

Abstract
The type III secretion systems are contact-activated secretion systems that allow bacteria to inject effector proteins across eukaryotic cell membranes. The secretion apparatus, called injectisome or needle complex, includes a needle that terminates with a tip structure. The injectisome exports its own distal components, like the needle subunit and the needle tip. Upon contact, it exports two hydrophobic proteins called translocators (YopB and YopD in Yersinia enterocolitica) and the effectors. The translocators, assisted by the needle tip, form a pore in the target cell membrane, but the structure of this pore remains elusive. Here, we purified the membranes from infected sheep erythrocytes, and we show that they contain integrated and not simply adherent YopB and YopD. In blue native PAGE, these proteins appeared as a multimeric 500- to 700-kDa complex. This heteropolymeric YopBD complex could be copurified after solubilization in 0.5% dodecyl maltoside but not visualized in the electron microscope. We speculate that this complex may not be stable and rigid but only transient.
INTRODUCTION
Type III secretion systems (T3SS) are found in several plant and animal pathogens, including Salmonella, Shigella, Yersinia, enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively), Pseudomonas, and others. They confer on these pathogens the ability to deliver effector proteins into the host cell cytosol (4, 9). The T3SS, also called the injectisome or needle complex, is composed of a basal body anchored in the bacterial membranes and an external needle. This apparatus is sufficient to secrete proteins into the culture supernatant. However, the delivery of effectors into the host cell cytosol requires additional proteins called translocators, themselves exported by the T3SS (5). In animal pathogens, there are three such proteins, and they are encoded in the same large operon. Two of the translocators have hydrophobic domains (1, 11, 12) while the third one is hydrophilic. Animal pathogens endowed with a T3SS have been shown to form pores in different cell types, including erythrocytes, and even in liposomes. Formation of these native pores is strictly dependent on the presence of the three translocators (1, 12, 21, 24, 34). Osmoprotection experiments lead to an estimated pore size of around 2.3 nm (1, 6, 12, 14, 15, 18, 28, 34), which fits with the 2.5-nm inner diameter of the needle (3). In Yersinia, the hydrophilic translocator (LcrV) forms a pentameric structure at the tip of the needle (2, 19, 20). In Shigella, the needle tip structure was shown to be composed of the hydrophilic translocator IpaD and the hydrophobic translocator IpaB, proposed to exist in a 4:1 ratio (33).
Generally, only live bacteria endowed with a functional T3SS including the needle tip structure have been shown to form functional pores (8, 17, 21, 23, 31), suggesting that the needle tip could act as a scaffold for the formation of a pore by the hydrophobic translocators (19). In good agreement with this, antibodies directed against LcrV impair pore formation but not insertion of YopB and YopD in the target cell membrane (7, 10). Nevertheless, oligomeric structures have been observed after incubation of liposomes with purified hydrophobic translocators from Pseudomonas aeruginosa (27) or after incubation of sheep red blood cells (RBCs) with the supernatant (SN) of EPEC bacteria grown under secretion-permissive conditions (15). Even more, hydrophobic translocators alone were shown to synergistically form pores in the membrane of lipids vesicles (7, 25). Ivanov and colleagues (15a) could also purify “translocon complexes,” composed of YopB, YopD and YopE, from culture supernatant of Yersinia pseudotuberculosis bacteria using a soluble His-tagged variant of YopB. They determined the complex stoichiometry by densitometric scans of SDS-PAGE to be 1:2:1 for His-YopB/YopD/YopE. However, there is no direct evidence that the observed structures were similar to those inserted by live bacteria.
Attempts to get some insight on the pore stoichiometry point toward a stoichiometry in the range of six to eight translocators in EPEC (15) and six translocators, one IpaB and five IpaC, in Shigella (33). More recently, Thorslund and colleagues infected red blood cells with Y. pseudotuberculosis, and they determined the YopB/YopD ratio in erythrocyte membranes by autoradiography after protein separation by SDS-PAGE (32). The determined ratio is ∼2.4 YopD proteins per YopB.
We present here a characterization of the translocation pore formed by live Yersinia enterocolitica in sheep red blood cell membranes. We show that both YopB and YopD are integral membrane proteins and form together a complex with a mass of between 500 and 700 kDa.
MATERIALS AND METHODS
Bacterial strains, plasmids, and genetic constructs.
Y. enterocolitica strains and plasmids are listed in Table 1.
Table 1.
Vectors used in this study
| Vector | Current strain designation | Genotype or description | Source or reference |
|---|---|---|---|
| pYV plasmids | |||
| pYV40 | wt | Wild-Type virulence plasmid from Y. enterocolitica strain E40 | 29 |
| pIM417 | ΔHOPEMN | pYV40 yopE21 yopH(Δ1-325) yopO(Δ65-558) yopP23 yopM23 yopN45 | 22 |
| pCN4008 | ΔHOPEMNB | pYV40 yopE21 yopH(Δ1-325) yopO(Δ65-558) yopP23 yopM23 yopN45 yopB(Δ89-217) | 22 |
| pCAM4002 | ΔHOPEMND | pYV40 yopE21 yopH(Δ1-325) yopO(Δ65-558) yopP23 yopM23 yopN45 yopD | Catherine Mueller, unpublished |
| pCHA4002 | ΔHOPEMN-Strep-YopD | pYV40 yopE21 yopH(Δ1-325) yopO(Δ65-558) yopP23 yopM23 yopN45 Strep-10aa-yopD (pIM417 mutated with pCHA3)a | This study |
| pCHA4003 | ΔHOPEMN-AsnStrep-YopD+ | pYV40 yopE21 yopH(Δ1-325) yopO(Δ65-558) yopP23 yopM23 yopN45 Asn-Strep-10aa-yopD (pIM417 mutated with pCHA4) | This study |
| Suicide vectors and mutators | |||
| pKNG101 | oriR6K sacBR+oriTRK2 strAB+ (suicide vector) | 16 | |
| pCHA3 | pYV4016256-16013-Strep-G-G-A-G-G-A-G-G-A-G-pYV4015991-15697, constructed by overlapping PCR using the primer pair 3969/5418 and the pair 5199/5419 on pYV40 of MRS40, ligated into pKNG101 using restriction sites XbaI/SalI | This study | |
| pCHA4 | pYV4016256-16013-Asn-Strep-G-G-A-G-G-A-G-G-A-G-pYV4015991-15697, constructed by overlapping PCR using the primer pair 3969/5420 and the pair 5199/5419 on pYV40 of MRS40, ligated into pKNG101 using restriction sites XbaI/SalI. The Asn-10aa-Strep tag was cloned in frame at the N terminus of YopD | This study |
10aa, a 10-amino-acid linker used with the Strep tag.
E. coli BW19610, used for plasmid purification and cloning, and E. coli Sm10 λ pir, used for conjugation, were routinely grown on LB agar plates and in LB broth. Streptomycin was used at a concentration of 50 μg/ml to select for suicide vectors. Plasmids were generated using Phusion polymerase (Finnzymes, Espoo, Finland). The mutator pCHA4 used for insertion of a Strep-tag with an Asn residue and a 10-amino-acid (aa) linker at the N terminus of YopD was constructed by overlapping PCR using the purified pYV40 plasmid as a template. In the first PCR, primer 3969 (ATCATGGTCGACTGTGGGCAGCGGAATAACTCA) and 5420 (ACCACCAGCACCACCTTTTTCAAATTGTGGATGACTCCAATTCATTGTTATTCCTCCTTAAACTT) were used, amplifying a 340-bp fragment corresponding to the upstream 5′ region of yopD. Primer 3969 contains an SalI restriction site (underlined in the sequence). The second PCR, using primers 5199 (ATCATGTCTAGACCATTTCTCGCGCTTTACGTGCC) and 5419 (TTTGAAAAAGGTGGTGCTGGTGGTGCTGGTGGTGCTGGTACAATAAATATCAAGACAGACA) amplified a 310-bp product corresponding to the downstream 5′ region of yopD. The XbaI restriction site is underlined in the sequence of primer 5199. The third overlapping PCR was performed with the products of the two first PCR rounds as a template, using primers 5199 and 3969. The overlapping regions are italicized in primers 5419 and 5420. The last product was then cloned into XbaI and SalI restriction sites of the suicide vector pKNG101. The construct was checked by sequencing using a 3100-Avant Genetic Analyser (Applied Biosystems, Rotkreuz, Switzerland). Allelic exchange was selected by plating diploid bacteria on sucrose (16).
Growth conditions.
Bacteria were routinely grown on Luria-Bertani agar plates with 400 μM arsenite and 35 μg/ml nalidixic acid and in brain heart infusion ([BHI] Remel R452472) liquid medium with 35 μg/ml nalidixic acid. For hemolysis experiments, bacterial cultures of Y. enterocolitica bacteria were started at an optical density at 600 nm (OD600) of 0.3 in 300 ml of BHI medium and cultivated for 3 h at 37°C at 150 rpm in a dry shaker for induction of the yop regulon.
Infection of erythrocytes.
Hemolysis assay and red blood cell membrane isolation were carried out using an improved protocol, based on the assay described by Broz et al. (2). RBCs were prepared from 400 ml of sheep blood (SB) (SB070; Oxoid AG). Fifty-milliliter conical tubes containing 20 ml of SB were filled up to 45 ml with Tris-saline (TS) (30 mM Tris-Cl, 150 mM NaCl, pH 7.4), vortexed, and centrifuged for 10 min at 1,000 × g and 4°C to pellet the RBCs. The supernatant was discarded, and the RBCs were washed two more times with Tris-saline. The RBC pellets were then pooled and resuspended in 4 ml of Tris-saline. RBCs were then diluted to 6 × 109 RBCs/ml in TS-BHI medium (3:1) containing a protease inhibitor cocktail (complete EDTA free, item 11873 580 001; Roche). For the infection Y. enterocolitica bacteria were inoculated at an OD600 of 0.3 in 300 ml of BHI medium for 3 h at 37°C. Defined amounts of bacteria (multiplicity of infection [MOI] of 1) were transferred to 400-ml bottles, and the bacteria were pelleted by centrifugation at 3,220 × g for 10 min at 4°C. The supernatant was then discarded, and the bacterial pellets were resuspended with 25 ml of RBC suspension in TS-BHI medium at 3:1. The infection assay was centrifuged for 10 min at 1,000 × g and 37°C and incubated for 10 min at 37°C. It was then resuspended by vortexing, centrifuged to trigger new contact, and again incubated for 10 min at 37°C. The procedure was repeated four times in total. The release of hemoglobin was measured, and the percentage of hemolysis was calculated as described previously by (1).
RBC membrane isolation.
The RBC/bacteria pellet was resuspended by vortexing and poured into 50-ml conical tubes. It was then centrifuged for 10 min at 1,500 × g at 4°C to pellet bacteria and intact RBCs. The infected RBC membranes in the supernatant were isolated by floatation on a sucrose gradient: 15 ml of the supernatant was mixed with 13.5 g of sucrose to obtain 60% sucrose. This suspension was overlaid with 50% and 25% sucrose solutions in TS, and the sucrose density gradient was centrifuged at 15,000 × g for 16 h at 4°C in a Centrikon T-1075 ultracentrifuge (Kontron) in a AH-629 rotor (Thermo scientific) and TST 41.14 swinging bucket rotor (Kontron). The next day, the membranes were harvested at the boundaries in between the 50% and 25% sucrose solutions. The membranes were then transferred to 94-ml polyallomer tubes (Centrikon) and washed in either 50 ml of 100 mM Tris-HCl, 150 mM NaCl, and 1 mM EDTA, pH 8 (buffer S), when used for purification or in blue native (BN)-PAGE sample buffer (50 mM Bis-Tris, 50 mM NaCl, 1 mM EDTA, 5 mM 6-aminocaproic acid, 10% glycerol [vol/vol], pH 7.2). Membranes were pelleted by centrifugation at 18,000 × g for 30 min at 4°C, using a TFT 45.94 rotor (Kontron) in a Centrikon T-075 ultracentrifuge (Kontron). The membranes were washed twice using this procedure. They were then stored at −20°C or used immediately.
Purification of the pore.
Isolated infected RBC membranes were first incubated for 10 min on ice with avidin at a final concentration of 0.3 mg·ml−1. Membranes were solubilized for 1 h on a slow wheel at 4°C in 0.5% n-dodecyl-β-d-maltopyranoside ([DDM] Sol-Grade; Anatrace) in buffer S. Nonsolubilized material was then pelleted by centrifugation at 20,000 × g for 1 h at 4°C. The supernatant (SN) was then applied onto 200 μl (1 column volume [CV]) of Strep-Tactin affinity resin (IBA) overnight in a cold room with a peristaltic pump. The column was then washed four times with 5 ml of 0.05% DDM in buffer S. Elution was done with five times 0.5 CV of 100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 2.5 mM desthiobiotin, and 0.05% DDM, pH 8. Elution fractions were dialyzed against 100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 0.05% DDM, pH 8, with Slide-A-Lyser MINI dialysis units (Thermo scientific) (cutoff, 10 kDa) for 3 h at 4°C. Fifty microliters was used for SDS-PAGE and Western blot analysis.
Immunoblotting.
Immunoblotting detection was carried out using monoclonal antibodies directed against YopB (MIPA98; 1:500) or YopD (MIPA96; 1:500). Detection was performed with secondary antibody coupled to horseradish peroxidase (1:2,000; Southern Biotech) before development with ECL Plus (Amersham). When immunoblotting was performed after transfer of BN-PAGE on polyvinylidene difluoride (PVDF) membranes, incubation with primary antibodies directed against YopD (MIPA96; 1:250) and YopB (MIPA98; 1:250) was performed overnight.
BN-PAGE.
The 3 to 12% and 4 to 16% NativePAGE Novex bis-Tris gels used for blue native PAGE were purchased from Invitrogen.
The gels were run on ice in a cold room at a constant 40 V for 1 h and then at a constant 12 mA for the rest of the run (∼4 h). The anode running buffer was 50 mM bis-Tris, 50 mM Tricine, pH 6.8. Two different cathode buffers were used. For the first third of the run, the cathode buffer was 50 mM bis-Tris, 50 mM Tricine, and 0.02% Coomassie brilliant blue (CBB) G250 (Serva), pH 6.8. This buffer was then replaced by a light blue cathode buffer (50 mM bis-Tris, 50 mM Tricine, 0.002% Coomassie brilliant blue G250, pH 6.8). The run was stopped when the migration front reached the bottom of the gel. When infected RBC membranes were loaded onto these gels, the total protein concentration was first measured using a Bradford assay and normalized to 2.22 mg·ml−1 in BN-PAGE sample buffer (50 mM bis-Tris, 50 mM 6-aminocaproic acid, 1 mM EDTA, 10% glycerol, pH 7.2). Infected RBC membranes were solubilized for 15 min on ice with DDM in BN-PAGE sample buffer and then centrifuged for 30 min at 20,000 × g and 4°C. Coomassie brilliant blue (5%, wt/vol) in double-distilled H2O (ddH2O; BN-PAGE sample buffer additive) was added to 30 μl of the supernatant to reach a final concentration equal to 25% of that of the detergent. Fifteen microliters of each sample was loaded onto the gel and 15 μl of molecular weight markers (Native Mark Invitrogen).
For the elution fractions, the procedure was as follows: the pooled fractions were first concentrated on an Amicon 4-ml unit (3 kDa cutoff) for 25 min by centrifugation at 7,500 × g at 4°C. BN-PAGE sample buffer was added up to 900 μl and then concentrated again for 30 min by centrifugation at 7,500 × g and 4°C. The samples were then aliquoted (40 μl) to be directly used or frozen in liquid nitrogen and stored at −80°C for further use.
Before proteins were transferred onto PVDF membranes, the BN-PAGE gels were first incubated for 10 min at room temperature in BN-PAGE transfer buffer (25 mM Tris-Cl, 192 mM glycine, 10% methanol, 0.1% SDS). Proteins were transferred for 1 h at 25 V using a semidry electrophoretic transfer cell (Bio-Rad). After transfer, the membranes were incubated for 10 min in BN-PAGE PVDF membrane staining solution (0.2% Coomassie brilliant blue G250, 40% methanol, 7% acetic acid) and destained for 5 to 10 min in BN-PAGE PVDF membrane destaining solution (50% methanol, 1% acetic acid) until the protein marker bands were visible. The membranes were then completely destained in 100% methanol before incubation with the primary antibodies.
RESULTS AND DISCUSSION
The contact hemolysis protocol from Broz et al. (2) was adapted in order to enrich the RBC ghosts in YopB and YopD. First, the assay was scaled up, infecting 12 × 1011 sheep RBCs at once. Second, the hemolysis rate was increased by shortening but repeating the contact periods between RBCs and Y. enterocolitica bacteria (see Materials and Methods). Third, the nonlysed RBCs were not lysed by ddH2O, as described before (1, 10), but were discarded with the bacteria by low-speed centrifugation. The ghosts from lysed RBCs were pelleted, resuspended, and floated on a sucrose gradient. The floated membranes were then harvested and washed.
To assess whether YopB and YopD were inserted or peripherally associated to the membranes, the ghosts were treated with 2 M NaCl or 0.1 M Na2CO3 at pH 11.2 or with 8 M urea before they were floated on a sucrose density gradient. YopB and YopD were assayed by immunoblotting after the total protein concentration of each sample had been normalized. As shown in Fig. 1 A, neither YopB nor YopD could be extracted by salt, carbonate, or urea, indicating that they were integrated into the RBC membranes. This suggests that translocation pore features differ from one T3SS to the other, implying a stability and structure peculiar to each of them.
Fig. 1.

(A) Association of YopB and YopD to RBC membranes after contact with ΔHOPEMN bacteria. The membranes from infected RBCs were stripped or not (Untreated) with 2 M NaCl, 0.1 MNa2CO3, pH 11, or with 8 M urea and subsequently floated on sucrose gradients before SDS-PAGE and immunoblotting. The total protein concentration was normalized using a Bradford assay. The boxed area shows a solubilization test of infected red blood cell membranes with 1% DDM. The immunoblots show membranes before solubilization (Untreated) and the solubilized YopB and YopD. Even though only a small fraction of inserted YopB and YopD could be recovered, DDM was the most effective detergent to solubilize both YopB and YopD (data not shown). (B) Calibration curve of BN-PAGE (BNP) with a soluble standard protein set. The curve was fitted with a linear regression. Apparent molecular weights (in thousands) of monomers (I) and dimers (II) of YopB and YopD are plotted as a function of their calculated molecular weights (inset). Mw aa, theoretical molecular weight calculated on the basis of the protein's amino acid sequence. The data set comes from at least three independent experiments. (C) Immunoblots of anti-YopB and -YopD after BN-PAGE of membranes from RBCs infected with ΔHOPEMN bacteria. Isolated membranes were solubilized with several concentrations of DDM, and proteins were separated by BN-PAGE on a 4 to 16% gradient gel. Arrows show the monomers and dimers of YopB and YopD. (D) YopB and YopD form 500- to 700-kDa oligomers in RBC membranes. Immunoblots of anti-YopB and anti-YopD after BN-PAGE on infected RBC membranes by ΔHOPEMN bacteria solubilized with 1.5% DDM or 1% SDS are shown. The molecular weight markers (in thousands) indicate the apparent molecular weights of the observed protein bands.
To get some insight into the size of the translocation pore, we first analyzed membranes from infected RBCs by blue native polyacrylamide gel electrophoresis (BN-PAGE) on gradient gels. The BN-PAGE behaves as a molecular sieve allowing protein complexes to migrate according to their size (26). The size can be calibrated with a set of soluble markers, but binding of Coomassie brilliant blue (CBB) may vary from protein to protein, changing the apparent molecular mass. To calculate the correction factor for YopB and YopD, we first applied on BN-PAGE gels a set of soluble markers together with membranes of infected erythrocytes treated with increasing concentrations of DDM in order to obtain monomers and dimers of YopB and YopD. YopB and YopD dimers migrated at different rates, indicating that they were homodimers (Fig. 1C). Retardation factor (Rf) values for the markers (running distance of the protein divided by the total running distance on the gel) were plotted as a function of their molecular masses (Fig. 1B), and the apparent molecular masses of YopB and YopD were inferred from their Rf values, reported on the calibration curve (Fig. 1B). The correction factor (Mw in BN-PAGE/Mwaa) appeared to be very similar, around 2.1, for both proteins (Fig. 1B, inset), very close to the one that was experimentally determined by Heuberger et al. (13) for some other membrane proteins (1.8). When 1% SDS was used for solubilization instead of DDM, only one protein band could be detected for both YopB and YopD (Fig. 1D). The proteins migrated at 81 kDa and 68 kDa, respectively, corresponding to 39 kDa and 32 kDa after correction for CBB binding (Fig. 1B, inset), consistent with the size of YopB and YopD monomers and thus validating the calibration of the BN-PAGE. Floated membranes from infected RBCs were then solubilized in 1.5% DDM, subjected to BN-PAGE, and immunoblotted with anti-YopB and anti-YopD antibodies (Fig. 1D). Several protein bands appeared, and the highest molecular mass complex that could be observed was composed of both YopB and YopD. It migrated with an apparent molecular mass of 1,200 kDa, which would correspond to an ∼600-kDa complex after correction for CBB binding. This size estimation should be taken with care since the CBB/protein binding ratio could be slightly different for monomers and oligomeric complexes. Furthermore, the higher the mass of an oligomeric complex, the higher the error in its mass estimation on a gel. Therefore, we interpret that the mass of this complex should lie in between 500 to 700 kDa, which would correspond to 15 to 20 YopB/YopD subunits/monomers. This ∼600-kDa complex was the largest that we could observe, and we assumed it corresponded to the translocation pore. The second complex was a homocomplex of YopD migrating at 700,000 apparent MW, corresponding to an actual MW around 340,000, which would correspond to 10 molecules of YopD. A third protein band for YopD, below 240 kDa, could also be observed, fitting with either YopD dimers or monomers. To purify the pore complex, a sequence encoding a Strep-tag II and a 10-amino-acid linker was then introduced at the 5′ end of yopD by homologous recombination in multimutant Y. enterocolitica ΔHOPEMN bacteria. An AAT codon, encoding asparagine, was introduced at the +2 position to increase the expression level of the YopD variant (AsnStrep-YopD) (30) (Fig. 2 A). The expression level of YopD was then controlled by inducing Yop secretion by temperature shift to 37°C. As shown in Fig. 2B, AsnStrep-YopD and wild-type (wt) YopD were detected in comparable amounts in the culture supernatants of ΔHOPEMN-AsnStrep-YopD+ and ΔHOPEMN Y. enterocolitica. As shown in Fig. 2C, the hemolytic capacity of ΔHOPEMN-AsnStrep-YopD+ was comparable to that of ΔHOPEMN bacteria expressing wt YopD. Furthermore, the tag had little influence on the amount of YopB and YopD inserted into erythrocyte membranes (Fig. 2C). The calculated hemolysis rate was in the same range as the one observed with ΔHOPEMN bacteria. Bacteria expressing AsnStrep-YopD were able to permeabilize red blood cell membranes (Fig. 2C). Strep-tagged YopD could thus be used as bait for the translocation pore purification.
Fig. 2.

(A) Location of yopD on the translocator operon on the pYV plasmid. DNA and amino acid sequence of the Strep tag and the 10-amino-acid linker. The +2 codon corresponding to an asparagine is depicted in red, the Strep tag is in green, and the 10-amino-acid linker is in blue. (B) Immunoblots of anti-YopB and anti-YopD from total cell (TC) and supernatant (SN) of bacterial cultures shifted for 4 h at 37°C to induce secretion. (C) Lytic activity on red blood cells after four periods of 10 min of contact with Y. enterocolitica ΔHOPEMN, ΔHOPEMNB, ΔHOPEMND, and ΔHOPEMN-AsnStrep-YopD bacteria. One hundred percent hemolysis corresponds to RBCs lysed with 0.1% Triton X-100. The immunoblotting of anti-YopB and anti-YopD was performed on the floated infected RBC membranes.
Erythrocytes were infected with ΔHOPEMN-AsnStrep-YopD, and solubilized membranes were applied onto a Strep-Tactin column. After elution, YopD was detected, quite pure and in the nanogram range, on a silver-stained SDS-PAGE gel (Fig. 3 A). Silver staining did not show a band corresponding to YopB, but YopB could be detected by immunoblotting in the same fractions, showing copurification of the two translocators (Fig. 3B, fractions E2 to E5). This indicated, first, that the method used for purification did not disrupt the interaction in between the two translocators. Second, this could suggest that YopB was substoichiometric in the pore complex. No component of the injectisome and especially no basal body component could be detected by mass spectrometry, indicating that there was no sample contamination due to bacterial lysis upon isolation of the infected red blood membranes and further purification. The fractions containing purified YopD and YopB are thus quite pure (data not shown).
Fig. 3.
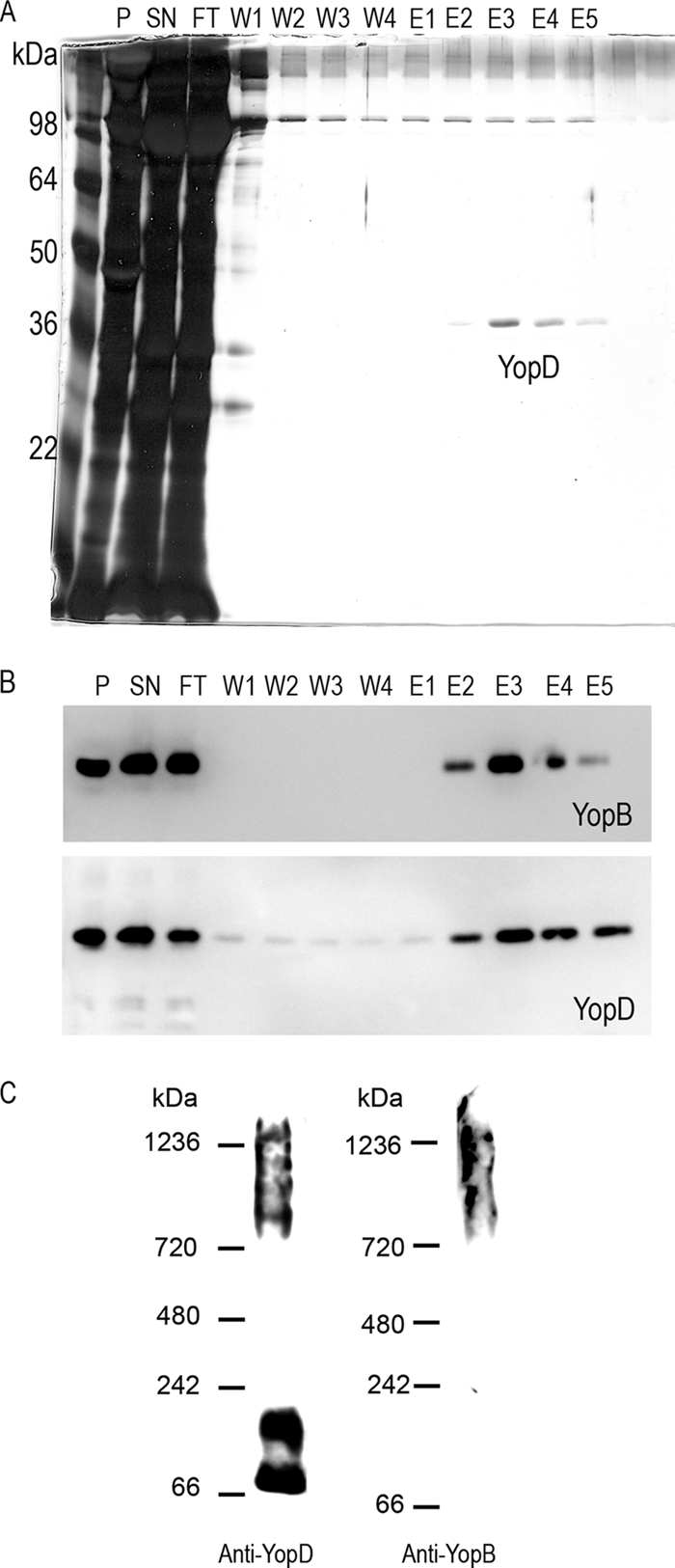
(A) Silver-stained 12% SDS-PAGE gel of fractions from Strep purification. P, pellet of unsolubilized proteins; SN, fraction applied onto the column (proteins solubilized with 0.5% DDM for 1 h at 4°C); FT, flowthrough; W1 to W4, washing steps; E1 to E5, elution fractions. (B) Immunoblots of anti-YopB and anti-YopD of the corresponding fractions. (C) Immunoblots of anti-YopB and anti-YopD after BN-PAGE on a 3 to 12% gradient gel. Elution fraction E2 was loaded onto the BN-PAGE gel.
BN-PAGE of the elution fractions followed by immunoblotting against YopB and YopD revealed that hetero-oligomeric complexes were partially preserved over the purification process (Fig. 3C). They had an apparent molecular mass ranging from ∼720 kDa to ∼1,200 kDa, corresponding to an actual molecular mass of ∼350 kDa to ∼600 kDa, which is in good correlation with what was observed with solubilized erythrocyte membranes (Fig. 1D), meaning that the purification process did preserve the YopB/YopD interaction and the complexes they form in the red blood cell membranes. Monomers and dimers of YopD could also be observed after purification (Fig. 3C) as well as after simple membrane solubilization (Fig. 1D), indicating some instability of the translocation pore.
In conclusion, in good agreement with previous work, when erythrocytes were infected by multimutant Y. enterocolitica ΔHOPEMN, both YopB and YopD could be detected in the erythrocyte membranes. In addition, we provide evidence that both proteins were integrated into the membrane and not simply associated to it, which is different from what was observed in Shigella (1). Both proteins formed large multimeric complexes with a molecular mass around 600 to 700 kDa. There is evidence that the complexes are heteropolymeric, but there is no evidence that these complexes have a fixed size and stoichiometry. Our attempts to visualize these complexes by electron microscopy did not reveal any clear structure (data not shown).
ACKNOWLEDGMENTS
This work was supported by grant 310000-313333/1 to G.R.C. from the Swiss National Science Foundation.
Footnotes
Published ahead of print on 14 October 2011.
REFERENCES
- 1. Blocker A., et al. 1999. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147:683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broz P., et al. 2007. Function and molecular architecture of the Yersinia injectisome tip complex. Mol. Microbiol. 65:1311–1320 [DOI] [PubMed] [Google Scholar]
- 3. Cordes F. S., et al. 2003. Helical structure of the needle of the type III secretion system of Shigella flexneri. J. Biol. Chem. 278:17103–17107 [DOI] [PubMed] [Google Scholar]
- 4. Cornelis G. R. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4:811–825 [DOI] [PubMed] [Google Scholar]
- 5. Cornelis G. R., Wolf-Watz H. 1997. The Yersinia Yop virulon: a bacterial system for subverting eukaryotic cells. Mol. Microbiol. 23:861–867 [DOI] [PubMed] [Google Scholar]
- 6. Dacheux D., Goure J., Chabert J., Usson Y., Attree I. 2001. Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol. Microbiol. 40:76–85 [DOI] [PubMed] [Google Scholar]
- 7. Faudry E., Vernier G., Neumann E., Forge V., Attree I. 2006. Synergistic pore formation by type III toxin translocators of Pseudomonas aeruginosa. Biochemistry 45:8117–8123 [DOI] [PubMed] [Google Scholar]
- 8. Fields K. A., Straley S. C. 1999. LcrV of Yersinia pestis enters infected eukaryotic cells by a virulence plasmid-independent mechanism. Infect. Immun. 67:4801–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galan J. E., Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573 [DOI] [PubMed] [Google Scholar]
- 10. Goure J., Broz P., Attree O., Cornelis G. R., Attree I. 2005. Protective anti-v antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J. Infect. Dis. 192:218–225 [DOI] [PubMed] [Google Scholar]
- 11. Hakansson S., Bergman T., Vanooteghem J. C., Cornelis G., Wolf-Watz H. 1993. YopB and YopD constitute a novel class of Yersinia Yop proteins. Infect. Immun. 61:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hakansson S., et al. 1996. The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 15:5812–5823 [PMC free article] [PubMed] [Google Scholar]
- 13. Heuberger E. H., Veenhoff L. M., Duurkens R. H., Friesen R. H., Poolman B. 2002. Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J. Mol. Biol. 317:591–600 [DOI] [PubMed] [Google Scholar]
- 14. Holmstrom A., et al. 2001. LcrV is a channel size-determining component of the Yop effector translocon of Yersinia. Mol. Microbiol. 39:620–632 [DOI] [PubMed] [Google Scholar]
- 15. Ide T., et al. 2001. Characterization of translocation pores inserted into plasma membranes by type III-secreted Esp proteins of enteropathogenic Escherichia coli. Cell Microbiol. 3:669–679 [DOI] [PubMed] [Google Scholar]
- 15a. Ivanov M. I., et al. 2008. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1− Yersinia pestis. Infect. Immun. 76:5181–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaniga K., Delor I., Cornelis G. R. 1991. A wide-host-range suicide vector for improving reverse genetics in gram-negative bacteria: inactivation of the blaA gene of Yersinia enterocolitica. Gene 109:137–141 [DOI] [PubMed] [Google Scholar]
- 17. Marenne M. N., Journet L., Mota L. J., Cornelis G. R. 2003. Genetic analysis of the formation of the Ysc-Yop translocation pore in macrophages by Yersinia enterocolitica: role of LcrV, YscF and YopN. Microb. Pathog. 35:243–258 [DOI] [PubMed] [Google Scholar]
- 18. Miki T., Okada N., Danbara H. 2004. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 279:34631–34642 [DOI] [PubMed] [Google Scholar]
- 19. Mueller C. A., Broz P., Cornelis G. R. 2008. The type III secretion system tip complex and translocon. Mol. Microbiol. 68:1085–1095 [DOI] [PubMed] [Google Scholar]
- 20. Mueller C. A., et al. 2005. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310:674–676 [DOI] [PubMed] [Google Scholar]
- 21. Neyt C., Cornelis G. R. 1999. Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol. Microbiol. 33:971–981 [DOI] [PubMed] [Google Scholar]
- 22. Neyt C., Cornelis G. R. 1999. Role of SycD, the chaperone of the Yersinia Yop translocators YopB and YopD. Mol. Microbiol. 31:143–156 [DOI] [PubMed] [Google Scholar]
- 23. Pettersson J., et al. 1999. The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol. Microbiol. 32:961–976 [DOI] [PubMed] [Google Scholar]
- 24. Picking W. L., et al. 2005. IpaD of Shigella flexneri is independently required for regulation of Ipa protein secretion and efficient insertion of IpaB and IpaC into host membranes. Infect. Immun. 73:1432–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Romano F. B., et al. 2011. Efficient isolation of Pseudomonas aeruginosa type III secretion translocators and assembly of heteromeric transmembrane pores in model membranes. Biochemistry 50:7117–7131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schagger H., Cramer W. A., von Jagow G. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220–230 [DOI] [PubMed] [Google Scholar]
- 27. Schoehn G., et al. 2003. Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 22:4957–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shaw R. K., Daniell S., Ebel F., Frankel G., Knutton S. 2001. EspA filament-mediated protein translocation into red blood cells. Cell Microbiol. 3:213–222 [DOI] [PubMed] [Google Scholar]
- 29. Sory M. P., Boland A., Lambermont I., Cornelis G. R. 1995. Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc. Natl. Acad. Sci. U. S. A. 92:11998–12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stenstrom C. M., Jin H., Major L. L., Tate W. P., Isaksson L. A. 2001. Codon bias at the 3′-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene 263:273–284 [DOI] [PubMed] [Google Scholar]
- 31. Tardy F., et al. 1999. Yersinia enterocolitica type III secretion-translocation system: channel formation by secreted Yops. EMBO J. 18:6793–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thorslund S. E., et al. 2011. The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS One 6:e16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Veenendaal A. K., et al. 2007. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol. Microbiol. 63:1719–1730 [DOI] [PubMed] [Google Scholar]
- 34. Warawa J., Finlay B. B., Kenny B. 1999. Type III secretion-dependent hemolytic activity of enteropathogenic Escherichia coli. Infect. Immun. 67:5538–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]