for bacterial growth, their inhibition is not usually fatal to bacteria. The seven LMM PBPs of E. coli are involved in cell division, PG maturation, or recycling, and the major biochemical activities of six of them are monofunctional or bifunctional dd-carboxypeptidases or dd-endopeptidases (2, 16, 17, 21, 27). AmpH has been associated with PG recycling, although at the time of this study, it was the only LMM PBP that remained uncharacterized for enzymatic activity. Cell fractionation studies localized both forms of His-tagged AmpH protein exclusively in the membrane fraction. Furthermore, AmpH seems to be strongly anchored to the membrane, although no canonical hydrophobic anchoring sequences were found. This situation appears to be consistent with other well-characterized LMM PBPs, whose amino termini are cleaved as the proteins mature but remain membrane associated (6).
Analysis in silico of the amino acid sequence of AmpH predicts a periplasmic protein associated with the bacterial membrane by a signal-like peptide segment that functions as a membrane anchor. However, our results show that the signal peptide is cleaved in the mature forms of both AmpH-ENd2 (both N- and C-terminally His-tagged protein) and AmpH-ENc1 (only C-terminally His-tagged protein). So, it seems that the N-terminal signal peptide functions in translocation of the protein, but once AmpH is fully translocated to the periplasm, the signal sequence is removed and then the bulk of the protein binds to the outer surface of the inner membrane.
AmpH was previously reported to be closely related to AmpC, but no β-lactamase activity was detected (11). Interestingly, PG recycling has been related to the induction of particular class C β-lactamases that hydrolyze β-lactam compounds (20). The penicillin-binding characteristics and the phenotypes of ampH mutants suggested that AmpH (and AmpC) may play roles in the synthesis, remodeling, or recycling of PG (11). Here, we analyzed β-lactamase activity of both purified His-tagged AmpH derivatives and found a low but significant activity for nitrocefin, about 1/1,000 the rate measured for AmpC under the same conditions; therefore, these data suggest that the native form of AmpH does have β-lactamase activity.
In vitro assays with isolated muropeptides showed that His-tagged AmpH forms cleave the terminal d-Ala residue from disaccharide pentapeptide (M5) and from the synthetic tripeptide Nα,Nε-diacetyl-Lys-d-Ala-d-Ala, therefore exhibiting a typical dd-carboxypeptidase activity. Activity on the synthetic tripeptide indicated that His-tagged AmpH derivatives displayed a low dd-carboxypeptidase activity with rates similar to those obtained using the complete muropeptide as a substrate, but this activity was higher than the maximum enzymatic activity (0.75 × 10−3 nmol min−1 μg−1) on synthetic tripeptides reported for LMM PBPD2 (Lmo2812) of Listeria monocytogenes under the same assay conditions (13). AmpH-ENc1 dd-carboxypeptidase activity has an apparent Km (225 μM) for M5 that is lower than the Km of PBP5, the predominant dd-carboxypeptidase in E. coli, for diacetyl-l-lysyl-d-Ala-d-Ala (Km > 1 mM) (25). However, the dd-carboxypeptidase activity associated with E. coli PBP4, the archetypal class C LMM PBP, on N-acetylmuramyl-pentapeptide, a substrate structurally closer to M5, had a Km (20.4 μM) approximately 1/10 the value for AmpH (20).
Incubation of AmpH with dd-cross-linked muropeptides clearly demonstrated that this protein has an efficient dd-endopeptidase activity, and the kinetic analysis indicated that this is the predominant activity of the protein, at least in vitro. In fact, kcat was about 10 to 100 times higher for the dd-endopeptidase than for the dd-carboxypeptidase on natural substrates. Interestingly, the dd-endopeptidase activity was essentially unaffected by the presence of a d-Ala residue at the acceptor stem peptide of dimeric muropeptides. The facts that in the in vitro assays with D45 as the substrate, the amount of D44 detected was minimal and the final amount of M5 (17.8%) was close to equimolar with M4 (18.6%) eliminate the possibility that AmpH first converts D45 into D44 and then acts on the latter. However, the presence of the (1→6)anhydro form of muramic acid seems to have a significant influence on the reaction. Indeed, the presence of the anhydro form reduced kcat to 1/10 of the value for the normal muropeptides. It is important to note that the muropeptides used here had been subjected to NaBH4 reduction and therefore contained muramicitol instead of the reducing sugar. The ability of AmpH to accept cross-linked trimers and tetramers as substrates reveals a relatively relaxed substrate specificity.
An important property of AmpH was its ability to accept intact sacculi as substrates for both dd-peptidase activities. Furthermore, our results are consistent with AmpH acting in vivo predominantly as a dd-endopeptidase, although its potential to work as a dd-carboxypeptidase was also clearly manifested when pentapeptide-enriched sacculi were used as substrates.
We therefore conclude that AmpH is a bifunctional LMM PBP with dd-carboxypeptidase and dd-endopeptidase activities on solubilized muropeptides and on whole sacculi and with a marginal β-lactamase activity. These traits suggest that AmpH may play roles in the course of PG remodeling or recycling. PBP7, the other PBP dd-endopeptidase, accepts only high-molecular-mass murein sacculi as substrates in vitro (21), and PBP4 preferentially cleaves monomer or dimer muropeptides (4), which implies a possible difference in the functions of these two enzymes. AmpH enzyme activity appears to be comparable to that of another dd-endopeptidase (MepA), a penicillin-insensitive enzyme that has been shown to cleave muropeptide dimers and insoluble murein sacculi in vitro (15). The functional overlap between PBP7, PBP4, MepA, and AmpH in vivo is unknown. From a methodological point of view, purified His-tagged AmpH proved to be a rather sturdy protein, useful for releasing the shorter glycan chains from purified sacculi.
ACKNOWLEDGMENT
This work was supported by grant BFU 2009-09200 from the Ministry of Science and Innovation (MICINN) of Spain.
Footnotes
Published ahead of print on 14 October 2011.
REFERENCES
- 1. Ausubel F. M., et al. 2002. Current protocols in molecular biology, vol. 2 John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 2. Baquero M. R., Bouzon M., Quintela J. C., Ayala J. A., Moreno F. 1996. dacD, an Escherichia coli gene encoding a novel penicillin-binding protein (PBP6b) with dd-carboxypeptidase activity. J. Bacteriol. 178: 7106–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Birdsell D. C., Cota-Robles E. H. 1967. Production and ultrastructure of lysozyme spheroplasts of Escherichia coli. J. Bacteriol. 93: 427–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clarke T. B., Kawai F., Park S.-Y., Tame J. R. H., Dowson C. G., Roper D. I. 2009. Mutational analysis of the substrate specificity of Escherichia coli penicillin binding protein 4. Biochemistry 48: 2675–2683 [DOI] [PubMed] [Google Scholar]
- 5. Frère J.-M., Leyh-Bouille M., Ghysen J.-M., Nieto M., Perkins H. R. 1976. Exocellular dd-carboxypeptidases-transpeptidases from Streptomyces. Methods Enzymol. 45: 610–636 [DOI] [PubMed] [Google Scholar]
- 6. Ghuysen J.-M. 1991. Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 45: 37–67 [DOI] [PubMed] [Google Scholar]
- 7. Glauner B., Höltje J.-V., Schwarz U. 1988. The composition of the murein of Escherichia coli. J. Biol. Chem. 263: 10088–10095 [PubMed] [Google Scholar]
- 8. Glauner B. 1988. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 172: 451–464 [DOI] [PubMed] [Google Scholar]
- 9. Goffin C., Ghysen J.-M. 1998. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62: 1079–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayashi K. 1975. A rapid determination of sodium dodecyl sulphate with methylene blue. Anal. Biochem. 67: 503–506 [DOI] [PubMed] [Google Scholar]
- 11. Henderson T. A., Young K. D., Denome S. A., Elf P. K. 1997. AmpC and AmpH, proteins related to the class C β-lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli. J. Bacteriol. 179: 6112–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holtje J. V. 1998. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62: 181–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Korsak D., Markiewicz Z., Gutkind G. O., Ayala J. A. 2010. Identification of the full set of Listeria monocytogenes penicillin-binding proteins and characterization of PBPD2 (Lmo2812). BMC Microbiol. 10: 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koyasu S., Fukuda A., Okada Y. 1984. Penicillin-binding proteins in the soluble fraction of Caulobacter crescentus. J. Gen. Microbiol. 128: 1117–1124 [Google Scholar]
- 15. Marcyjaniak M., Odintsov S. G., Sabala I., Bochtler M. 2004. Peptidoglycan amidase MepA is a LAS metallopeptidase. J. Biol. Chem. 279: 43982–43989 [DOI] [PubMed] [Google Scholar]
- 16. Meberg B. M., Paulson A. L., Priyadarshini R., Young K. D. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J. Bacteriol. 186: 8326–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nelson D. E., Young K. D. 2001. Contributions of PBP5 and dd-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J. Bacteriol. 183: 3055–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nuñez de Castro I. 2001. Cinética enzimática, p. 101–123. In Nuñez de Castro I. (ed.), Enzimología. Ediciones Pirámide, Grupo Anaya SA, Madrid, Spain [Google Scholar]
- 19. O'Callaghan C. H., Moris A., Kirby S. M., Shingler A. H. 1972. Novel method for detection of β-lactamases by using a chromogenic cephalosporin substrate. Antimicrob. Agents Chemother. 1: 283–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park J. T. 1995. Why does Escherichia coli recycle its cell wall peptides? Mol. Microbiol. 17: 421–426 [DOI] [PubMed] [Google Scholar]
- 21. Romeis T., Holtje J. V. 1994. Penicillin-binding protein 7/8 of Escherichia coli is a dd-endopeptidase. Eur. J. Biochem. 224: 597–604 [DOI] [PubMed] [Google Scholar]
- 22. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 23. Sauvage E., Kerff F., Terrak M., Ayala J. A., Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32: 234–258 [DOI] [PubMed] [Google Scholar]
- 24. Spratt B. G., Pardee A. B. 1975. Penicillin-binding proteins and cell shape in Escherichia coli. Nature 254: 516–517 [DOI] [PubMed] [Google Scholar]
- 25. Stefanova M. E., Davies C., Nicholas R. A., Gutheil W. G. 2002. pH, inhibitor, and substrate specificity studies on Escherichia coli penicillin-binding protein 5. Biochim. Biophys. Acta 1597: 292–300 [DOI] [PubMed] [Google Scholar]
- 26. Tatusov R. L., et al. 2003. The COG database: an updated version includes eukaryotes, BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vega D., Ayala J. A. 2006. The dd-carboxypeptidase activity encoded by pbp4B is not essential for the growth of Escherichia coli. Arch. Microbiol. 185: 23–27 [DOI] [PubMed] [Google Scholar]
- 28. Vollmer W., Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim. Biophys. Acta 1778: 1714–1734 [DOI] [PubMed] [Google Scholar]
- 29. Vollmer W., Blanot D., de Pedro M. A. 2008. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32: 149–167 [DOI] [PubMed] [Google Scholar]
- 30. Work E. 1957. Reaction of ninhydrin in acid solution with straight-chain amino acids containing two amino groups and its application to the estimation of αε-diaminopimelic acid. Biochem. J. 67: 416–423 [DOI] [PMC free article] [PubMed] [Google Scholar]