

Abstract
Helicobacter pylori is a bacterial pathogen colonizing half of the world's human population. It has been implicated in a number of gastric diseases, from asymptomatic gastritis to cancer. It is characterized by an amazing genetic variability that results from high mutation rates and efficient DNA homologous recombination and transformation systems. Here, we report the characterization of H. pylori RecA (HpRecA), a protein shown to be involved in DNA repair, transformation, and mouse colonization. The biochemical characterization of the purified recombinase reveals activities similar to those of Escherichia coli RecA (EcRecA). We show that in H. pylori, HpRecA is present in about 80,000 copies per cell during exponential growth and decreases to about 50,000 copies in stationary phase. The amount of HpRecA remains unchanged after induction of DNA lesions, suggesting that HpRecA is always expressed at a high level in order to repair DNA damage or facilitate recombination. We performed HpRecA localization analysis by adding a Flag tag to the protein, revealing two different patterns of localization. During exponential growth, RecA-Flag presents a diffuse pattern, overlapping with the DAPI (4′,6-diamidino-2-phenylindole) staining of DNA, whereas during stationary phase, the protein is present in more defined areas devoid of DAPI staining. These localizations are not affected by inactivation of competence or DNA recombination genes. Neither UV irradiation nor gamma irradiation modified HpRecA localization, suggesting the existence of a constitutive DNA damage adaptation system.
INTRODUCTION
Helicobacter pylori, a microaerophilic Gram-negative bacterium, is the most common etiologic agent of gastric diseases. It colonizes the stomach mucosa of about half of the human population, causing chronic inflammation which can lead to peptic ulcers and, in a small proportion of cases, to adenocarcinoma. H. pylori persistence during the whole life of the host and its adaptation to changes within a host or to new hosts are attributed to its extreme genetic diversity. High mutation and recombination frequencies are at the origin of this extreme variability (33). Moreover, H. pylori is naturally competent for transformation, horizontal gene transfer between unrelated strains colonizing the same host, thus contributing to its genetic diversity (8, 32). DNA uptake occurs preferentially at cell poles and depends on the type IV secretion system ComB. After processing by nucleases, DNA is internalized through the inner membrane as single-stranded DNA (ssDNA) (30). The ssDNA is then thought to be bound by DprA, which subsequently mediates RecA loading, allowing DNA integration into the recipient chromosome by homologous recombination (HR). DprA was previously shown to be essential for transformation in H. pylori (7, 17).
It is clear that HR plays a crucial role in generating the diversity that is characteristic of H. pylori (3, 11, 31). HR is also required for genome integrity maintenance, and probably because of this role, it is crucial for colonization of the host (1, 2, 16, 37, 38). Strains lacking intact recombination systems exhibit sensitivity to DNA-damaging agents, hypermutability, and impaired growth rates.
In the two most-studied bacterial models, Escherichia coli and Bacillus subtilis, two HR initiation pathways coexist (9, 15). The RecBCD (AddAB in B. subtilis) pathway is essential for the repair of double-stranded DNA breaks and for resolving regressed forks. The RecFOR pathway is needed for postreplication gap repair and for replication restart after UV damage. It was recently shown that H. pylori harbors efficient RecOR and AddAB systems to initiate DNA recombinational repair (1, 17, 18). These ubiquitous initiation pathways metabolize the DNA break to generate single-stranded DNA on which the recombinase RecA is loaded and cooperatively forms a nucleoprotein filament (6). The filament is then aligned with a homologous duplex and promotes strand exchange. The product of this reaction is a branched DNA molecule named the Holliday junction that is processed by the RuvABC complex. Orthologues of these proteins are also present in H. pylori and were shown to be important for DNA repair and mouse colonization (12, 16).
RecA is found in all eubacteria, with the exception of a few intracellular symbionts (26). This key recombination enzyme is required for DNA pairing and homologous strand exchange. In E. coli, the RecA filament can also activate autocleavage of the LexA repressor to induce the SOS response (21). This coordinated response present in many bacteria induces about 40 genes, including recA itself, in response to DNA damage. All the induced genes are involved in DNA repair or lesion tolerance. In H. pylori, where SOS seems to be absent, it was recently shown that DNA damage does not induce the expression of DNA repair genes but rather that of genes encoding proteins involved in the uptake of exogenous DNA. Surprisingly, this induction of the transformation machinery is RecA dependent since disruption of recA (hp0153) prevents induction of competence genes by DNA-damaging agents (7). The mechanism mediating this activation is unknown.
As it is the case for the E. coli protein, H. pylori RecA (HpRecA) was shown to be necessary for DNA repair. Indeed, H. pylori recA mutants are hypersensitive to DNA-damaging agents such as metronidazole or UV or ionizing radiation (18, 25, 35). As expected, intrachromosomal recombination decreases drastically and transformation by an integrative marker is totally abolished in the absence of HpRecA (18, 25). Surprisingly, neither RecOR nor AddAB inactivation reduced transformation efficiencies.
HpRecA has a predicted molecular mass of 37.7 kDa, very similar to that of E. coli RecA (EcRecA; 37.8 kDa), with which it shares 58% identity. The protein detected in H. pylori cell extracts was shown to have a 40-kDa apparent molecular mass, while EcRecA migrates as a 38-kDa protein, leading to the proposal that in H. pylori, HpRecA is posttranslationally modified by glycosylation (10, 25). This glycosylation was shown to be the result of the action of the two genes downstream of HprecA. The role of this glycosylation is not yet fully understood since, although it affects metronidazole resistance, UV survival is not impaired in its absence (10). Because the recombinant protein expressed in E. coli was insoluble, no biochemical characterization of HpRecA has been reported thus far (10, 25).
Here, we describe the purification of HpRecA and its biochemical characterization. Thus, we conclude that in vitro, the H. pylori recombinase has activities similar to those of EcRecA. In contrast, we show that expression of HpRecA is not induced following UV or gamma irradiation, but it is constitutive and leads to high levels of the protein in the cells. We also analyzed the cellular localization of HpRecA by following a tagged version of the protein.
MATERIALS AND METHODS
Oligonucleotides, enzymes, and reagents.
Oligonucleotides used in this work were from Eurogentec. Their sequences are listed in Table S1 in the supplemental material. Restriction endonucleases, DNA polymerases, and DNA-modifying enzymes were purchased from New England BioLabs (NEB) or Fermentas. EcRecA was from NEB. Culture media and antibiotics used were from AES Chemunex and Sigma-Aldrich, respectively.
Strains and growth conditions.
All Helicobacter pylori strains were in the strain 26695 background (36) and are listed in Table S2 in the supplemental material. Plate cultures were grown at 37°C under microaerophilic conditions (5% O2, 95% CO2, using the MAC-MIC system from AES Chemunex) on blood agar base medium (BAB) supplemented with 10% defibrillated horse blood (AES) and an antibiotics mix. Plates were incubated from 24 h to 5 days depending on the experiment or the mutant selected. Liquid cultures were grown at 37°C with gentle shaking under microaerophilic conditions in brain heart infusion medium (BHI) supplemented with 10% decomplemented fetal bovine serum (Invitrogen).
To generate the corresponding mutant derivatives, the gene of interest cloned into pILL570 was disrupted, leaving the 5′ and 3′ ends (300 bp) of the gene, by a nonpolar cassette carrying either kanamycin (Kn), apramycin (Apr), or chloramphenicol (Cm) resistance genes. DNA was introduced into H. pylori strains by natural transformation, and mutants were selected by growth on either 20 μg/ml Kn, 12.5 μg/ml Apr, or 8 μg/ml Cm. Allelic replacement was verified by PCR. Double or triple mutant strains were obtained by plasmid or genomic DNA transformation of the single mutant or by mixing two mutant strains together before plating the mix on double or triple antibiotic selection plates. In the case of multiple mutations, all the disruptions were verified by PCR. At least two independent clones for each construction were studied for each phenotype.
Escherichia coli strain BL21(DE3)(pLysS) (Invitrogen) was grown at 37°C in Luria-Bertani medium (LB) in the presence of 34 μg/ml chloramphenicol.
Construction of RecA-Flag fusion gene.
A fragment containing the last 300 bp of open reading frame (ORF) hp0153 and the downstream 300 bp was amplified by PCR from the strain 26695 genome with oligonucleotides Ohp153A and Ohp154B as primers. The 622-bp fragment was cloned into pJET1.2 (Fermentas), leading to a plasmid named pPR938. pPR938 was subjected to PCR analysis using oligonucleotides Osf136 and Osf137 to amplify the whole plasmid, which was disrupted before the stop codon of hp0153. The fragment obtained was digested with EcoRI and BamHI. A Flag tag was joined to the nonpolar Kan gene (27) using oligonucleotides Osf172 and Osf173 and cloned into pJET1.2. The fragment of interest was then excised with EcoRI and BamHI and purified. The plasmid and the fragment harboring the Flag tag and the Kan gene were ligated to obtain plasmid pPR943. DNA was introduced into H. pylori strains as described above. Genetic and microscopy analyses were performed on two independent clones. Allelic replacement and protein expression were verified by PCR and Western blot analysis, respectively.
Sensitivity assay.
Bacterial cells were serially diluted, and 10 μl of each dilution was spotted on BAB. For UV-induced DNA damage, cells were irradiated with 0, 15, 30, 45, or 60 J of 264 nm UV light delivering 1 J/m2/s. Gamma irradiation was performed using a 137Cs source delivering 30 Gy/min. Survival was determined as the number of cells forming colonies on plates after a given irradiation dose divided by the number of colonies from nonirradiated cells.
Natural transformation assay.
Genomic DNA (200 ng) from strain LR133 (streptomycin [Str] resistant) was mixed with 15 μl of cells resuspended in peptone water (2.5 × 105 cells) from exponential- or stationary-phase cultures. Mixes were incubated for 1 h at 37°C, and dilutions of the cells were plated on BAB with and without the appropriate antibiotic (50 μg/ml Str) and incubated for 3 to 5 days. Transformation frequency was calculated as the number of resistant colonies per recipient CFU. P values were calculated using the Mann-Whitney U test.
Cloning, expression, and purification of the recombinant HpRecA protein.
H. pylori RecA (HpRecA) was overexpressed in E. coli strain BL21(DE3)(pLysS) (Invitrogen) as an N-terminal maltose-binding protein (MBP)-tagged protein. For this purpose, ORF hp0153 was amplified from H. pylori 26695 genomic DNA using the Osf107 and Osf158 primers and the amplification product was cloned into pMALc2x (NEB). E. coli BL21(pLysS) cells transformed with pMALc2x-HprecA were grown in LB plus 34 μg/ml chloramphenicol, 100 μg/ml ampicillin, and 2 g/liter glucose at 37°C to an optical density at 600 nm (OD600) of 0.4. Isopropyl-β-d-thio-galactopyranoside (IPTG) was added to a final concentration of 0.3 mM. After incubation for 2 h at 37°C, cells were harvested, resuspended in lysis buffer (50 mM Tris [pH 8], 200 mM NaCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 0.5 mg/ml lysozyme), and disrupted by sonication. After centrifugation at 100,000 × g for 20 min at 4°C, the soluble fraction was diluted to 150 mM NaCl and applied on a QMA anion-exchange column. The QMA flowthrough was loaded on an amylose column (NEB) equilibrated with lysis buffer. HpRecA-MBP was eluted in lysis buffer containing 10 mM maltose. The MBP-HpRecA pooled protein was treated with 2.5% factor Xa protease (NEB) for 3.5 h at 30°C, resulting in the cleavage of the fusion protein to generate two products, MBP (42 kDa) and HpRecA (38 kDa). The products of proteolysis were desalted on a PD10 column (GE Healthcare) in buffer A (25 mM phosphate [pH 6], 50 mM NaCl, 1 mM EDTA, 1 mM DTT) and loaded on a HiTrap heparin column (GE Healthcare) equilibrated with buffer A. Elution was performed with a 50% gradient of buffer B (25 mM phosphate [pH 6], 2 M NaCl, 1 mM EDTA, 1 mM DTT). Fractions containing HpRecA were concentrated by Amicon (Millipore) filtration and pooled. The recombinant protein was conserved in 50% glycerol.
ATP hydrolysis assay.
ATPase activity was measured by linking ATP hydrolysis to the oxidation of NADH as described previously (23). Assays were performed at 37°C in buffer containing 50 mM phosphate (pH 6), 750 μM phospho-enolpyruvate trisodium, and 5 mM MgCl2 in the presence of 330 ng φX174 single-stranded DNA (10 μM nucleotides; NEB), 0.8 mM NADH, and 0.6 μM HpRecA. Reactions were started by the addition of ATP at the indicated concentrations. Values for the Michaelis-Menten constants kcat and Km for ATP at saturating amounts of ssDNA were derived by fitting data directly to the Michaelis-Menten equation.
DNA strand exchange reaction.
M13CEF3 ssDNA (4.5 μM nucleotides; NEB) was incubated in 30 mM phosphate buffer (pH 6), 9 mM MgCl2, 1.8 mM DTT, 1.1 mM ATP, 7.2 mM phosphocreatine, and 9 U/ml phosphocreatine kinase for 3 min at 37°C before the addition of 0.26 μM EcSsb and variable amounts of HpRecA protein. The reaction mixtures were kept at 37°C for 10 min. Then linearized M13CEF3 double-stranded DNA (dsDNA) (4.5 μM nucleotides) was added. After 90 min of incubation, the reaction mixture was deproteinized and analyzed by electrophoresis in 0.8% agarose gel in Tris-acetate-EDTA (TAE) buffer. The gels were stained with SYBR gold (Molecular Probes) and quantified with ImageJ software.
Western blot analysis of HpRecA or EcRecA.
Proteins from total cell extracts were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated in blocking buffer (1× Tris-buffered saline [TBS] and 0.1% Tween 20 with 5% nonfat dry milk) for 1 h at room temperature. Incubations with the primary antibodies against either EcRecA (AB63797 from Abcam) or HpRecA (AK263 provided by R. Haas [10]) were carried out for 1 h at room temperature, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody under the same conditions. Proteins were revealed by adding ECL Plus Western blotting detection reagents (GE Healthcare) and by exposure to X-ray film (CL-XPosure film; Pierce). The intensities of the bands were quantified using ImageJ software. To estimate RecA protein amounts before and after irradiation in H. pylori or E. coli, liquid cultures in exponential or stationary growth phase were irradiated at 10 J or at 75 Gy. Samples were collected before and at different times after irradiation and analyzed by Western blotting. Quantifications were performed with at least three independent experiments.
Microscopy assays.
Samples from liquid cultures were collected, and the cells were washed once with phosphate-buffered saline (PBS). Cell membranes were stained with FM64-X (Invitrogen) at a 1:500 dilution in PBS for 10 min and washed with PBS. Samples were then spotted on coated gelatin glass coverslips and fixed for 1 h with 4% formaldehyde. Slides were washed four times with PBS containing 0.1% glycine and once with PBS and incubated 1 h at 37°C with mouse anti-Flag antibody (Sigma-Aldrich) at 0.1 μg/ml in blocking solution (PBS, 0.1% Tween, 1% normal goat serum, 3% bovine serum albumin [BSA]). Cells were then washed three times (10 min each) with PBS and incubated with Alexa-Fluor 488 goat anti-mouse IgG antibodies (Molecular Probes) at a 1:1,000 dilution in blocking solution for 1 h at 37°C. DNA was stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI). Coverslips were mounted in Dako fluorescent mounting medium. Image acquisition was performed with a Leica SPE confocal microscope (Wetzlar, Germany) using an ACS APO 63.0×, 1.30 oil lense. Image treatment and analysis were performed using Leica and ImageJ software programs. Pearson's coefficients were calculated with the JACoP version 2.0 plug-in from ImageJ. The results are the median of at least 3 fields and 2 independent experiments corresponding to more than 500 bacteria. Standard deviations were calculated.
RESULTS
Expression and purification of recombinant HpRecA.
In order to determine the biochemical properties of the HpRecA protein, we attempted the purification of the protein. His-tagged RecA was previously reported as an insoluble protein found in inclusion bodies when expressed in E. coli (10). By expressing HpRecA from 26695 with the maltose-binding protein (MBP) fused to its N terminus, we obtained a soluble protein (Fig. 1A). The fusion protein was then cleaved, and the recombinant HpRecA was purified to near homogeneity as described in Materials and Methods. No nuclease activity was found associated with the purified protein (data not shown).
Fig. 1.

HpRecA in vitro characterization. (A) Recombinant HpRecA expression and purification. SDS-PAGE of purification fractions. Lane 1, uninduced cells; lane 2, induced cells; lane 3, RecA-MBP pool after amylose elution; lane 4, products of factor Xa cleavage; lanes 5 to 9, different HpRecA fractions from heparin column. (B) Size comparison between HpRecA and EcRecA. RecA Western blot analysis was performed with antibodies against EcRecA as follows: total E. coli cell extract (lane 1), total 26695 cell extract (lane 2), purified HpRecA (lane 3). (C) Catalytic constants for ATP hydrolysis of HpRecA, EcRecA (19), and BsRecA (29). Assays were performed as described in Materials and Methods. (D) DNA three-strand exchange reactions promoted by HpRecA. Assays were performed as described in Materials and Methods, with HpRecA at the indicated concentrations. The reactions were analyzed by electrophoresis on an 0.8% agarose gel in TAE running buffer. dsL, linear double-stranded DNA; Sc, circular single-stranded DNA; jm, partially strand-exchanged joint molecules; P, fully strand-exchanged nicked circular dsDNA.
As mentioned above, it was reported that HpRecA was glycosylated when expressed in the H. pylori P1 strain but not in E. coli (10). Surprisingly, in our experiments, Western blot analysis of the RecA proteins revealed that both the recombinant and endogenous proteins from the H. pylori 26695 strain have an apparent size of 40 kDa, whereas EcRecA migrates as a 38-kDa protein (Fig. 1B), suggesting that in the 26695 genetic background, HpRecA is not modified. Moreover, no changes of the recombinant protein were observed after incubation with the N-glycosylase PNGase F (data not shown).
HpRecA enzymatic activities are similar to those of EcRecA.
ATP hydrolysis by the purified HpRecA was monitored using a coupled spectrophotometric assay. The catalytic constants were determined as described in Materials and Methods. Despite the similarities between HpRecA and EcRecA, HpRecA seems to be less efficient in vitro than its homolog, since its kinetic constants are reduced 2- to 4-fold (Fig. 1C) (19). Nevertheless, the constants are in the same range as those of Bacillus subtilis RecA (BsRecA) (29). RecA proteins are recombinases, which are able to promote strand exchange reactions between linear double-stranded DNA and homologous circular single-stranded DNA to yield a nicked circular product. When such an activity was tested on HpRecA, as expected, we confirmed that the H. pylori recombinase was capable of mediating DNA strand exchange in vitro (Fig. 1D).
HpRecA is an abundant and constitutively expressed protein in H. pylori.
In E. coli under normal growth conditions, the amount of EcRecA is known to be between 7,000 and 10,000 monomers per cell, increasing up to 70,000 copies after SOS induction (6, 28). Western blot quantification of HpRecA allowed us to estimate that HpRecA is present at about 80,000 monomers per cell during exponential growth and about 50,000 in stationary phase (Fig. 2B, compare lanes E1 and E2 to lanes S1 and S2). This result shows that HpRecA is expressed in H. pylori at a level similar to the SOS-induced level in bacteria harboring this response system.
Fig. 2.

HpRecA expression analysis. (A) H. pylori 26695 strain growth curve. Black arrows indicate when samples were collected. E1, 0.5 OD; E2, 1 OD; S1, 3.5 OD; S2, 2.5 OD. (B and C) Western blot analysis of HpRecA expression. Total cell lysates were separated by SDS-PAGE and immunoblotted with AK263. Equal amounts of cells were loaded in each lane. (B) Quantification of HpRecA during growth phases. For quantification controls, we used 25 ng and 50 ng of purified HpRecA. (C) RecA expression before and after irradiations. Exponential- or stationary-phase liquid cultures were irradiated at 10 J or 75 Gy. E. coli samples were collected after 1 h of incubation. H. pylori samples were collected after 3 h of incubation. Ni, nonirradiated; UV, ultraviolet irradiation; IR, ionizing irradiation.
In order to explore whether the cellular levels of HpRecA are modified after a genotoxic insult, we exposed H. pylori to UV (10 J/m2) or gamma (75 Gy) irradiation and quantified the amount of HpRecA after 3 h, the time of one generation (Fig. 2C). No changes in HpRecA levels were observed after induction of DNA damage. We confirmed the induction of EcRecA in E. coli after one generation (Fig. 3C) and estimated that the amount of protein per cell increased from around 5,000 to about 50,000 monomers per cell after UV and to about 80,000 after gamma irradiation, confirming previously published data (28). In H. pylori, we tested shorter and longer times of recovery after irradiation, as well as exposures of up to 60 J/m2 and 300 Gy without detecting differences in the levels of HpRecA (data not shown). Therefore, we conclude that HpRecA is constitutively expressed at high levels and its expression is not influenced by DNA damage.
Fig. 3.

Analysis of strains harboring the RecA-Flag gene instead of recA. (A) UV irradiation sensitivities; (B) gamma irradiation sensitivities. Irradiations were performed as described in Materials and Methods. Averages from three experiments are shown. (C) Transformation frequencies.
HpRecA is relocalized during stationary phase.
In order to follow the localization of HpRecA in the cell, we expressed a C-terminal fusion of HpRecA to the green fluorescent protein (GFP) from the endogenous HprecA locus. We analyzed four independent clones, and all four of them presented ΔrecA phenotypes, probably due to GFP interference with HpRecA functions (data not shown). We therefore decided to use another tagging system to follow the localization of the protein and fused the 3′ end of the endogenous HprecA gene to the sequence encoding the Flag peptide. Three independent clones were analyzed for survival to UV and ionizing radiation. As shown in Fig. 3A and B, the mutants presented a wild-type (wt) phenotype, indicating that addition of a Flag tag to HpRecA did not perturb its ability to repair lesions caused by UV or ionizing radiation. Moreover, the transformation efficiencies of these strains remained unmodified (Fig. 3C) compared to that of the wild type, confirming the functionality of the tagged protein. Western blot analyses using antibodies against RecA showed that the protein level was not modified by the addition of the Flag tag (data not shown).
RecA-Flag was then followed by confocal microscopy using immunofluorescence. As expected from the high-level expression of HpRecA (see above), the protein was easily detectable (Fig. 4). No immunofluorescence could be detected in strains devoid of Flag sequence (data not shown), confirming the specificity of the signal. Images issued from samples collected from exponential-phase cultures (Fig. 4A, expo) presented a diffuse pattern of RecA-Flag that coincided with the DAPI staining pattern corresponding to DNA. When images were obtained from stationary-phase cultures (after 30 h) (see Fig. 2A), the RecA-Flag signal was weaker, confirming the lower levels of protein observed using Western blots (Fig. 2B). RecA-Flag was more concentrated in large foci mostly in midcell or, less often, at the poles, corresponding to areas with essentially no DAPI staining (Fig. 4A, stat). To rule out exclusion of DAPI staining by the formation of RecA foci, we reversed the order of the staining protocol. Even when DNA was stained before the antibody was applied to the samples, the foci were devoid of the DAPI signal, strongly suggesting the accumulation of HpRecA in DNA-poor regions. The difference in localization between exponential and stationary phases was confirmed by analyses using ImageJ software and Pearson's coefficient method for quantifying the degree of colocalization between DNA and HpRecA. Results are presented in Fig. 5. By computer analysis of more than 500 cells in each case, we confirmed a better colocalization of RecA-Flag with DAPI in exponential-phase cells. Occasionally, some H. pylori bacteria from stationary-phase cultures presented a circular shape corresponding to coccoids, a physiological form known to appear under starvation conditions. RecA-Flag was also detected in these cells (Fig. 4).
Fig. 4.

Localization of RecA-Flag in Helicobacter pylori. A Flag tag was fused to HpRecA, and the fusion protein was expressed at the HprecA locus. Images were obtained with a confocal microscope as described in Materials and Methods. (FM64-X and DAPI are membrane and DNA stainings, respectively.). Samples were obtained from liquid cultures in exponential (expo) or stationary (stat) phases. Coccoid forms are indicated by the letter C. White arrows indicate RecA-Flag foci excluded from DNA. RecA-Flag in 26695 wild-type (A), ΔdprA (B), and ΔcomB6 (C) strains.
Fig. 5.
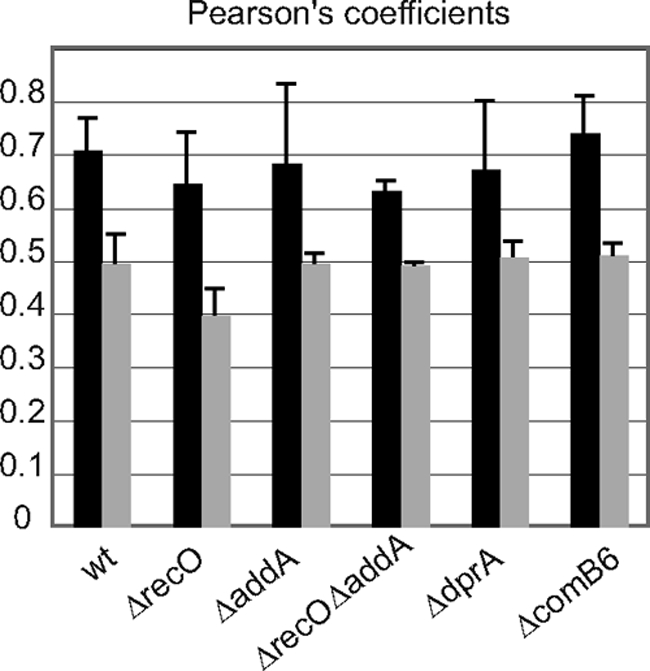
RecA-Flag localization analysis. Pearson's coefficient between DNA (DAPI) and RecA-Flag were determined as described in Materials and Methods. Black bars correspond to exponential phase. Gray bars correspond to stationary phase. The different mutant strains analyzed are indicated.
HpRecA localization is not affected by disruption of dprA, comB6, or recombination mediator genes.
As mentioned in the introduction, during DNA strand break repair by HR, RecA can be loaded onto ssDNA by two different pathways, RecBCD (or AddAB in B. subtilis and H. pylori) or RecFOR (or RecOR in H. pylori). We replaced the HprecA gene with the RecA-Flag gene in H. pylori strains deleted for either recO or addA or both. Western blot analysis using antibodies against RecA confirmed that the level of RecA-Flag expression is not modified by the inactivation of the recombination genes studied (data not shown). Analysis of RecA-Flag localization (Fig. 5; see also Fig. S1 in the supplemental material) showed that the RecA-Flag localization pattern in the mutants is identical to that found in the wild-type strain. This was true for both the exponential-phase culture (a diffuse pattern) and the stationary-phase culture (formation of discrete foci).
The concentration of RecA-Flag at the pole of some cells in stationary phase evoked the relocalization of BsRecA in Bacillus subtilis cells where competence is induced (14). To test whether the pattern of relocalization of HpRecA described for stationary-phase cultures was linked to DNA uptake, we disrupted either dprA or comB6 genes in the strain expressing RecA-Flag. DprA is the mediator protein loading RecA on transforming DNA, and ComB6 is part of the membrane ComB complex responsible for transforming DNA entrance into the cell. In both cases, the transformation capacity was completely abolished (data not shown). Analysis of RecA-Flag localization in both mutant strains revealed that RecA-Flag relocalization during stationary phase is not altered (Fig. 4B and C). Computer analysis of images confirmed these observations (Fig. 5). Therefore, localization of RecA-Flag at the pole of some cells during stationary phase seems to be independent of the transformation process.
HpRecA distribution is unchanged after UV or gamma irradiation.
EcRecA relocalization after DNA damage formation was described in E. coli by following the expression of EcRecA fused to GFP from the endogenous promoter (24). Both the SOS-dependent induction of its expression and the relocalization into foci were observed after a genotoxic stress. In B. subtilis, BsRecA fused to GFP and expressed from a strong promoter was also shown to relocalize in response to mitomycin C treatment (13). We showed above that HpRecA expression is not modified by DNA damage. We wanted to test whether HpRecA changes its distribution pattern after exposure of the cells to UV or gamma irradiation. Liquid cultures in exponential growth or in stationary phase were submitted to irradiation at various doses and allowed to recover under normal growth conditions. Samples were taken at different times and analyzed. Between 15 min and 30 h after irradiation with up to 60 J/m2 of UV or 300 Gy of ionizing radiation, we were not able to see any relocalization of the protein (Fig. 6). This absence of relocalization after DNA damage was also observed in the recombination mutants studied (see Fig. S1 in the supplemental material).
Fig. 6.

Localization of RecA-Flag after UV (10 J/m2) or gamma (75 Gy) irradiation. See the Fig. 4 legend for details. Samples were collected 4 h after irradiation of liquid cultures in exponential (A) or stationary (B) phases.
DISCUSSION
We present here the biochemical and cellular characterization of the HpRecA protein. The purification of a recombinant protein allowed the first analyses of its enzymatic properties. The determination of the kinetics parameters revealed ATPase and recombinase activities for the recombinant protein similar to but somewhat lower than those of EcRecA and BsRecA (Fig. 1). It was first reported that HpRecA is modified in vivo by glycosylation, leading to a molecular mass increase of 2 kDa (25). Our comparison of the purified HpRecA with EcRecA confirmed the size difference. However, recombinant HpRecA expressed in E. coli migrated as the endogenous HpRecA (Fig. 1B), suggesting that either the protein expressed in E. coli is modified as in H. pylori or that it simply displays an aberrant migration in PAGE. The genes involved in HpRecA glycosylation (hp0156 and hp0158 [10]) do not have homologues in E. coli, but the presence of functional analogues that may modify the ectopically expressed HpRecA may not be excluded a priori. The smaller size reported by Schmitt et al. (25) for the recombinant HpRecA expressed in E. coli may in that case be explained by the fact that the protein was insoluble and therefore not recognized by the glycosylation machinery. However, the absence of apparent variation in molecular size after a glycosylase treatment of the E. coli-expressed HpRecA strongly suggests that in the 26695 background, the bulk of the protein is not modified and that the higher apparent molecular size is due to a migration aberration.
Although HpRecA enzymatic activities do not show significant differences with those of other well-characterized RecA proteins, some striking differences were revealed by the determination of the HpRecA protein expression pattern in H. pylori cells.
First, we showed that the protein is present in about 80,000 copies per cell in exponential-phase cultures, a level similar to that of EcRecA in SOS-induced bacteria (Fig. 2). We observed that the number of HpRecA molecules per cell decreases to 50,000 copies during stationary phase (Fig. 2). This result is consistent with the reported 2-fold decrease of HprecA transcripts in stationary cultures (7). This high expression, together with the predicted absence of an SOS system in H. pylori, suggests that HpRecA is always highly expressed to protect chromosome integrity. This hypothesis may be related to H. pylori infectivity. Indeed, it was reported that strains disrupted for the HprecA gene are impaired in mouse colonization capacity (1). Recombination is therefore essential for colonization and probably also for survival in the host, where bacteria are constantly subjected to the inflammatory response of the host (22). Consistently, induction of DNA damage does not increase the expression of DNA repair genes, including HprecA, but rather that of genes coding for the competence machinery components (7). The induction of competence by genotoxic agents was previously reported for other pathogens, such as Streptococcus pneumoniae (5), Coxiella burnetii (20), and Legionella pneumophila (4). Induction of competence genes instead of the SOS system, absent in these organisms, may then be an adaptation to pathogenicity.
Since a critical function of RecA proteins is the rescue of stalled or collapsed replication forks, it can be expected that higher levels of the protein are required during exponential growth. The lower levels of HpRecA observed in stationary phase can then be correlated with the reduced need for RecA in nonreplicating cells.
Furthermore, the use of a tagged version of the protein (RecA-Flag) that is fully active (Fig. 3) allowed us to follow the localization of the protein in cells. We showed that RecA-Flag colocalizes with DAPI-stained DNA during exponential growth. This pattern is modified during stationary phase when the protein is present in one to three foci per cell, preferentially in areas excluded from DAPI staining (Fig. 4A and 5). This change can be related both to the reduced amount of HpRecA and to the absence of replication. RecA-Flag is found on DNA in exponential-phase cultures when replication is active. It may not be needed there once DNA synthesis is complete. On the other hand, the relocalization may also be explained by a competence signal that leads to HpRecA migration to the membrane and the DNA uptake machinery (10). However, the same pattern of RecA-Flag in wild-type cells was observed in both ΔdprA and ΔcomB6 strains, indicating that the relocalization during stationary phase is not dependent on these proteins. Even if RecA-Flag relocalization is due to a competence signal, it is not linked to the entrance or formation of a DprA filament on the entering DNA.
Perhaps more striking is the absence of RecA-Flag relocalization after induction of DNA damage (Fig. 6). We tested UV and gamma radiations, and neither of them induces changes in RecA-Flag localization pattern. This absence of redistribution may be due to the large amount of HpRecA present in the cell at all times, even in the absence of DNA damage, in contrast to what occurs in E. coli or B. subtilis. It can be proposed that the protein is always present in sufficient quantity to ensure the repair of chromosomal DNA, thus avoiding the requirement for a specific recruitment to the damaged region. The RecA-Flag localization is also independent of the HR initiators RecO and AddA. Localization is indeed not affected in nonirradiated ΔrecO, ΔaddA, or ΔrecA ΔaddA mutants (Fig. 5). Moreover, after different times of recovery and irradiation doses, no modifications were observed in HpRecA localization (see Fig. S1 in the supplemental material).
Interestingly, H. pylori does not seem to be particularly resistant to DNA damage, compared to E. coli and B. subtilis, but presents a high spontaneous mutation rate and an efficient transformation system. In response to modifications of environmental conditions, mutations can appear and spread among the entire infecting population by transformation and recombination, explaining the panmictic characteristics of H. pylori populations (8, 34). The constant presence of a large amount of available RecA in the cell may contribute to the adaptation capacity of this pathogen.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rainer Haas for his gift of antibodies AK263 against HpRecA. We thank T. Kortulewski from the IRCM Microscopy Facility for technical assistance with microscopy analyses. We thank P. Servant for suggestions regarding the manuscript.
This work was supported by a grant from the Agence Nationale de la Recherche (ANR-09-BLAN-0271-01 to J.P.R.), the CNRS, and predoctoral fellowships from the CEA and the Fondation pour la Recherche Medicale (to E. Orillard).
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 23 September 2011.
REFERENCES
- 1. Amundsen S. K., et al. 2008. Helicobacter pylori AddAB helicase-nuclease and RecA promote recombination-related DNA repair and survival during stomach colonization. Mol. Microbiol. 69:994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amundsen S. K., Fero J., Salama N. R., Smith G. R. 2009. Dual nuclease and helicase activities of Helicobacter pylori AddAB are required for DNA repair, recombination, and mouse infectivity. J. Biol. Chem. 284:16759–16766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aras R. A., Small A. J., Ando T., Blaser M. J. 2002. Helicobacter pylori interstrain restriction-modification diversity prevents genome subversion by chromosomal DNA from competing strains. Nucleic Acids Res. 30:5391–5397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charpentier X., Kay E., Schneider D., Shuman H. A. 2011. Antibiotics and UV radiation induce competence for natural transformation in Legionella pneumophila. J. Bacteriol. 193:1114–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Claverys J. P., Prudhomme M., Martin B. 2006. Induction of competence regulons as a general response to stress in Gram-positive bacteria. Annu. Rev. Microbiol. 60:451–475 [DOI] [PubMed] [Google Scholar]
- 6. Cox M. M. 2003. The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 57:551–577 [DOI] [PubMed] [Google Scholar]
- 7. Dorer M. S., Fero J., Salama N. R. 2010. DNA damage triggers genetic exchange in Helicobacter pylori. PLoS Pathog. 6:e1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falush D., et al. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. U. S. A. 98:15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fernández S., Ayora S., Alonso J. C. 2000. Bacillus subtilis homologous recombination: genes and products. Res. Microbiol. 151:481–486 [DOI] [PubMed] [Google Scholar]
- 10. Fischer W., Haas R. 2004. The RecA protein of Helicobacter pylori requires a posttranslational modification for full activity. J. Bacteriol. 186:777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Israel D. A., et al. 2001. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc. Natl. Acad. Sci. U. S. A. 98:14625–14630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang J., Blaser M. J. 2008. Repair and antirepair DNA helicases in Helicobacter pylori. J. Bacteriol. 190:4218–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kidane D., Graumann P. L. 2005. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J. Cell Biol. 170:357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kidane D., Graumann P. L. 2005. Intracellular protein and DNA dynamics in competent Bacillus subtilis cells. Cell 122:73–84 [DOI] [PubMed] [Google Scholar]
- 15. Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63:751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loughlin M. F., Barnard F. M., Jenkins D., Sharples G. J., Jenks P. J. 2003. Helicobacter pylori mutants defective in RuvC Holliday junction resolvase display reduced macrophage survival and spontaneous clearance from the murine gastric mucosa. Infect. Immun. 71:2022–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marsin S., et al. 2010. Genetic dissection of Helicobacter pylori AddAB role in homologous recombination. FEMS Microbiol. Lett. 311:44–50 [DOI] [PubMed] [Google Scholar]
- 18. Marsin S., Mathieu A., Kortulewski T., Guerois R., Radicella J. P. 2008. Unveiling novel RecO distant orthologues involved in homologous recombination. PLoS Genet. 4:e1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Menge K. L., Bryant F. R. 1992. Effect of nucleotide cofactor structure on recA protein-promoted DNA pairing. 1. Three-strand exchange reaction. Biochemistry 31:5151–5157 [DOI] [PubMed] [Google Scholar]
- 20. Mertens K., Lantsheer L., Ennis D. G., Samuel J. E. 2008. Constitutive SOS expression and damage-inducible AddAB-mediated recombinational repair systems for Coxiella burnetii as potential adaptations for survival within macrophages. Mol. Microbiol. 69:1411–1426 [DOI] [PubMed] [Google Scholar]
- 21. Michel B. 2005. After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol. 3:e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Rourke E. J., et al. 2003. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc. Natl. Acad. Sci. U. S. A. 100:2789–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pullman M. E., Penefsky H. S., Datta A., Racker E. 1960. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J. Biol. Chem. 235:3322–3329 [PubMed] [Google Scholar]
- 24. Renzette N., et al. 2005. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol. Microbiol. 57:1074–1085 [DOI] [PubMed] [Google Scholar]
- 25. Schmitt W., Odenbreit S., Heuermann D., Haas R. 1995. Cloning of the Helicobacter pylori recA gene and functional characterization of its product. Mol. Gen. Genet. 248:563–572 [DOI] [PubMed] [Google Scholar]
- 26. Sharples G. J. 2009. For absent friends: life without recombination in mutualistic gamma-proteobacteria. Trends Microbiol. 17:233–242 [DOI] [PubMed] [Google Scholar]
- 27. Skouloubris S., Thiberge J. M., Labigne A., De Reuse H. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sommer S., Boudsocq F., Devoret R., Bailone A. 1998. Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol. Microbiol. 28:281–291 [DOI] [PubMed] [Google Scholar]
- 29. Steffen S. E., Bryant F. R. 2000. Purification and characterization of the RecA protein from Streptococcus pneumoniae. Arch. Biochem. Biophys. 382:303–309 [DOI] [PubMed] [Google Scholar]
- 30. Stingl K., Muller S., Scheidgen-Kleyboldt G., Clausen M., Maier B. 2010. Composite system mediates two-step DNA uptake into Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 107:1184–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suerbaum S., Achtman M. 1999. Evolution of Helicobacter pylori: the role of recombination. Trends Microbiol. 7:182–184 [DOI] [PubMed] [Google Scholar]
- 32. Suerbaum S., Achtman M. 2004. Helicobacter pylori: recombination, population structure and human migrations. Int. J. Med. Microbiol. 294:133–139 [DOI] [PubMed] [Google Scholar]
- 33. Suerbaum S., Josenhans C. 2007. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 5:441–452 [DOI] [PubMed] [Google Scholar]
- 34. Suerbaum S., et al. 1998. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 95:12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thompson S. A., Blaser M. J. 1995. Isolation of the Helicobacter pylori recA gene and involvement of the recA region in resistance to low pH. Infect. Immun. 63:2185–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tomb J. F., et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 37. Wang G., Maier R. J. 2008. Critical role of RecN in recombinational DNA repair and survival of Helicobacter pylori. Infect. Immun. 76:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang G., Maier R. J. 2009. A RecB-like helicase in Helicobacter pylori is important for DNA repair and host colonization. Infect. Immun. 77:286–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.