

Abstract
Chlamydia trachomatis is a medically important pathogen that encodes a relatively high percentage of proteins with unknown function. The three-dimensional structure of a protein can be very informative regarding the protein's functional characteristics; however, determining protein structures experimentally can be very challenging. Computational methods that model protein structures with sufficient accuracy to facilitate functional studies have had notable successes. To evaluate the accuracy and potential impact of computational protein structure modeling of hypothetical proteins encoded by Chlamydia, a successful computational method termed I-TASSER was utilized to model the three-dimensional structure of a hypothetical protein encoded by open reading frame (ORF) CT296. CT296 has been reported to exhibit functional properties of a divalent cation transcription repressor (DcrA), with similarity to the Escherichia coli iron-responsive transcriptional repressor, Fur. Unexpectedly, the I-TASSER model of CT296 exhibited no structural similarity to any DNA-interacting proteins or motifs. To validate the I-TASSER-generated model, the structure of CT296 was solved experimentally using X-ray crystallography. Impressively, the ab initio I-TASSER-generated model closely matched (2.72-Å Cα root mean square deviation [RMSD]) the high-resolution (1.8-Å) crystal structure of CT296. Modeled and experimentally determined structures of CT296 share structural characteristics of non-heme Fe(II) 2-oxoglutarate-dependent enzymes, although key enzymatic residues are not conserved, suggesting a unique biochemical process is likely associated with CT296 function. Additionally, functional analyses did not support prior reports that CT296 has properties shared with divalent cation repressors such as Fur.
INTRODUCTION
Chlamydia trachomatis is an obligate intracellular bacterial pathogen that is the leading sexually transmitted bacterial infection and cause of nonheritable blindness worldwide (6, 32). These phylogenetically distant bacteria are maintained through a characteristic biphasic developmental cycle that is intrinsically linked to these organisms' ability to cause disease. Despite its immense impact on public health, the factors that control the growth and pathogenesis of Chlamydia are still relatively poorly understood. Progress to gain a better understanding of these factors has been hindered by several experimental constraints, including the absence of an established method for genetic exchange and inability to cultivate the organism axenically. Another critical factor is the inherent restraint presented by the phylogenetic distance between C. trachomatis and better-characterized bacterial systems, such as Escherichia coli and Bacillus subtilis. This distance dramatically reduces the utility of protein function assignments as inferred by sequence similarity. As a result, Chlamydia trachomatis encodes a relative overabundance (∼25%) of proteins with little or no sequence similarity to functionally defined proteins (i.e., hypothetical proteins) (5, 47, 50, 53).
The three-dimensional structure of a protein can be very informative and useful in regard to understanding functional characteristics of proteins with unknown function. This is because the structure of a protein provides the precise molecular details that often facilitate experimental characterization of an expected function. In a case in which there is no expected function, the structure of a protein can be used to facilitate functional predictions by being used as a search template for better-characterized proteins that share regions of structural similarity. A problem with this approach is that obtaining high-resolution, three-dimensional structural information can be challenging in certain cases, and this proves to be a major constraint to using protein structure to facilitate functional studies. The development and validation of computational methods that can predict protein structure to a relatively high level of accuracy and thereby facilitate functional annotation, biochemical analyses, and biological characterization are a high priority (67).
I-TASSER (Iterative Threading ASSEmbly Refinement) is a computational method that has been successful in accurately modeling protein structures. I-TASSER uses a combinatorial approach, employing all three conventional methods for structure modeling: comparative modeling, threading, and ab initio modeling (44). Its effectiveness in ab initio modeling on small proteins with low primary sequence similarity is particularly striking (61). Ab initio modeling is the most technically challenging method because it cannot leverage structural components from proteins that share sequence similarity for protein structure modeling. Therefore, this method must be relied upon when modeling structures for proteins with very little or no shared similarity evident by their primary sequence.
To provide further support for the utility of protein structure analyses to facilitate functional characterization of hypothetical proteins encoded by Chlamydia, both computational and experimental approaches were applied to a chlamydial hypothetical protein for which experimental information regarding the function of the protein had been previously obtained. The Chlamydia trachomatis open reading frame (ORF) encoding CT296 has very limited sequence similarity to any proteins outside chlamydiae, and CT296 was initially annotated as a protein with unknown function (47). Subsequent functional studies indicated that CT296 exhibits properties of a divalent cation transcription repressor (DcrA), with functional similarity to the Fur repressor (41, 63). Fur (short for ferric uptake regulator) is an iron-responsive transcriptional repressor that regulates genes involved in iron acquisition in an iron-dependent fashion (14). Iron concentrations have been demonstrated to have a profound effect on the growth of Chlamydia by inducing a persistent growth phenotype that is expected to be clinically relevant (42). As such, it was anticipated that structural studies of CT296 would also be instrumental in understanding the molecular mechanisms critical for a transcription factor associated with chlamydial pathogenesis.
In this study, the structure of CT296 was determined computationally using I-TASSER and experimentally using X-ray crystallography. These two structures were compared to assess the ability of I-TASSER to model CT296 ab initio with a relatively high level of accuracy. Each of the structures was used as a search template to identify proteins with shared structural features. The functional properties of the proteins with structural similarity to CT296 were compared to the previous experimental observations of CT296. As subsequently described, given the structural observations and potential functional disparity, functional capabilities of CT296 as a Fur-like divalent cation repressor were analyzed.
MATERIALS AND METHODS
Cloning, expression, and purification of CT296.
The open reading frame encoding CT296 (GenBank accession no. CAP03988.1) was PCR amplified with Phusion high-fidelity DNA polymerase (Finnzymes, Lafayette, CO), using Chlamydia trachomatis LGV (serovar L2/484/Bu) genomic DNA and primers 5′-CTGTACTTCCAATCGCGAATGAGGGCAGTTTTACACCTAGAG-3′ and 5′-GAATTCGGATCCTCGCGATTAGTTAGGAAATCCCGCTGAGGAG-3′. The amplified DNA was inserted into a modified pET21b (Novagen, San Diego, CA) vector at the EcoRI restriction site, using the In-Fusion Advantage kit (Clontech, Mountain View, CA), following the manufacturer's protocol. The resulting plasmid contained the CT296 gene downstream of DNA encoding a polyhistidine tag and a tobacco etch virus (TEV) protease recognition sequence. The plasmid insert was sequenced (ACGT, Wheeling, IL) and confirmed to be completely present and without mutation. The plasmid was then transformed into E. coli Acella cells (EdgeBio, Gaithersburg, MD). For native CT296 overexpression, the cells were grown in LB medium supplemented with 100 μg/ml ampicillin to an optical density at 600 nm (OD600) of 0.8 before the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). For selenomethionine (SeMet)-labeled CT296, the cells were grown and labeled using a previously described method (11). The cells were harvested after an overnight growth at 15°C through centrifugation (10 min at 5,000 × g at 4°C). The resulting pellet was resuspended in purification buffer (50 mM Na2HPO4, pH 7.0, 300 mM NaCl) and disrupted by sonication. Cell debris was removed through centrifugation (30 min at 14,000 × g, 4°C). CT296 was purified by Co2+-affinity chromatography (Clontech), following the manufacturer's protocol. The eluted fractions containing CT296 were pooled, and a polyhistidine-tagged TEV protease was added to a final concentration of 0.6 μM in order to cleave the polyhistidine tag from the protein. Following an overnight dialysis in the purification buffer, the protein mixture was added to a Co2+ column to remove the polyhistidine-tagged TEV protease and the cleaved polyhistidine fragments.
Size exclusion chromatography.
Purified recombinant CT296 at concentrations of 1000, 100, and 10 μM was analyzed by size exclusion chromatography using a BioLogic DuoFlow system (Bio-Rad). One milliliter of protein solution was applied to a Sephacryl S-200 HR column (GE Healthcare), equilibrated with 50 mM Na2HPO4 (pH 7.0) and 300 mM NaCl. A protein standard mixture containing bovine gamma globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.4 kDa) (Bio-Rad) was used to generate a standard curve.
EMSA.
Chlamydia trachomatis LGV (L2/484/Bu) genomic DNA was used as a template to amplify ∼400-bp fragments of DNA by PCR. These fragments consist of a region spanning ∼100 bp upstream of the transcription start site and ∼300 bp downstream of the transcriptional start site. These putative promoter regions of DNA were amplified for the genes CT248 and CT709 using the following IR800-labled primers (Integrated DNA Technologies): for CT248, 5′-GATGCCGATTGAGGAGTCTG-3′ and 5′-GCAACTTTTGCTGTACATTCC-3′; and for CT709, 5′-CTTAGTTTCTTTAAAAGCTGGAG-3′ and 5′-GGAAAAAAAATTAAATACACGATC-3′. These genes were previously identified as DNA binding targets of CT296 (41). To serve as a negative control in the electrophoretic mobility shift assay (EMSA) reaction, ∼400 bases from the region of DNA between the two converging genes CT032 and CT033 was amplified using the IR800-labeled primers (5′-ACATTCCTTAGATCTAGGTTCCC-3′ and 5′-GCTATTGCTGTTCGTAATAATAAGG-3′).
EMSA reaction mixtures were set up for a total volume of 20 μl each, using the methods and buffers previously described (41). Reaction mixtures contained EMSA buffer, DNA fragments (48 pmol), and purified recombinant CT296 (at DNA/protein molar ratios of 1:100, 1:500, and 1:2,500). Reactions without protein were used as a negative control. Once protein was added, the solution was gently mixed and incubated for 30 min at room temperature. Five microliters of 5× DNA dye (90 mM Tris, 90 mM boric acid, 2 mM EDTA, 20% glycerol, 1% xylene cyanol, 1% bromphenol blue) was added to the reaction mixtures and mixed gently. Twenty microliters of each reaction mixture was loaded on a precast 6% Tris-borate-EDTA (TBE) gel (Invitrogen) and run at 150 V for 75 min. Bands were visualized using an Odyssey infrared imaging system (Li-Cor Biosciences).
Fur complementation analyses.
To produce untagged recombinant CT296 for functional complementation, the open reading frame encoding CT296 (GenBank accession no. CAP03988.1) was PCR amplified with Taq DNA polymerase (Bioline, Taunton, MA), using Chlamydia trachomatis LGV (L2/484/Bu) genomic DNA and primers 5′-ACAGGACCTCGAGCGCATGAGGGCAGTTTTACACC-3′ and 5′-CTATTAGTTAGGAAATCCCGCTG-3′. The E. coli K-12 fur gene (GenBank accession no. NC_000913) was amplified from plasmid pMH15 (a gift from Klaus Hantke) using primers 5′-ACAGGACCTCGAGCGCATGACTGATAAC-3′, and 5′-CTATTATTTGCCTTCGTGCGCG-3′. The amplified DNA products were purified by gel electrophoresis and inserted into the pGEM-T Easy vector (Promega, Madison, WI) at the EcoRV site, following the manufacturer's protocol. The plasmid insert was sequenced (ACGT, Wheeling, IL) and confirmed to be completely present and without mutation. The plasmids were then transformed into E. coli H1780 cells (a gift from Klaus Hantke). As a negative control, a pGEM-T Easy plasmid containing no insert was also transformed into this cell line.
H1780 cells containing plasmid were freshly inoculated in triplicate into LB medium supplemented with 100 μg/ml ampicillin, 25 μg/ml kanamycin, and either 200 μM the iron chelator 2,2-dipyridyl, 40 μM FeSO4, or 100 μM FeCl2. These cultures were incubated for 3 h at 37°C with shaking at 200 rpm until an OD600 of 0.25 to 0.5 was reached. β-Galactosidase activity in the cultures was measured as described previously (38).
Target selection and I-TASSER structure modeling method.
The following amino acid sequence from the genome of Chlamydia trachomatis LGV (serovar L2/484/Bu) was selected as the target: MRAVLHLEHKRYFQNHGHILFEGLAPVSDCKQLEAELKLFLKEVAVVKDRHLQRWRENVHRTLPGVQMIVKRVRLDHLAAELTHRSRVALVRDLWVQKQEEILFDDCDCSVLLCLSGEKAGWGLFFSGEYPQDVFDWGAGDTAIILRFSSAGFPN. Using the nomenclature of the L2 genome, the name of this protein is CTLon_0544. However, in this report, the more widely used nomenclature of the serovar D genome is used, and the protein is referred to as CT296.
The I-TASSER server has been described previously (44, 66). Briefly, the target sequence is first threaded through a representative Protein Data Bank (PDB) library by LOMETS, a locally installed meta-threading program (62). The continuous fragments (>5 residues) are excised from the LOMETS alignments and used to reassemble the structure by replica-exchange Monte Carlo simulations (62). The simulation trajectories are then clustered by SPICKER (68) and are used as the starting state of the second round I-TASSER assembly simulation. Finally, the structures of the lowest energy are selected, which are then refined by a fragment-guided molecular dynamic procedure, with the purpose of optimizing the hydrogen-binding network and removing steric clashes.
The experimentally determined protein structure of CT296 was analyzed for putative DNA binding regions using PreDs (51) and HTHQuery (16).
CT296 crystallization.
Purified recombinant CT296 was concentrated to 6.3 mg/ml in 50 mM Na2HPO4 (pH 7.0) plus 300 mM NaCl and screened for crystallization with commercially available sparse matrix screens. One microliter of the protein was mixed with 1 μl of the crystallization solution in Compact Jr. (Emerald Biosystems, Bainbridge Island, WA) sitting drop vapor diffusion plates and equilibrated against 100 μl of crystallization solution. CT296 crystals were observed after 3 days at 20°C from the Wizard 4 screen condition 48 (Emerald Biosystems) (100 mM HEPES, pH 7.0, 15% [wt/vol] polyethylene glycol 20000 [PEG 20000]). Single crystals were transferred to a cryoprotectant solution containing the crystallization condition supplemented with 20% PEG 400.
Cocrystallization was attempted with purified CT296 at 8.6 mg/ml and 5 mM α-ketoglutarate (Sigma-Aldrich, St. Louis, MO) and/or 5 mM FeCl2 (Sigma-Aldrich). The resulting mixture was screened for crystallization as described above; however, no crystals were obtained. Additionally, apo-CT296 crystals were soaked in the crystallization solution supplemented with 50, 5, 1, or 0.1 mM α-ketoglutarate and/or FeCl2. The crystals dissolved within seconds to minutes of exposure to either compound or combination.
Data collection and processing.
Data sets for the native and SeMet-labeled CT296 crystals were collected at 100K at the Advanced Photon Source (APS) IMCA-CAT, sector 17ID. Data for the native and the SeMet-labeled crystals were collected using an ADSC Quantum 210r charge-coupled device (CCD) detector and Dectris Pilatus 6 M pixel array detector, respectively. Native diffraction data were processed with HKL2000, and SeMet data were integrated and scaled using XDS (24) and SCALA (15), respectively, via the AUTOPROC (57) software package. Structure solution using the SAD phasing method and preliminary model building were performed using AUTOSOLVE (48), contained within the PHENIX suite (2). Automated model building was performed using AUTOBUILD (49). The SeMet-derived CT296 model was used as the search model for molecular replacement against the native data set using PHASER (33). Manual model building and subsequent refinement of the models were performed using COOT (13) and PHENIX (2), respectively.
RESULTS
The I-TASSER-modeled structure of CT296 shows no significant structural similarity to Fur family members and has no apparent DNA-binding motifs.
The protein encoded by ORF CT296 has been reported to contain very limited sequence similarity to the Fur protein but exhibits metal-dependent DNA binding properties comparable to those of Fur (41, 63). To better understand the molecular basis for its metal and DNA binding functions as well as test the accuracy and impact of I-TASSER protein structure modeling, both computational and experimental structural analyses were performed on CT296.
Initially, computational modeling of the structure of CT296 using I-TASSER was performed. Five models of CT296 were computationally generated using the I-TASSER algorithm with C-scores ranging from −5 to −2.25. The C-score is a confidence score and ranges from −5 to 2, with higher scores representing higher confidence in the model (44). Although −1.5 has been used previously as a cutoff for confidence in models, there can still be significant modeling accuracy in models with C-scores below this limit (44). Model 1 (C-score of −2.25) was used for all analyses described (Fig. 1A) as the remaining models had C-scores of −5. The overall structure of CT296 model 1 is primarily α-helical (5 helices; 46%) with a significant number of unstructured loops (48%) and two relatively short β-strands (6%).
Fig. 1.

Tertiary and secondary structure comparisons of the I-TASSER model and the experimentally determined structure of CT296. (A) Ribbon depiction of the I-TASSER model of CT296. The topology of the CT296 model is as follows (with residue numbers in parentheses): α1 (H6 to H16)–α2 (V27 to V47)–α3 (H51 to R61)–α4 (P64 to K71)–α5 (R74 to L82)–α6 (R87 to W95)–β1 (D105 to D106)–β2 (S110 to C114)–β3 (A143 to F148). (B) The experimentally determined structure of CT296. The topology of the crystal structure of CT296 is as follows (with residue numbers in parentheses): α1 (L7 to H16)–β1 (H18 to F21)–α2 (V27 to F40)–α3 (G65 to V73)–α4 (L75 to T83)–β2 (A89 to V96)–β3 (D108 to C114)–β4 (W122 to F126)–β5 (A143 to S149). Secondary structure elements that are conserved between the two structures (four α-helices and two β-sheets) are labeled and highlighted in red. (C) Secondary structure topology comparison of the I-TASSER model and the experimentally determined structure of CT296.
Despite the low overall primary sequence similarity between CT296 and Fur, it was anticipated that the two would share significant structural characteristics, largely based on the previously reported ability of CT296 to functionally complement Fur in E. coli (63). However, the I-TASSER-generated model of CT296 bears little resemblance to the structures of Fur family members (PDB no. 2W57, 2O03, 2FU4, and 1MZB) as no significant similarity was identified using DaliLite pairwise comparisons (with a Z-score of >2 used as a cutoff for significance; data not shown) (21). Additionally, no DNA binding motifs (e.g., helix-turn-helix, a leucine zipper, a zinc finger, winged helix-turn-helix, etc.) were apparent in the I-TASSER model of CT296. To identify potential regions of DNA-binding sites based on electrostatics and curvatures on the protein surface, the model of CT296 was subjected to computational analysis by PreDs (51). These protein characteristics are summarized as a P-score, the value of which ranges from 0.0 to 1.0 and is predictive of a protein with the necessary structural characteristics to bind to DNA. The results of this analysis give a P-score of 0.03 to the model of CT296. A P-score of 0.12 is typically used as a cutoff point, because DNA-binding proteins almost always have P-scores above this threshold. As a control, the effector domain of ChxR, a well-characterized chlamydial DNA-binding protein, was subjected to the same analysis (19, 28). It was given a P-score of 0.22, correctly predicting that it is capable of binding to DNA. Altogether, the I-TASSER-generated model of CT296 is in contrast with the previous observations that CT296 is a DNA-binding protein with structural similarity to Fur.
Homology searches using the I-TASSER-generated model of CT296 support its functional prediction as a non-heme Fe(II) 2-oxoglutarate-dependent enzyme.
The model of CT296 generated by I-TASSER was used to search for proteins with regions of structural homology within the PDB (Table 1). This assessment was carried out using both the PSFloger server (http://zhanglab.ccmb.med.umich.edu/PSFloger/) and the DALI server (http://ekhidna.biocenter.helsinki.fi/dali_server/). PSFloger was developed in part to complement I-TASSER; it detects the structural and functional analogs from a representative PDB library by the global structural alignment algorithm, TM-align (69), assisted with local motif matches (69). DALI is a well-established, widely utilized server for protein structure alignment (22). Although the overall goal of the PSFloger and the DALI servers is to identify structural homologs, both servers were employed in order to give a deeper insight into the relationship between the modeled structure of CT296 and other proteins of known structure.
Table 1.
Top 10 structures with homology to the CT296 I-TASSER model as measured by PSFloger and DALI
| Rank | PSFloger |
DALI |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TM-scorea | PDB file | Protein name | Species | Classification (reference) | Z-scoreb | PDB file | Protein name | Species | Classification (reference) | |
| 1 | 0.6619 | 2OPW | PHYHD1 | Homo sapiens | Hydroxylase (37) | 6.3 | 2G1M | PHD2c | Homo sapiens | Hydroxylase (35) |
| 2 | 0.6564 | 2HBT | PHD2 | Homo sapiens | Hydroxylase (58) | 5.7 | 3GJB | CytC3c | Streptomyces sp. | Halogenase (60) |
| 3 | 0.6365 | 2CSG | YbiU | Salmonella enterica serovar Typhimurium | Putative oxidoreductase | 5.5 | 3EMR | ECTD | Virgibacillus salexigens | Dioxygenase (43) |
| 4 | 0.6131 | 2FCU | SyrB2 | Pseudomonas syringae | Halogenase (4) | 5.3 | 3KT7 | Tpa1c | Saccharomyces cerevisiae | Hydroxylase (27) |
| 5 | 0.6063 | 1GP6 | ANS | Arabidopsis thaliana | Dioxygenase (59) | 5.1 | 3OBZ | PHYHD1c | Homo sapiens | Hydroxylase |
| 6 | 0.6060 | 1UNB | DAOCS | Streptomyces clavuligerus | Oxidoreductase (55) | 5.1 | 3NN1 | Halc | Nitrospira defluvii | Halogenase (25) |
| 7 | 0.6019 | 2DBN | JW0805 | Escherichia coli | Unknown function | 4.5 | 2RG4 | Q2CBJ1_9RHOB | Oceanicola granulosus | Unknown function |
| 8 | 0.5998 | 2A1X | PHYHD1 | Homo sapiens | Hydroxylase (34) | 4.4 | 3DKQ | Sbal_3634c | Shewanella baltica | Putative hydroxylase |
| 9 | 0.5917 | 1W3X | IPNS | Emericella nidulans | Oxidoreductase (9) | 4.2 | 3BVC | Ism_01780c | Roseovarius nubinhibens | Unknown function |
| 10 | 0.5820 | 2RDN | PtlH | Streptomyces avermitilis | Hydroxylase (65) | 4.0 | 2FCU | SyrB2 | Pseudomonas syringae | Halogenase (4) |
The TM-score is a measurement of similarity between two protein structures, and it varies from 0 to 1, with 1 indicating a perfect alignment. A score of 0.5 or higher indicates significant similarity (64).
Structures with Z-scores above 2 are considered to be significant matches (20).
This protein was listed multiple times on the DALI output, with various minor differences (ligands, etc.); these redundant structures are not listed here.
The PSFloger search indicated that the 10 proteins that share structural similarity to the CT296 model are from diverse sources (e.g., Homo sapiens [mammal], Pseudomonas syringae [bacteria], and Arabidopsis thaliana [plant]) but have similar functions as non-heme Fe(II) and 2-oxoglutarate-dependent enzymes. The closest structural match (2OPW) is the enzyme PHYHD1 from Homo sapiens, which carries out a hydroxylation reaction using a non-heme Fe(II) cofactor and a 2-oxoglutarate cosubstrate (37, 58). The next closest match (2HBT) is also from Homo sapiens and is called PHD2. PHD2 is a prolyl hydroxylase and likewise makes use of non-heme Fe(II) and 2-oxoglutarate (35). The third structural match to the model of CT296 (2CSG) is the protein YbiU from the bacterium Salmonella enterica serovar Typhimurium and is a putative oxidoreductase with iron bound in the crystal structure. As a final example, the fourth closest structural match to CT296 (2FCU) is the halogenase SyrB2 from Pseudomonas syringae; like the first two matches, it requires non-heme Fe(II) and 2-oxoglutarate (4). Although these structural homologs differ in their precise enzymatic function (e.g., hydroxylase or oxidoreductase), the similarities among them predict that CT296 is an enzyme that utilizes iron as a cofactor and may bind 2-oxoglutarate.
Like PSFloger, the DALI server also identified structural homologs from diverse sources (e.g., H. sapiens [mammal], Saccharomyces cerevisiae [fungus], and S. salexigens [bacteria]). Also similar to the PSFloger results, all matches made by DALI function as non-heme Fe(II) and 2-oxoglutarate-dependent enzymes. Despite the similar functional characteristics of the PSFloger and DALI matches, only 3 of the top 10 structures identified as homologous to the CT296 model by PSFloger were also identified by DALI. This is partly because PSFloger uses a nonredundant PDB library (with pairwise sequence identity of <95%), while DALI search utilizes all available PDB structures. The top-ranked DALI structure (2G1M) is the protein PHD2, the same protein that was listed as the second-highest-ranked homolog by PSFloger (PDB file 2HBT; same protein, different ligands bound). The second-closest structural match (3GJB) is the halogenase CytC3 from Streptomyces sp., and it was not ranked in the top 10 structure homologs by PSFloger. This enzyme also uses Fe(II) and 2-oxoglutarate for catalysis (60). The third match (3EMR) is ECTD from Salibacillus salexigens and is a non-heme Fe(II)-dependent dioxygenase (43). As a final example, the number 4 structural match to the CT296 model by DALI is PDB code 3KT7. It is the protein Tpa1 from Saccharomyces cerevisiae, a hydroxylase that binds iron and 2-oxoglutarate (27). Overall, the observations of the two alignment programs support the prediction of CT296 as an Fe(II) 2-oxoglutarate-dependent enzyme.
A relatively conserved feature of the non-heme Fe(II) 2-oxoglutarate-dependent enzymes is the presence of a double-stranded beta helix (DSBH) core in which the catalytic events typically occur (8). This DSBH fold is formed by two opposing β-sheets, composed of at least four β-strands each (8). The CT296 model appears to contain an incomplete DSBH core with only one relatively small β-sheet present containing 3 β-strands (Fig. 1A and Fig. 2A). In order to examine the conserved structural characteristics of the CT296 model and its homologs, the model was aligned with three homologous structures (2OPW, 2FCU, and 2HBT) from the PDB using the CCP4 Molecular Graphics program (39) (Fig. 2). The three models selected for alignment represent proteins that were identified as homologs to CT296 by both PSFloger and DALI. There are five (2HBT) or six (2OPW and 2FCU) secondary structures that are present and similarly oriented among the three models: three or four α-helices and two β-strands. The three or four conserved α-helices are oriented immediately adjacent to one of the β-sheets that forms the canonical DSBH core containing the known (2FCU [4] and 2HBT [35]) and proposed (2OPW [37]) ligand-binding and catalytic site. There are two β-strands in the CT296 model that are immediately adjacent to the structurally conserved α-helices. These β-strands are oriented similar to the β-sheets that comprise the typical DSBH in Fe(II), 2-oxoglutarate-dependent enzymes. It was also apparent that the structures of the three proteins used for this comparison (2HBT, 2OPW, and 2FCU) have only minimal opposing β-strands that compose the canonical DSBH typical of this family of non-heme Fe(II) 2-oxoglutarate-dependent enzymes (4, 3, and 6 short strands, respectively).
Fig. 2.
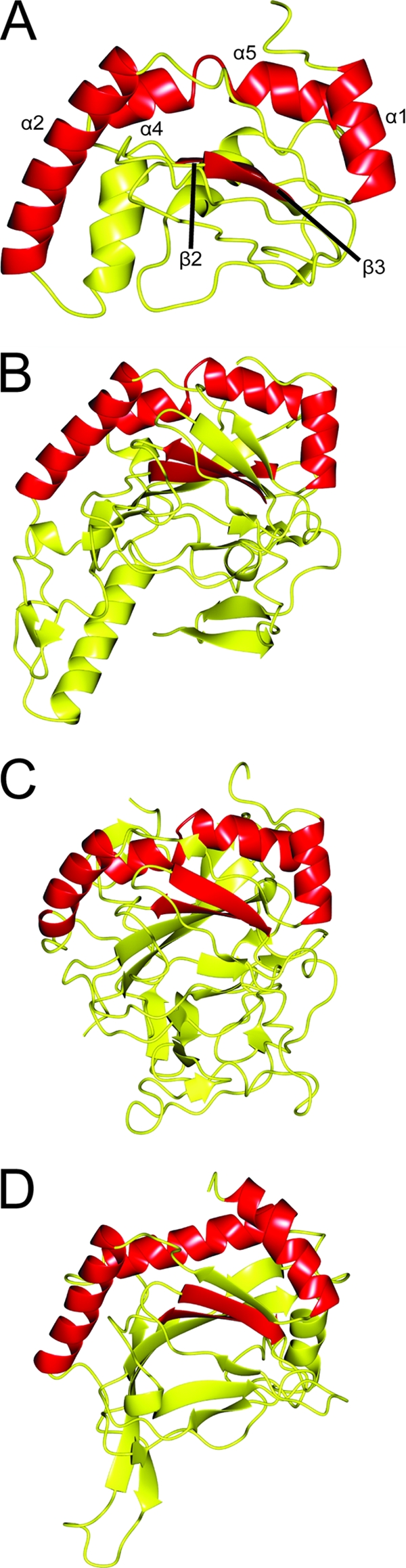
Ribbon depiction of the I-TASSER model for CT296 aligned with three proteins exhibiting the highest structural homology. Regions of structure that are conserved between all four structures are highlighted in red. (A) I-TASSER model of CT296. Four α-helices are highlighted (with residue numbers in parentheses): α1 (L7 to H16), α2 (V27 to F40), α3 (G65 to V73), and α4 (L75 to T83). Two β-strands are highlighted: β3 (D108 to C114) and β4 (W122 to F126). (B) Crystal structure of H. sapiens PHYD1 (PDB no. 2OPW). Four α-helices (similarly oriented to α1, α2, α3, and α4 from the I-TASSER model of CT296) and two β-strands (similar to β3 and β4 of CT296) are high-lighted. (C) Crystal structure of P. syringae SyrB2 (PDB no. 2FCU). Four α-helices (similarly oriented to α1, α2, α3, and α4 from the I-TASSER model of CT296) and two β-strands (similar to β3 and β4 of CT296) are highlighted. (D) Crystal structure of H. sapiens PHD2 (PDB no. 2HBT). Three α-helices (similarly oriented to α1, α2, and α3 + 4 from the I-TASSER model of CT296) and two β-strands (similar to β3 and β4 of CT296) are highlighted.
The crystal structure of CT296 shows no significant structural similarity to Fur family members and has no apparent DNA-binding motifs.
In addition to computational prediction of the structure of CT296, recombinant CT296 was expressed, purified, and crystallized. A data set was collected and processed to a resolution of 1.8 Å, with one molecule of CT296 in the asymmetric unit within the crystal lattice (Table 2). The structure of CT296 was solved using SAD phasing with selenomethionine-labeled recombinant protein. The crystal structure of CT296 is composed of a single 5-stranded antiparallel β-sheet, supported by 4 α-helices (Fig. 1B). The region between α2 and α3 (E43 to R54) was not modeled due to the lack of electron density, indicating that this region is flexible. Overall, the structure is 19% β-sheet, 32% α-helix, and 49% unstructured. No additional molecules, such as divalent cations, were evident in the crystallized protein.
Table 2.
Data collection and refinement statistics for CT296
| Parameter | Value(s) for: |
|
|---|---|---|
| Apo-CT296 | SeMet-CT296 | |
| Data collection statistics | ||
| Unit cell dimensions a, b, c (Å) | 51.2, 64.0, 46.3 | 50.8, 63.7, 46.3 |
| Space group | P21212 | P21212 |
| Resolution (Å)a | 30.0–1.8 (1.86–1.8) | 63.66–2.5 (2.5–2.5) |
| Wavelength (Å) | 1.000 | 0.979 |
| No. of reflections: | ||
| Observed | 101,642 | 65,969 |
| Unique | 14,832 | 5,585 |
| Mean 〈I/σI〉a | 25.4 (2.3) | 18.0 (6.1) |
| Completeness (%)a | 99.8 (99.9) | 99.9 (99.8) |
| Redundancya | 6.8 (6.2) | 11.8 (12.3) |
| Rmerge (%)a,b | 10.2 (59.4) | 11.8 (48.0) |
| Refinement statistics | ||
| Resolution (Å) | 23.1–1.8 | 39.7–2.5 |
| Rfactor/Rfree (%)c | 20.5/23.8 | 20.4/25.7 |
| No. of atoms (protein/water) | 1,212/68 | 1,147/20 |
| Model quality | ||
| Bond length (Å) | 0.015 | 0.011 |
| Bond angle (°) | 1.423 | 1.159 |
| Avg B factor (Å2) | ||
| Protein | 28.6 | 35.7 |
| Water | 31.7 | 32.1 |
| Coordinate error based on maximum likelihood (Å) | 0.19 | 0.26 |
| Ramachandran plot | ||
| Favored regions (%) | 98.5 | 97.8 |
| Allowed regions (%) | 1.5 | 2.2 |
| Outliers (%) | 0.0 | 0.0 |
| PDB ID no. | 3QH6 | 3QH7 |
Values in parentheses are for the highest-resolution shell.
Rmerge = ΣhklΣi |Ii(hkl) − 〈I(hkl)〉|/ΣhklΣi Ii(hkl), where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
Rfactor = Σhkl ||Fobs (hkl) | − |Fcalc (hkl) ||/Σhkl |Fobs (hkl)|. Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement.
As observed for the I-TASSER model of CT296, the experimentally determined crystal structure does not contain similarity to Fur family members, as indicated by a DaliLite pairwise comparison (data not shown). Additionally, the structure of CT296 contains no apparent DNA-binding motifs, such as a helix-turn-helix, a leucine zipper, a zinc finger, or a winged helix turn helix. As before, the structure of CT296 was subjected to analysis by PreDs, resulting in a P-score of 0.06, well below the typical cutoff point for DNA-binding proteins (0.12). Together, these analyses of the crystal structure of CT296 are in contrast with the precious observations that CT296 is a DNA-binding protein with structural similarity to Fur.
The structure of CT296 predicted by I-TASSER is similar to the CT296 high-resolution structure solved using X-ray crystallography.
Because CT296 lacks homologs by primary sequence, template-based homology modeling cannot be used to generate a model of the structure of CT296. Therefore, ab initio modeling must be employed. This type of modeling is a particular challenge in the field of protein structure prediction (30). Impressively, the I-TASSER-generated model of CT296 has significant overall structural similarity (Cα root mean square deviation [RMSD] of 2.72 Å for 101/137 residues) to the experimentally determined structure of CT296 (Fig. 1). A notable difference between the two structures is a helix (α3; H51 to R94) modeled by I-TASSER that is unstructured in the experimentally determined structure. The most prominent difference between the two structures is the presence of a five-stranded β-sheet (β1 to β5) in the experimentally determined structure, whereas I-TASSER modeled only two β-strands (β2 and β3).
Structural homology searches using the high-resolution X-ray crystallography CT296 structure yield almost identical results as the CT296 I-TASSER model.
To determine the impact on functional predictions due to the structural differences between experimentally determined and modeled CT296, the crystal structure of CT296 was used to search for structural homologs in the PDB using PSFloger and DALI (Table 3). As expected, the scores for top matches improved dramatically (TM-score range, 0.7806 to 0.6764; DALI Z-score range, 10.5 to 8.7). Strikingly, all of the top 10 PSFloger homologs and all but one (PutA; PDB no. 3ITG) of the DALI homologs are retained in the comparable homology search of the I-TASSER model for CT296 (Table 1), reinforcing the prediction that CT296 has a structural scaffold similar to non-heme Fe(II) 2-oxoglutarate enzymes. The top hit for both PSFloger and DALI was PHYHD1 (2OPW), an enzyme from H. sapiens. Similar to the I-TASSER CT296 model homology search, only three of the proteins (PHYD1, PHD2, and SyrB2) were shared between the top CT296 structure homology matches of PSFloger and DALI.
Table 3.
Top 10 homologous structures to the crystal structure of CT296 as measured by PSFloger and DALI
| Rank | PSFloger |
DALI |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TM-scorea | PDB file | Protein name | Species | Classification (reference) | Z-scoreb | PDB file | Protein name | Species | Classification | |
| 1 | 0.7806 | 2OPW | PHYHD1 | Homo sapiens | Hydroxylase (37) | 10.5 | 2OPW | PHYHD1c | Homo sapiens | Hydroxylase (37) |
| 2 | 0.7537 | 2FCU | SyrB2 | Pseudomonas syringae | Halogenase (4) | 10.5 | 3GJB | CytC3 | Streptomyces sp. | Halogenase (60) |
| 3 | 0.7473 | 2A1X | PHYHD1 | Homo sapiens | Hydroxylase (34) | 10.3 | 3NN1 | Hal | Nitrospira defluvii | Halogenase (25) |
| 4 | 0.7221 | 2CSG | YbiU | S. enterica serovar Typhimurium | Putative oxidoreductase | 9.9 | 3EMR | ECTD | Virgibacillus salexigens | Dioxygenase (43) |
| 5 | 0.7138 | 2DBN | JW0805 | Escherichia coli | Unknown function | 9.7 | 3HQU | PHD2c | Homo sapiens | Hydroxylase (7) |
| 6 | 0.7113 | 2HBT | PHD2 | Homo sapiens | Hydroxylase (58) | 9.4 | 2FCU | SyrB2 | Pseudomonas syringae | Halogenase (4) |
| 7 | 0.6905 | 1UNB | DAOCS | Streptomyces clavuligerus | Oxidoreductase (55) | 9.1 | 3GZE | P4Hc | Chlamydomonas reinhardtii | Hydroxylase (29) |
| 8 | 0.6802 | 2RDN | PtlH | Streptomyces avermitilis | Hydroxylase (65) | 9.0 | 2RG4 | Q2CBJ1_9RHOB | Oceanicola granulosus | Unknown function |
| 9 | 0.6793 | 3GJB | CytC3 | Streptomyces sp. | Halogenase (60) | 8.8 | 3MGU | Tpa1 | Saccharomyces cerevisiae | Hydroxylase (27) |
| 10 | 0.6764 | 1GP6 | ANS | Arabidopsis thaliana | Dioxygenase (59) | 8.7 | 3ITG | PutA | Escherichia coli | Oxioreductase (46) |
The TM-score is a measurement of similarity between two protein structures, and it varies from 0 to 1, with 1 indicating a perfect alignment. A score of 0.5 or higher indicates significant similarity (69).
Structures with Z-scores above 2 are considered to be significant matches (20).
This protein was listed multiple times on the DALI output, with various minor differences (ligands, etc.); the redundant structures are not listed here.
Based upon the conservation of proteins with structural homology identified between the two search processes and increase in homology scores, it was expected that additional structural elements would be shared. The experimentally determined structure of CT296 was aligned with three of its structural homologs from the PDB, in order to determine which structural regions are similar (Fig. 3). The three structural homologs selected are the same three that were aligned to the I-TASSER model of CT296 (2OPW, 2FCU, and 2HBT) (Fig. 1); these proteins can be found on the PSFloger and the DALI lists of both the model and the experimental structure of CT296. A total of seven secondary structure elements are equivalent between CT296 and the three selected homologs, four α-helices (α1, α2, α3, and α4) and three β-sheets (β3 to -5). Importantly, six (α1, α2, α4, α5, β2, and β3) of these seven structural elements were also found to be equivalent between the model of CT296 and the homologs. This suggests that these elements are the underlying reason for the large number of proteins that contain structural homology to both the model and the experimental structure of CT296.
Fig. 3.
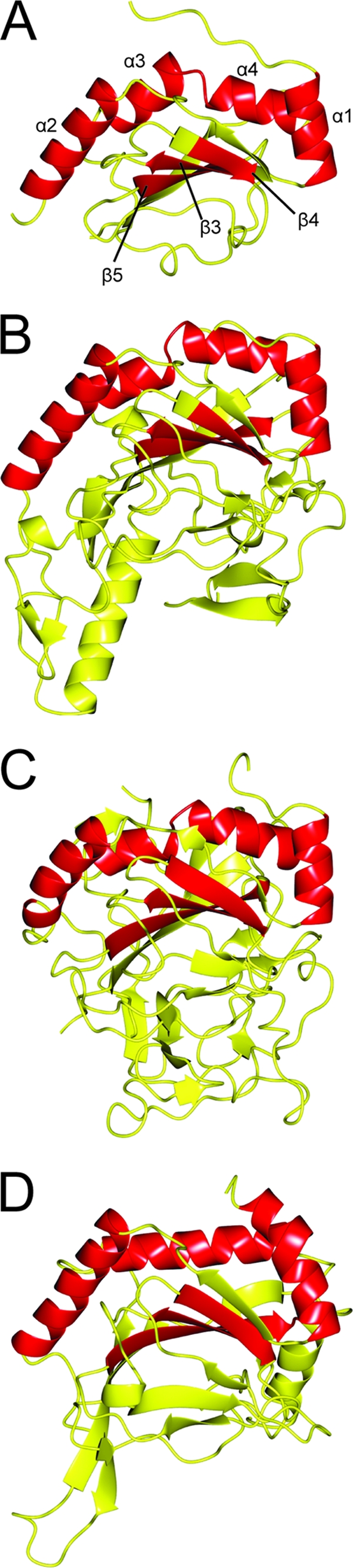
Ribbon depiction of experimentally determined structure of CT296 aligned with three proteins exhibiting the highest structural homology. Regions of structure that are conserved between all four structures are highlighted in red. (A) Crystal structure of CT296. Four α-helices are highlighted (with residue numbers in parentheses): α1 (L7 to H16), α2 (V27 to F40), α3 (G65 to V73), and α4 (L75 to T83). Three β-strands are highlighted (with residue numbers in parentheses): β3 (D108 to C114), β4 (W122 to F126), and β5 (A143 to S149). (B) Crystal structure of H. sapien PHYD1 (PDB no. 2OPW). Four α-helices (similarly oriented to α1, α2, α3, and α4 from the I-TASSER model of CT296) and three β-strands (similar to β3, β4, and β5 of CT296) are highlighted. (C) Crystal structure of P. syringae SyrB2 (PDB no. 2FCU). Four α-helices (similarly oriented to α1, α2, α3, and α4 from the I-TASSER model of CT296) and three β-strands (similar to β3, β4, and β5 of CT296) are highlighted. (D) Crystal structure of H. sapiens PHD2 (PDB no. 2HBT). Three α-helices (similarly oriented to α1, α2, and α3 + 4 from the I-TASSER model of CT296) and three β-strands (similar to β3, β4, and β5 of CT296) are highlighted.
Recombinant CT296 is predominantly monomeric at physiological concentrations.
Recombinant Fur has been shown to exist predominantly as a homodimer in solution (10, 36). At high protein concentrations, Fur has been shown to form other higher-order structures, but the predominant species present at physiological concentrations (1 to 17 μM) of the protein is that of a dimer (10). Likewise, recombinant CT296 has previously been reported to form homodimers (63). Given the structure of CT296 and its lack of similarity to Fur, the quaternary structure of recombinant CT296 was reassessed, including a study of the effects of protein concentration. To accomplish this, recombinant CT296 was purified and subjected to size exclusion chromatography (SEC). Although SEC is not an appropriate technique for precise quantification of molecular mass, a standard curve can be constructed based on the elution of profiles of proteins of known molecular mass. A protein standard solution containing gamma globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.4 kDa) was used to approximate the molecular masses of CT296 at various concentrations. The molecular mass of CT296 is 17.9 kDa, and so a dimer of CT296 would be 35.8 kDa.
At a high protein concentration (1,000 μM), a predominant peak that corresponds to the approximate molecular mass of a CT296 dimer was observed (48.0 kDa) (Fig. 4A). Other peaks that correspond to even-higher-order structures were also observed at this concentration (109 kDa) (Fig. 4A). At a lower protein concentration, (100 μM), there was a single peak which corresponds to the molecular mass of a monomer of CT296 (17.0 kDa) (Fig. 4B). At the lowest concentration tested (10 μM), an approximate monomer of CT296 was observed (12.1 kDa) (Fig. 4C). The physiological concentration of CT296 is not known, but physiological concentrations of Fur have been reported to be between 1 and 17 μM (10). These data show that, unlike Fur, CT296 exists predominantly as a monomer in solution at concentrations of 10 to 100 μM. This suggests that CT296 may lack other functional properties of Fur or other Fur-like repressors.
Fig. 4.

Size exclusion chromatograms of recombinant CT296 at various concentrations. The molecular mass of monomeric CT296 is 17.9 kDa. The molecular mass of a CT296 dimer would be 35.8 kDa. The components of a molecular mass ladder are marked at their respective elution volumes on a line above the chromatograms. (A) At a concentration of 1,000 μM, CT296 is predominantly present in a peak that approximately corresponds to the molecular mass of a dimer. Other, even-higher-order, structures can also be seen, in lower abundance. (B) At a concentration of 100 μM, CT296 elutes within fractions that correspond to the molecular mass of a monomer. (C) At a concentration of 10 μM, CT296 elutes as a peak that corresponds to the molecular mass of a monomer.
CT296 does not bind to previously reported chlamydial promoter DNA by electrophoretic mobility shift assay.
In previous reports, CT296 was shown to bind to DNA in the context of an electrophoretic mobility shift assay (EMSA) (41, 63). Given the apparent absence of DNA-binding motifs in both the modeled and the experimentally determined structures of CT296, the DNA binding properties of CT296 were reexamined. The promoters of two genes that were previously shown to bind to CT296 by EMSA were selected for analysis (41), namely, CT248 and CT709. The exact sequences of DNA from these promoters that were used in the previous study were not reported. However, the recent determination of the transcription start site (TSS) for these genes (3) allowed for the design of ∼400-bp regions that include at least 150 bases upstream and downstream of the TSS. To serve as a negative control, ∼400 bp from the region of DNA between the two converging genes CT032 and CT033 was used. No shifts in DNA were detected with any of the promoter regions analyzed at any of the DNA/protein molar ratios used, even at the highest ratio of 1:2,500 (Fig. 5). These findings are in direct contrast with the previous observations that CT296 binds to chlamydial promoter DNA.
Fig. 5.

Electrophoretic mobility shift assay (EMSA) using recombinant CT296. Three different promoter regions of DNA were tested with various DNA/protein molar ratios: 1:100, 1:500, and 1:2,500 (− indicates the absence of protein). CT032/033 is an intergenic region between two converging genes and is used as a negative control. CT248 and CT709 had been previously reported to be bound by recombinant CT296 in the context of an EMSA.
CT296 does not functionally complement Fur in E. coli.
In a previous report, CT296 was characterized as a chlamydial Fur homolog by its ability to functionally complement Fur (63). In light of the lack of structural similarity between CT296 and Fur, the ability of CT296 to functionally complement Fur was reassessed (Fig. 6). In this assay, E. coli cells that are fur negative and have the lacZ gene preceded by a Fur-responsive promoter element are used (17). When these cells are transformed with a vector control (pGEM-T Easy), they show constitutive expression of β-galactosidase. The expression levels are not significantly different between iron-depleted (2,2-dipyridyl) and iron-replete (FeSO4 and FeCl2) conditions. However, when these cells harbor a plasmid containing fur (Fig. 6; pGEM-T Easy +Fur), repression of the lacZ gene is observed in an iron-dependent manner, as previously demonstrated (40, 52). In contrast, in cells with a plasmid containing gene CT296 (pGEM-T Easy +CT296) (Fig. 6), lacZ expression is not repressed. These data indicate that CT296 is not functionally complementing the fur deficiency and further supports that CT296 is not a chlamydial Fur homolog.
Fig. 6.

CT296 does not functionally complement Fur in E. coli. The H1780 strain of E. coli, containing the indicated plasmids, was grown in LB medium under iron-limiting (2,2-dipyridyl; black bars) or iron-replete (FeSO4, white bars; or FeCl2, gray bars) conditions. β-Galactosidase activity was measured and is displayed in Miller units. Assays were performed in triplicate with standard deviations displayed as error bars.
DISCUSSION
One of the major observations of this study is that the experimentally determined properties of CT296 are not consistent with previous reports regarding the function of this protein. CT296 had been characterized previously (41, 63) as a chlamydial homolog to the E. coli Fur protein—a metal-responsive transcriptional repressor termed DcrA (divalent cation-dependent regulator A). The evidence for this functional assignment came primarily from E. coli Fur cross-reactive antibodies, the ability of CT296 to functionally complement Fur in E. coli, and the ability of CT296 to bind to chlamydial promoter DNA sequences via EMSA. However, the experimentally determined structure of CT296 bears no structural similarity to Fur and does not contain an identifiable DNA binding motif, and putative regions for DNA binding were not identified using PreDs. No divalent cations were observed in the crystal structure, and a metal-binding site could not be identified. The previous studies amplified the CT296 gene ortholog from C. trachomatis serovar E DNA, while DNA from serovar L2/434/Bu was used in this study. Importantly, the CT296 gene sequences are 100% identical between these two serovars.
Attempts to demonstrate that CT296 can bind to the chlamydial DNA targets reported previously (41) were unsuccessful (Fig. 5). In the previous analysis, EMSA was used to validate targets isolated from a screen designed to find regions of chlamydial DNA that bound E. coli Fur. In that analysis, the variable-size regions of chlamydial DNA directly from a screen were analyzed for CT296 binding. Importantly, the molar ratios of DNA to protein in reaction mixtures were not stated, and precise regions of the chlamydial genome tested were not reported. In this report, the analyses used regions of DNA designed based on the recently reported transcription start sites for these genes (3). At a wide range of DNA/protein ratios and with the inclusion of different divalent cations (data not shown), DNA binding by CT296 could not be detected. Finally, CT296 was previously reported to form dimers that are resistant to denaturation by SDS-PAGE (63). During protein purification, only monomers of CT296 were observed by SDS-PAGE. Using gel filtration chromatography, higher-order structures corresponding roughly to the molecular mass of a dimer can be seen, but this is only at very high concentrations (1,000 μM) (Fig 4) of protein, where even-higher-order structures are observed.
Attempts to demonstrate functional complementation of Fur by CT296, as previously reported (63), were also unsuccessful. In the previous analysis, polyhistidine-tagged proteins (on the pBAD plasmid) were used in functional complementation assays. While it may seem unlikely that this small tag would have an effect on the function of the proteins, this may explain why their findings differ from ours, which used untagged protein. Additionally, the levels of background repression in the previously reported analysis were quite high (28% reduction in activity with an empty plasmid), especially compared to the moderate repression levels observed by CT296 (53% reduction). This may have resulted from their system of comparing β-galactosidase activity levels between induced and noninduced conditions. Other groups researching Fur homologs have allowed for constitutive expression of protein and have compared β-galactosidase activity levels between iron-depleted and iron-supplemented conditions (40, 52, 54). Utilizing this approach, we were able to assay the repression activities of Fur and CT296 with a very low level of background repression. In this experimental setup, we did not observe functional complementation of Fur by CT296. A direct explanation for these apparently discordant observations is not evident, but not without significant efforts.
One of the primary purposes for elucidating the structure (experimentally or computationally) of a protein of unknown function is to enable functional prediction and facilitate further analyses to confirm the predicted function. Functional assignment typically begins by searching for structural homologs to the protein of unknown function (1). In the case of CT296, this search yielded many structures, all with significant structural similarity (Tables 1 and 3). According to the protein family database, all of the proteins with structural similarity are members of the cupin superfamily of proteins. This superfamily is functionally diverse, but all members share a characteristic β-barrel (12, 26). The proteins with structural similarity to CT296 have relatively different enzymatic functions (hydroxylase, halogenase, dioxygenase, and oxidoreductase), but all are members of the predominant cupin subclass of 2-oxoglutarate-Fe(II)-dependent enzymes (12). These proteins have a well-defined fold termed a double-stranded β-helix (DSBH) in which the enzymatic processes typically occur, including the binding site of the enzymatic cofactors Fe(II) and 2-oxoglutarate.
Despite the correlation of protein function and structural similarity, there are many observations that do not support functional annotation of CT296 as a 2-oxoglutarate-Fe(II)-dependent enzyme in the cupin superfamily. First, the structure of CT296 contains an incomplete DSBH fold, the structural motif that is characteristic of cupin superfamily proteins. The structure of CT296 contains one side of the β-helix consisting of five β strands; however, it lacks an opposing β-sheet that would form the typical DSBH fold. Second, no Fe(II) or 2-oxoglutarate was identified within the CT296 protein structure. Furthermore, attempts to soak CT296 protein crystals with these compounds (independently or in combination) or to generate new CT296 protein crystals in the presence of these compounds did not yield stable crystals (data not shown). Lastly, the vast majority of key residues that function in coordinating Fe(II) and 2-oxoglutarate for the proteins with structural homology are not conserved in CT296. For instance, CT296 shares structural homology to the prolyl hydroxylase PHD2 from Homo sapiens (Tables 1 and 3); however, the residues critical to the enzymatic function of PHD2 do not appear to be retained in CT296. The enzymatic function of PHD2 requires Fe(II) and 2-oxoglutarate, which are coordinated by four residues (His 313, Asp 315, His 374, and Arg 383) (7). The corresponding residues in CT296 (Leu 103, Asp 105, Pro 131, and Asp 141) would likely not coordinate an iron atom or a 2-oxoglutarate molecule. Similar comparisons have been made for EctD from Virgibacillus salexigens (43) and CytC3 from Streptomyces (60), with the same conclusion as PHD2.
In contrast, there are many structural elements of CT296 that are remarkably similar to the arrangements of its structural homologs, as illustrated in Fig. 3. In fact, several secondary structure elements of CT296— four α-helices (α1 to α4) and three β-sheets (β2 to β4)—are all localized together and similarly aligned with homologs. The four α-helices have not been shown to play a role in ligand binding or catalysis but appear to serve as structural support for the double-stranded β-helix (4, 34). While CT296 appears to contain an incomplete double-stranded β-helix, this region of structural homology may be stabilizing the β-sheet region of CT296. Use of this α-helical scaffold to support a function other than non-heme iron-mediated catalysis has not been previously described and may represent a unique application of this structural support. Additionally, CT296 could function as a part of a higher-order structure and the conserved region of surface-exposed α-helices could stabilize an interaction interface with another protein(s). Importantly, additional experimental evidence is needed to appropriately define the function of CT296.
As typically performed, annotation of the Chlamydia trachomatis genomes was done using primary sequence homology to assign function. After application of this strategy, chlamydial genomes have an abundance of ORFs of unknown function—approximately 25% of encoded proteins (5, 47, 50, 53). For Chlamydia pneumoniae, another chlamydial species with an immense impact on public health, the situation is worse, with approximately 35% of encoded proteins characterized as hypothetical. In addition to this report, there are limited data on Chlamydia to demonstrate the utility of protein structure to facilitate functional assignment for uncharacterized proteins. However, a highly supportive and recent example is an analysis of protein encoded by open reading frame CT670. CT670 has very limited primary sequence similarity to any other protein, but the X-ray crystal structure supports that this protein functions in the chlamydial type 3 secretion system and is homologous to YscO from Yersinia pestis (31). Furthermore, this protein was demonstrated to interact in vivo with many other type III secretion components in Chlamydia (31), and functional prediction of CT670 is supported by the inclusion of the encoding gene in an operon that contains type 3 secretion system homologs (18). Outside of Chlamydia, there is an abundance of reports on hypothetical proteins having function assigned based on structure (23, 45, 56). Therefore, more thorough annotations of bacterial genomes (and the chlamydial genome in particular) would benefit greatly from the incorporation of computation and/or experimentally determined structural information.
Another important observation is the ability of I-TASSER to accurately model the three-dimensional structure and predict the function of a protein encoded by C. trachomatis that does not share primary sequence homology with any proteins with functional assignment. While accuracy can be a subjective measurement, the Cα RMSD (2.72 Å) between the CT296 model and experimentally determined structure provides a relative measurement that the model generated by I-TASSER is a very close representation of the physical structure of the protein. It is especially encouraging that none of the templates used in the I-TASSER modeling had sequence identity to the target higher than 25%, and all of the initial templates in threading had a different topology from the experimental structure (the highest TM-score of the template is 0.37), although a substantial portion of the superstructure motifs in templates is close to the target. Proteins with such low similarity to any of the templates represent a “twilight zone” in the structural genome, which is one of the most challenging categories of targets for protein structure prediction.
This study demonstrates that I-TASSER is effective at predicting the structures of proteins that have no function homologs by primary sequence and would therefore be very effective as a complement to primary sequence annotation. While this study also highlights the limitations of structure-based functional annotation, the structural information provides a wealth of molecular information that can be leveraged to elucidate the biochemical function and biological role of a given protein about which relatively little was previously known.
ACKNOWLEDGMENTS
We are very grateful for the technical assistance of Zane Jaafar. We thank Klaus Hantke (University of Tübingen) for bacterial strains and plasmids used in the Fur functional complementation assay.
This research was supported primarily by the J. R. and Inez Jay fund. K.K. was supported by a National Institutes of Health T32 training grant (AI070089), and P.S.H. was supported by National Institutes of Health grant AI079083. Y.Z. is supported by the Alfred P. Sloan Foundation, the National Science Foundation (Career Award 1027394), and the National Institute of General Medical Sciences (GM083107 and GM084222). Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357. Use of the KU COBRE-PSF Protein Structure Laboratory was supported by NIH grant no. P20 RR-17708 from the National Center for Research Resources.
Footnotes
Published ahead of print on 30 September 2011.
REFERENCES
- 1. Adams M. A., Suits M. D., Zheng J., Jia Z. 2007. Piecing together the structure-function puzzle: experiences in structure-based functional annotation of hypothetical proteins. Proteomics 7:2920–2932 [DOI] [PubMed] [Google Scholar]
- 2. Adams P. D., et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol. Crystallogr 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albrecht M., Sharma C. M., Reinhardt R., Vogel J., Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38:868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blasiak L. C., Vaillancourt F. H., Walsh C. T., Drennan C. L. 2006. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 440:368–371 [DOI] [PubMed] [Google Scholar]
- 5. Carlson J. H., Porcella S. F., McClarty G., Caldwell H. D. 2005. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect. Immun. 73:6407–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Centers for Disease Control and Prevention 2010. Sexually transmitted disease surveillance 2009. Centers for Disease Control and Prevention, Atlanta, GA [Google Scholar]
- 7. Chowdhury R., et al. 2009. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 17:981–989 [DOI] [PubMed] [Google Scholar]
- 8. Clifton I. J., et al. 2006. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J. Inorg. Biochem. 100:644–669 [DOI] [PubMed] [Google Scholar]
- 9. Daruzzaman A., Clifton I. J., Adlington R. M., Baldwin J. E., Rutledge P. J. 2006. Unexpected oxidation of a depsipeptide substrate analogue in crystalline isopenicillin N synthase. Chembiochem 7:351–358 [DOI] [PubMed] [Google Scholar]
- 10. D'Autreaux B., et al. 2007. Reversible redox- and zinc-dependent dimerization of the Escherichia coli fur protein. Biochemistry 46:1329–1342 [DOI] [PubMed] [Google Scholar]
- 11. Doublie S. 2007. Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol. Biol. 363:91–108 [DOI] [PubMed] [Google Scholar]
- 12. Dunwell J. M., Purvis A., Khuri S. 2004. Cupins: the most functionally diverse protein superfamily? Phytochemistry 65:7–17 [DOI] [PubMed] [Google Scholar]
- 13. Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 14. Escolar L., Perez-Martin J., de Lorenzo V. 1999. Opening the iron box: transcriptional metalloregulation by the Fur protein. J. Bacteriol. 181:6223–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62:72–82 [DOI] [PubMed] [Google Scholar]
- 16. Ferrer-Costa C., Shanahan H. P., Jones S., Thornton J. M. 2005. HTHquery: a method for detecting DNA-binding proteins with a helix-turn-helix structural motif. Bioinformatics 21:3679–3680 [DOI] [PubMed] [Google Scholar]
- 17. Hantke K. 1987. Selection procedure for deregulated iron transport mutants (fur) in Escherichia coli K 12: fur not only affects iron metabolism. Mol. Gen. Genet. 210:135–139 [DOI] [PubMed] [Google Scholar]
- 18. Hefty P. S., Stephens R. S. 2007. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J. Bacteriol. 189:198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hickey J. M., Weldon L., Hefty P. S. 2011. The atypical OmpR/PhoB response regulator ChxR from Chlamydia trachomatis forms homodimers in vivo and binds a direct repeat of nucleotide sequences. J. Bacteriol. 193:389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holm L., Kaariainen S., Rosenstrom P., Schenkel A. 2008. Searching protein structure databases with DaliLite v. 3. Bioinformatics 24:2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holm L., Park J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16:566–567 [DOI] [PubMed] [Google Scholar]
- 22. Holm L., Sander C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233:123–138 [DOI] [PubMed] [Google Scholar]
- 23. Janowski R., Panjikar S., Eddine A. N., Kaufmann S. H., Weiss M. S. 2009. Structural analysis reveals DNA binding properties of Rv2827c, a hypothetical protein from Mycobacterium tuberculosis. J. Struct. Funct. Genomics 10:137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26:795–800 [Google Scholar]
- 25. Khare D., et al. 2010. Conformational switch triggered by alpha-ketoglutarate in a halogenase of curacin A biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 107:14099–14104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khuri S., Bakker F. T., Dunwell J. M. 2001. Phylogeny, function, and evolution of the cupins, a structurally conserved, functionally diverse superfamily of proteins. Mol. Biol. Evol. 18:593–605 [DOI] [PubMed] [Google Scholar]
- 27. Kim H. S., et al. 2010. Crystal structure of Tpa1 from Saccharomyces cerevisiae, a component of the messenger ribonucleoprotein complex. Nucleic Acids Res. 38:2099–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koo I. C., Walthers D., Hefty P. S., Kenney L. J., Stephens R. S. 2006. ChxR is a transcriptional activator in Chlamydia. Proc. Natl. Acad. Sci. U. S. A. 103:750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koski M. K., et al. 2009. The crystal structure of an algal prolyl 4-hydroxylase complexed with a proline-rich peptide reveals a novel buried tripeptide binding motif. J. Biol. Chem. 284:25290–25301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee J., Wu S., Zhang Y. 2009. Ab initio protein structure prediction, p. 3–25 In Rigden J. D. (ed.), From protein structure to function with bioinformatics. Springer, Dordrecht, Netherlands [Google Scholar]
- 31. Lorenzini E., et al. 2010. Structure and protein-protein interaction studies on Chlamydia trachomatis protein CT670 (YscO homolog). J. Bacteriol. 192:2746–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mariotti S. P., Pascolini D., Rose-Nussbaumer J. 2009. Trachoma: global magnitude of a preventable cause of blindness. Br. J. Ophthalmol. 93:563–568 [DOI] [PubMed] [Google Scholar]
- 33. McCoy A. J., et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McDonough M. A., et al. 2005. Structure of human phytanoyl-CoA 2-hydroxylase identifies molecular mechanisms of Refsum disease. J. Biol. Chem. 280:41101–41110 [DOI] [PubMed] [Google Scholar]
- 35. McDonough M. A., et al. 2006. Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U. S. A. 103:9814–9819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Michaud-Soret I., et al. 1997. Electrospray ionization mass spectrometry analysis of the apo- and metal-substituted forms of the Fur protein. FEBS Lett. 413:473–476 [DOI] [PubMed] [Google Scholar]
- 37. Mihalik S. J., et al. 1997. Identification of PAHX, a Refsum disease gene. Nat. Genet. 17:185–189 [DOI] [PubMed] [Google Scholar]
- 38. Miller J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 39. Potterton L., et al. 2004. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 60:2288–2294 [DOI] [PubMed] [Google Scholar]
- 40. Quatrini R., Lefimil C., Holmes D. S., Jedlicki E. 2005. The ferric iron uptake regulator (Fur) from the extreme acidophile Acidithiobacillus ferrooxidans. Microbiology 151:2005–2015 [DOI] [PubMed] [Google Scholar]
- 41. Rau A., Wyllie S., Whittimore J., Raulston J. E. 2005. Identification of Chlamydia trachomatis genomic sequences recognized by chlamydial divalent cation-dependent regulator A (DcrA). J. Bacteriol. 187:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raulston J. E. 1997. Iron and micronutrients, p. 171–194 In Bavoil P. M., Wyrick P. B. (ed.), Chlamydia: genomics and pathogenesis. Horizon Bioscience, Wymondham, United Kingdom [Google Scholar]
- 43. Reuter K., et al. 2010. Synthesis of 5-hydroxyectoine from ectoine: crystal structure of the non-heme iron(II) and 2-oxoglutarate-dependent dioxygenase EctD. PLoS One 5:e10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roy A., Kucukural A., Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sim D. W., et al. 2009. HP0902 from Helicobacter pylori is a thermostable, dimeric protein belonging to an all-beta topology of the cupin superfamily. BMB Rep. 42:387–392 [DOI] [PubMed] [Google Scholar]
- 46. Srivastava D., et al. 2010. The structure of the proline utilization a proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction. Biochemistry 49:560–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stephens R. S., et al. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759 [DOI] [PubMed] [Google Scholar]
- 48. Terwilliger T. C., et al. 2009. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65:582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Terwilliger T. C., et al. 2008. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64:61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomson N. R., et al. 2008. Chlamydia trachomatis: genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 18:161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsuchiya Y., Kinoshita K., Nakamura H. 2005. PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics 21:1721–1723 [DOI] [PubMed] [Google Scholar]
- 52. Uebe R., et al. 2010. Deletion of a fur-like gene affects iron homeostasis and magnetosome formation in Magnetospirillum gryphiswaldense. J. Bacteriol. 192:4192–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Unemo M., et al. 2010. The Swedish new variant of Chlamydia trachomatis: genome sequence, morphology, cell tropism and phenotypic characterization. Microbiology 156:1394–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vajrala N., Sayavedra-Soto L. A., Bottomley P. J., Arp D. J. 2011. Role of a Fur homolog in iron metabolism in Nitrosomonas europaea. BMC Microbiol. 11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Valegard K., et al. 2004. The structural basis of cephalosporin formation in a mononuclear ferrous enzyme. Nat. Struct. Mol. Biol. 11:95–101 [DOI] [PubMed] [Google Scholar]
- 56. van Staalduinen L. M., et al. 2010. Structure-based annotation of a novel sugar isomerase from the pathogenic E. coli O157:H7. J. Mol. Biol. 401:866–881 [DOI] [PubMed] [Google Scholar]
- 57. Vonrhein C., et al. 2011. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67:293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Warshakoon N. C., et al. 2006. Structure-based design, synthesis, and SAR evaluation of a new series of 8-hydroxyquinolines as HIF-1alpha prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett. 16:5517–5522 [DOI] [PubMed] [Google Scholar]
- 59. Wilmouth R. C., et al. 2002. Structure and mechanism of anthocyanidin synthase from Arabidopsis thaliana. Structure 10:93–103 [DOI] [PubMed] [Google Scholar]
- 60. Wong C., Fujimori D. G., Walsh C. T., Drennan C. L. 2009. Structural analysis of an open active site conformation of nonheme iron halogenase CytC3. J. Am. Chem. Soc. 131:4872–4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu S., Skolnick J., Zhang Y. 2007. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu S., Zhang Y. 2007. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35:3375–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wyllie S., Raulston J. E. 2001. Identifying regulators of transcription in an obligate intracellular pathogen: a metal-dependent repressor in Chlamydia trachomatis. Mol. Microbiol. 40:1027–1036 [DOI] [PubMed] [Google Scholar]
- 64. Xu J., Zhang Y. 2010. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. You Z., Omura S., Ikeda H., Cane D. E., Jogl G. 2007. Crystal structure of the non-heme iron dioxygenase PtlH in pentalenolactone biosynthesis. J. Biol. Chem. 282:36552–36560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Y. 2009. I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77(Suppl. 9):100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Y. 2008. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 18:342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang Y., Skolnick J. 2004. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25:865–871 [DOI] [PubMed] [Google Scholar]
- 69. Zhang Y., Skolnick J. 2005. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33:2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]