

Abstract
The metabolically versatile purple bacterium Rhodobacter sphaeroides 2.4.3 is a denitrifier whose genome contains two periplasmic nitrate reductase-encoding gene clusters. This work demonstrates nonredundant physiological roles for these two enzymes. One cluster is expressed aerobically and repressed under low oxygen while the second is maximally expressed under low oxygen. Insertional inactivation of the aerobically expressed nitrate reductase eliminated aerobic nitrate reduction, but cells of this strain could still respire nitrate anaerobically. In contrast, when the anaerobic nitrate reductase was absent, aerobic nitrate reduction was detectable, but anaerobic nitrate reduction was impaired. The aerobic nitrate reductase was expressed but not utilized in liquid culture but was utilized during growth on solid medium. Growth on a variety of carbon sources, with the exception of malate, the most oxidized substrate used, resulted in nitrite production on solid medium. This is consistent with a role for the aerobic nitrate reductase in redox homeostasis. These results show that one of the nitrate reductases is specific for respiration and denitrification while the other likely plays a role in redox homeostasis during aerobic growth.
INTRODUCTION
Nitrate reduction is widespread among bacteria, where it is used for both assimilatory and dissimilatory processes (14, 37). Assimilatory nitrate reduction produces nitrite, which is further reduced to ammonia and incorporated into cell biomass. Dissimilatory processes include denitrification, the respiration of nitrate to nitrogen gas, and ammonification, i.e., the respiration of nitrate to ammonia. These varied uses for nitrate make its reduction an important reaction in the global nitrogen cycle (5). The use of chemically fixed sources of nitrogen as fertilizer has led to nitrate being a common cause of eutrophication, increasing the significance of nitrate reduction in global biogeochemical cycles (5). Understanding the regulation and physiological roles of nitrate reductases in diverse organisms will allow more accurate modeling and predictions of nitrate fate in the environment.
There is one type of assimilatory nitrate reductase, known as Nas, but there are two dissimilatory types, referred to as Nar and Nap. Nas is cytoplasmic, Nar is membrane bound, and Nap is localized to the periplasm (37). Environmental surveys for the dissimilatory Nap- and Nar-type genes demonstrate an almost equal representation of both groups (4). Many bacteria with Nap have NapABC as the functional core, where NapA is the catalytic subunit and NapB and NapC are heme-containing subunits involved in electron transfer (14). NapA has a molybdenum cofactor as the site of nitrate reduction. NapA and NapB form a periplasmic complex while NapC is membrane associated to mediate electron transfer from quinol to NapAB. While Nap turnover is not directly associated with generation of a proton motive force (PMF), electron flow through NADH dehydrogenase along with the further reduction steps, as in denitrification, will generate a PMF (14, 41).
In some bacteria, Nap activity is not used to support anaerobic respiration. Instead, these Nap enzymes have been shown to play a role in redox homeostasis (14). Redox imbalances can occur as a consequence of rapid catabolism of preferred substrates, the Crabtree effect, or as a consequence of growth on electron-rich substrates (38, 47, 51). Recent work has shown that growth on substrates at oxidation levels similar to or greater than the oxidation level of cell biomass can generate excess reducing equivalents (27). This “overflow” of excess electrons is frequently dissipated via enzymes that, like Nap, are directly uncoupled from energy conservation. The Rhodobacter sphaeroides 2.4.3 genome has two related but nonidentical Nap-encoding gene clusters (40, 43). While many denitrifiers have multiple nitrate reductases, it is rare for one bacterium to have two versions of the same type. While possible, it seems unlikely that these Nap enzymes are fulfilling the same physiological functions in 2.4.3. To gain insight into how a cell might utilize multiple Nap enzymes, the expression patterns and functional roles of each enzyme were determined. As predicted, the two Naps are not redundant since one is used under oxic conditions while the other is used under microoxic conditions.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Three strains of R. sphaeroides were used in this study. Strain 2.4.3 can reduce nitrate to the gaseous end product N2O, while strains 2.4.1 and 2.4.9 are partial denitrifiers, lacking the full suite of enzymes required for reduction of nitrate to gaseous end products, and are used for comparison to strain 2.4.3. Derivatives of these strains are listed in Table 1. Rhodobacter strains were grown in Sistrom's medium (25) at 30°C, and, when necessary, antibiotics were added at the following concentrations: kanamycin, 25 μg/ml; tetracycline, 1 μg/ml; and streptomycin, 50 μg/ml. Flask culture conditions have been previously described (49). For denitrifying growth KNO3 was added to 10 mM. For photosynthetic culture, 15-ml serum vials were filled and sealed. The vials were then incubated at ∼30 cm from two incandescent 75-W bulbs at room temperature with vigorous shaking. For microoxic plate growth, plates were incubated in anaerobic jars with a headspace that was alternately evacuated and N2 flushed for three cycles.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| DH5αF′ | Host for E. coli cloning; F′ 80dlacZΔM15 recA endA1 gyrA96 thi-1 hsdR17(rK− mK−) supE44 relA1 deoR Δ(lacZYA-argF)U169 | 15 |
| JM109 | Host for E. coli cloning | 52 |
| S17-1 | For conjugational transfer of plasmids; recA thi pro hsdRM+ RP4-2-Tc::Mu-Km::Tn7 | 42 |
| S17-1λpir | For conjugational transfer of plasmids | 42 |
| 2.4.3 | Wild-type strain of R. sphaeroides | ATCC 17025 |
| 2.4.1 | Wild-type strain of R. sphaeroides | Type strain |
| 2.4.9 | Wild-type strain of R. sphaeroides | ATCC 17029 |
| prrA strain | prrA::aph, 2.4.3 derivative; Kmr | 24 |
| fnrL strain | 2.4.3 with insertional inactivation of fnrL | 18 |
| ΔccoN strain | 2.4.3 with ΩSm/Sp in ccoN; deletion mutant | 16 |
| nap-β strain | 2.4.3 with insertional inactivation of napA-β; Kmr | This study |
| nap-α strain | 2.4.3 with insertional inactivation of napA-α; Tcr | This study |
| nap-α nap-β strain | 2.4.3 with inactivation of both nap-α and nap-β; Kmr Tcr | This study |
| fnrX strain | 2.4.3 with insertional inactivation of gene 3486; Tcr | This study |
| qsrR strain | 2.4.3 with insertional inactivation of gene 0408; Tcr | This study |
| Plasmids | ||
| pUC19 | Used for cloning in E. coli DH5α (Apr) | 52 |
| pRK415 | Broad-host-range plasmid (Tcr) | 19 |
| pSUP202-1 | Mobilizable suicide vector (Tcr Apr Cmr) | 42 |
| pKOK-6 | Source of lacZ-Km cassette (Tcr Kmr Apr) | 21 |
| pJP5603 | Mobilizable suicide vector (Kmr) | 33 |
| pNAP-βZ | pRK415 with the nap-β promoter fused to lacZ, transcriptional reporter (Tcr Kmr) | This study |
| pNAP-αZ | pRK415 with the nap-α promoter fused to lacZ, transcriptional reporter (Tcr Kmr) | This study |
| pNAP-βKO | pJP5603 with napA internal region (nap-β) (Kmr) | This study |
| pNAP-αKO | pSUP202-1 with napA internal region (nap-α) (Tcr) | This study |
| pFNRXKO | pSUP202-1 with fnrX internal region (Tcr) | This study |
| pQSRRKO | pSUP202-1 with qsrR internal region (Tcr) | This study |
Escherichia coli strain DH5α or JM109 was used for cloning and transformation purposes, and strain S17-1 or S17-1λpir was used for biparental conjugations. E. coli was cultured in Luria-Bertani (LB) medium (26) at 32°C, and, when necessary, antibiotics were added at the following concentrations: ampicillin, 100 μg/ml; kanamycin, 25 μg/ml; tetracycline, 10 μg/ml; streptomycin, 25 μg/ml.
Construction of plasmids and strains.
Chromosomal DNA was isolated from strain 2.4.3 using a Wizard Genomic DNA Purification Kit (Promega Corp.). All oligonucleotide primers were purchased from Integrated DNA Technologies (IDT). In most cases, restriction sites were added to the 5′ ends of the primers to facilitate cloning, and these are underlined in primer sequences. PCR, restriction digests, and ligations were done according to standard protocols. Plasmid isolation was done by the alkaline lysis method (26). Transformations were done using the transformation and storage solution (TSS) method (8). Plasmids are listed in Table 1.
lacZ expression fusions.
Expression fusions were constructed for the nap-β cluster (Rsph17025_3218 to Rsph17025_3224) (accession numbers YP_001169407 to YP_001169413) and the nap-α cluster (Rsph17025_3986 to Rsph17025_3992) (accession numbers YP_001170145 to YP_001170151). For the nap-β fusion, pNAP-βZ, the predicted promoter was amplified using primers 5′-CCCGAATTCCCATGACATCGACACCGAC-3′ and 5′-CCCGTCGACCAGGATCCGCAGAAGGTG-3′ (restriction sites are underlined). This region corresponds to 896 bases upstream of the predicted start of the open reading frame of the first gene in the Nap-encoding gene cluster. This amplicon was cloned into pUC19 via EcoRI and SalI and verified by sequencing. The promoter was cloned into the broad-host-range vector pRK415, which has a copy number of 4 to 6 in R. sphaeroides (9, 19). The lacZ cassette of pKOK-6 was cloned via PstI, and orientation was confirmed. The final pNAP-βZ construct was transformed into E. coli S17-1 and subsequently conjugated into Rhodobacter strains (2.4.3, 2.4.1, and 2.4.9 and the prrA, fnrL, nap-α nap-β, and fnrX strains). For the pNAP-αZ construct, the predicted nap-α promoter was amplified using primers 5′-GCGGAATTCACTATCTCGTGGTCGGCC-3′ and 5′-GCGCTGCAGCAGGAACGGCCAGATCAC-3′ (restriction sites are underlined). This corresponds to 682 bases upstream of the cluster start. The amplicon was cloned into pUC19 via EcoRI and PstI and verified by sequencing. The promoter was cloned into pRK415, and the lacZ cassette of pKOK-6 was cloned via PstI. The final pNAP-αZ construct was then transformed into E. coli S17-1 and subsequently conjugated into Rhodobacter strains (2.4.3, 2.4.1, and 2.4.9 and the prrA, ΔccoN, and qsrR strains).
Mutant strains.
The nap-α mutant was generated by homologous recombination using the suicide vector pSUP202-1. First, an internal region of napA was amplified using primers 5′-CGCGAATTCAGTGGACGATCTGGGAGG-3′ and 5′-CGGCTGCAGAGCGAGAAGGGCGAATTG-3′ (restriction sites are underlined). This amplicon was cloned into pUC19 and verified by sequencing. The gene fragment was then cloned into the suicide plasmid pSUP202-1 via EcoRI and PstI. The final construct, pNAP-αKO (where KO is knockout), was transformed into E. coli S17-1 and conjugated into R. sphaeroides 2.4.3. The gene disruption was confirmed by PCR using primers flanking the crossover region. This mutant causes a disruption of napA via homologous recombination and results in integration of the suicide plasmid into the genome.
The nap-β mutant was generated by homologous recombination using the suicide vector pJP5603. First, an internal region of napA was targeted for amplification using primers 5′-AACATGGCCGARATGCAYCC-3′ and 5′-CCSCGSACRTGCTGGTTGAASCC-3′. These primers are degenerate because at the time this gene was disrupted, the genome sequence of 2.4.3 was unavailable, and these primers were based on napA sequence from strain 2.4.1 and related proteobacteria. This amplicon was cloned into pUC19 via HincII and verified by sequencing. The gene fragment was then cloned into pJP5603 via BamHI and HindIII. The final construct, pNAP-βKO, was verified and transformed into E. coli S17-1λpir. This strain was used for conjugation with R. sphaeroides 2.4.3. Gene disruption was verified by PCR. This mutant causes a disruption of napA via homologous recombination and results in integration of the suicide plasmid into the genome. The double nap-α nap-β mutant was generated by starting with the nap-β mutant and conjugating in the construct to generate the nap-α disruption. Gene disruptions in this mutant were also verified by PCR.
Gene Rsph17025_3486 (accession number YP_001169666) encodes the Fnr-type regulator that was targeted as a potential regulator of nap-β. This gene was inactivated by insertional inactivation. A central region of the gene, not including either end, was amplified using primers 5′-CATCTGCAGAGATCGTGGACTTCCAGG-3′ and 5′-CACGAATTCGAGAGCGGGCAGGCATAG-3′ (restriction sites are underlined). This amplicon was cloned into pUC19 via EcoRI and PstI and verified by sequencing. The gene fragment was then moved into the suicide vector pSUP202-1 via EcoRI and PstI. The final pFNRXKO construct was transformed into E. coli S17-1 and then conjugated into strain 2.4.3 to generate the mutant. Inactivation of the gene was verified by PCR using primers flanking the Rsph17025_3486 open reading frame. The nap-β::lacZ construct, pNAP-βZ, was then conjugated into the mutant to test nap-β expression.
The gene at locus 0408 (accession number YP_001166619) encodes the LuxR-type regulator targeted as a potential regulator of nap-α. This gene has been designated qsrR in strain 2.4.1 (17). A central region of the gene was amplified using primers 5′-CACCTGCAGCTTCTCAACGAGACCTTC-3′ and 5′-CACGAATTCCACAGGGCCGCACCAAG-3′ (restriction sites are underlined). The amplicon was cloned into pUC19 via EcoRI and PstI and verified by sequencing. The fragment was then cloned into pSUP202-1 via EcoRI and PstI. The final pQSRRKO construct was transformed into E. coli S17-1 and then conjugated into strain 2.4.3, generating the mutant. Inactivation of the gene was verified by PCR using primers flanking the open reading frame. The nap-α::lacZ construct, pNAP-αZ, was then conjugated into the mutant to test nap-α expression.
Enzyme assays.
Gene expression was measured via β-galactosidase activity as previously described (26). Activity was determined based on three independently grown cultures. In all cases, expression was monitored throughout growth, and representative values for growth phases of interest were averaged. Error bars represent one standard deviation.
Nitrate reductase activity was measured using methyl viologen (0.2 mM) as an artificial electron donor (45). Nitrate (500 μM) was added to cells along with the artificial electron donor to initiate the assay. All assays were incubated for the same amount of time and then terminated by vigorous vortexing to oxidize the methyl viologen. A common assay incubation time allowed direct comparison of nitrite production. Activity was based on three independently grown cultures. Error is represented as one standard deviation.
Nitrate was measured in nitrate (10 mM)-amended Sistrom's medium using the Szechrome reaction (Polysciences, Inc.). The reaction uses 4-(phenylamino) benzenesulfonic acid dissolved in 1:1 phosphoric acid-sulfuric acid for colorimetric detection of nitrate. Samples of the culture medium were taken at various time points and diluted 20-fold. Szechrome reagent was then added, and the absorbance was recorded at 570 nm. When nitrate was no longer detectable, the assay was repeated without culture dilution to confirm the absence of nitrate. A nitrate standard curve (0 to 20 mM) was generated to convert absorbance values to concentrations. The correlation between absorbance and concentration was linear over the range of concentrations (0 to 10 mM) relevant to this study.
Nitrite was detected by the Griess reaction (29). The Griess reaction allows colorimetric detection of nitrite through the formation of an azo-dye upon addition of sulfanilic acid and N-naphthyl-ethylenediamine. For liquid culture, 50 μl of culture medium containing cells was used for assays. For solid medium, samples were taken adjacent to the thickest growth of cells by extracting agar plugs using the blunt end of a 200-μl pipette tip. The agar plugs were melted in 100 μl of H2O, and then 50 μl was used for assays. Since nitrite diffusion throughout the plate from the point of production was difficult to assess, this made quantification of nitrite produced by the cells difficult. Therefore, the data are simply reported as absorbance at 540 nm allowing qualitative comparisons between samples.
RESULTS
Sequence comparison of the two nap operons.
The genome of R. sphaeroides strain 2.4.3 contains two related but nonidentical nap clusters. The difference in gene order and gene content makes it unlikely that these two operons arose by a simple duplication event. One cluster is located on the largest plasmid (pRSPA02; 289,489 bp), extending from Rsph17025_3986 to Rsph17025_3992 and including seven genes identified as napKEFDABC. This gene organization is most similar to nap clusters from closely related alphaproteobacteria, such as Paracoccus pantotrophus, that utilize this enzyme for redox homeostasis (39). Following the previously established nomenclature for nap gene clusters in bacteria with multiple clusters, this nap cluster has been designated nap-α (43).
The second nap cluster is located on chromosome II (pRSPA01; 877,879 bp) from Rsph17025_3218 to Rsph17025_3224 and includes seven genes designated napFDAGHBC. The inclusion of napGH differentiates this cluster from the nap-α cluster and makes it more similar in organization to nap-β clusters from gammaproteobacteria, including several Shewanella species and E. coli (43). NapGH likely form a quinol dehydrogenase and may transfer electrons to NapBA (20). In many bacteria, this Nap supports anaerobic growth via nitrate respiration (14). This nap cluster has been designated nap-β. Genes common to both clusters produce products with high identity, for example, the two NapA proteins are 70% identical.
Expression and activity of nap-β.
Expression of nap-β increased 16-fold as cells transitioned from oxic to microoxic conditions (Fig. 1). Inclusion of nitrate did not impact expression nor did Nap activity (Fig. 1). Under photosynthetic conditions, on solid medium, and with various carbon sources, expression of nap-β followed the same oxygen-dependent pattern (data not shown).
Fig. 1.

Expression of the nap-β operon as measured by β-galactosidase activity in various R. sphaeroides strains and under various conditions. The prrA, fnrL, and nap-α nap-β strains are mutants of strain 2.4.3 in which these genes have been inactivated. Strains 2.4.1 and 2.4.9 are wild-type strains of R. sphaeroides. Black bars represent aerobic conditions, white bars represent microoxic conditions, and gray bars represent microoxic conditions supplemented with 10 mM nitrate.
The contribution of Nap-β to overall Nap activity was assessed by measuring nitrate levels in the culture during an oxic to microoxic transition under nonphotosynthetic conditions (Fig. 2). Nitrite production cannot be measured in this case since under microoxic conditions nitrite reductase will consume any nitrite produced. The first 15 to 20 h represents aerobic nondenitrifying growth during which there is no decrease in nitrate. During this time, the cell density of all cultures increased from an absorbance at 600 nm of less than 0.1 to an absorbance of 0.8 to 1.0, after which the cell density was stable. After the shift to microaerobic denitrifying growth, the wild type and the nap-α-deficient mutant showed the same rate of nitrate consumption, consistent with Nap-β playing a major role in denitrification (Fig. 2). The amount of nitrate reduced by the nap-β mutant was 50% of that reduced by wild-type cells under the same conditions over the same culture duration (Fig. 2). Nitrate levels did not change in the nap-α nap-β double mutant, demonstrating that there are no other nitrate-reducing enzymes expressed under these conditions (Fig. 2).
Fig. 2.
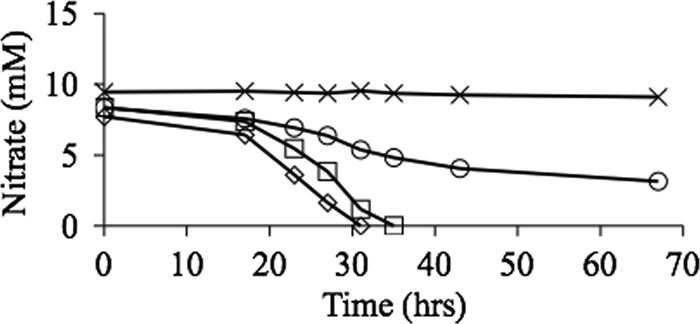
Nitrate consumption during growth of denitrifying liquid medium cultures of 2.4.3 or the various nap mutants. Cultures were started with 10 mM nitrate, and the consumption of nitrate was monitored over time using the Szechrome reaction to detect nitrate. ♢, wild-type 2.4.3; □, nap-α strain; ○, nap-β strain; ×, nap-α nap-β strain.
The relative contribution of each Nap to cellular nitrate reductase activity was also tested under photosynthetic conditions. Unexpectedly, nitrite was found to accumulate under these anoxic conditions. The nap-α mutant produced the same levels of nitrite as the wild type. The nap-β mutant produced significantly less nitrite while, as expected, the nap-α nap-β mutant produced no nitrite (data not shown). The absence of Nir activity was explained by an increase in the pH of the medium from 7.0 to >9.0 as cells transitioned from lag to stationary phase (data not shown). Nir activity has been shown to decrease significantly as pH increases (32). As the pH increased, whole-cell Nir activity decreased to nearly undetectable levels, demonstrating that Nap-β remains functional in this pH range while Nir does not (data not shown).
Expression and activity of nap-α.
In R. sphaeroides strain 2.4.1, expression of the orthologous nap-α cluster increases significantly during a transition from anoxic photosynthetic growth to aerobic growth (1). This growth pattern was simulated in 2.4.3 by initiating growth under anoxic photosynthetic conditions, followed by a switch to oxic conditions. Under this growth regime, there was a 7-fold increase in expression of nap-α after the shift to high oxygen levels (Fig. 3). Expression of a nap-α::lacZ fusion did not decrease significantly in an oxic to microoxic transition, probably because the stability of LacZ masks any decrease in transcription under these conditions.
Fig. 3.
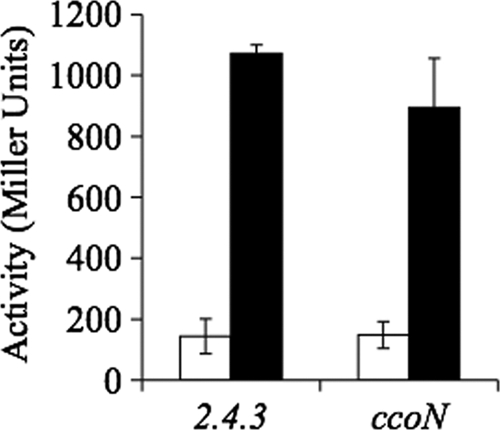
Expression of the nap-α operon in liquid culture as measured by β-galactosidase activity in R. sphaeroides 2.4.3. The ccoN strain is a mutant of strain 2.4.3 in which this gene has been inactivated. The white bars represent expression under anoxic photosynthetic conditions, and all cultures were started under this condition. Black bars represent expression in cultures that were transitioned from anoxic photosynthetic growth to oxic growth. Values represent the maximal expression value under each condition.
nap-α expression was also monitored in cells grown on agar under atmospheric levels of oxygen, and maximal expression was consistently about 4-fold higher than the maximal levels in highly aerated liquid culture (Fig. 4). An increase in expression on agar was unexpected because the bulk of the cells in a colony experience oxygen levels lower than atmospheric levels (34), and expression of nap-α in liquid culture was proportional to oxygen levels (Fig. 3). Addition of 10 mM nitrate to the agar decreased nap-α expression about 45% (Fig. 4). nap-α expression was also examined on solid medium containing different carbon sources. No significant differences in expression were observed during growth on 5 mM malate, 10 mM succinate, and 10 mM butyrate (data not shown). In all cases, expression was about 4-fold higher than during growth on succinate in liquid culture.
Fig. 4.
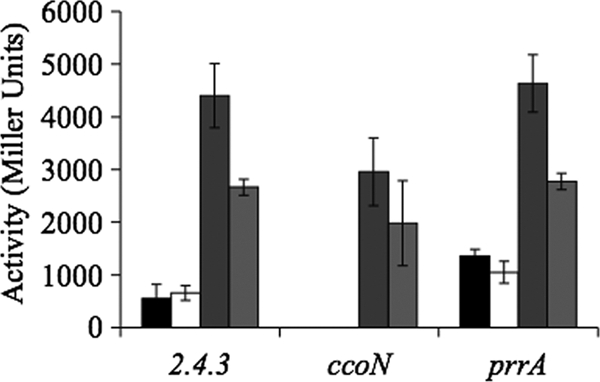
Expression of nap-α in cells growing on agar plates under various conditions as measured by β-galactosidase activity. The ccoN and prrA strains are mutants of strain 2.4.3 in which these genes have been inactivated. Black bars represent expression under microoxic growth, white bars represent expression under microoxic growth with 10 mM nitrate, dark gray bars represent expression under oxic growth, and light gray bars represent expression under oxic growth with 10 mM nitrate. There are no microoxic values for the ccoN strain because this mutant cannot grow under low-oxygen conditions.
Nap-α activity was monitored under various conditions to determine if expression levels correlated with activity. Strain 2.4.3 cells growing in liquid medium under oxic conditions did not produce nitrite until cultures had been in stationary phase for >24 h (data not shown). Importantly, no nitrite was produced during early log phase, when expression is maximal. These assays were done under oxic conditions, making nitrite a valid measure of Nap activity since any nitrite produced will accumulate in the medium as Nir is not expressed (50). To confirm that intact Nap-α was present in cells from liquid medium, the nitrate reductase activity of harvested cells was measured in the presence of dithionite and methyl viologen. With reduced methyl viologen as an electron source, wild-type cells were able to reduce nitrate to nitrite, demonstrating the presence of a nitrate reductase. In comparison tests, cells of the nap-α mutant had <20% of wild-type nitrate reductase activity (data not shown). The nap-β mutant had wild-type levels of Nap activity in cells grown under oxic conditions, while under the same condition cells of the nap-α nap-β double mutant had no detectable Nap activity. Together, these data indicate that Nap-α is expressed and functional but is not physiologically active in cells growing in liquid medium under oxic conditions.
It was postulated that the increase in nap-α expression on solid medium was correlated with an increased need for nitrate reductase activity for redox balancing and overflow metabolism. To test this, Nap activity during growth on solid medium was assessed by testing for nitrite accumulation in the agar. When colonies were incubated under atmospheric levels of oxygen, nitrite accumulated to significant levels (Fig. 5). Consistent with expression patterns, the nap-α mutant did not produce any detectable nitrite under these conditions. The nap-β mutant accumulated levels of nitrite comparable to the wild-type level (Fig. 5). Nap assays using artificial electron donors demonstrated that cells of the nap-α mutant growing on solid medium had very low nitrate reductase activity while the nap-β mutant had wild-type levels of Nap activity (data not shown). These results suggest that Nap-α is likely being used for redox homeostasis in cells growing on solid medium. Nap-β does not contribute to the nitrite production, and as there is no evidence of Nir activity, nitrogen oxide reduction is not being used for respiratory purposes.
Fig. 5.
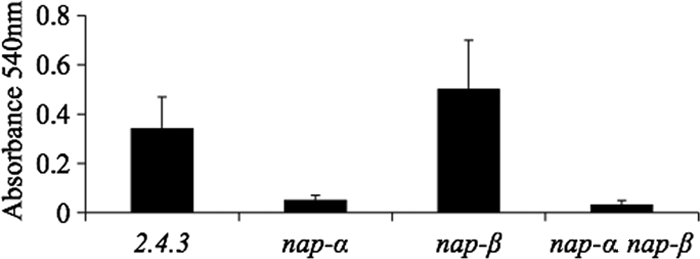
Nitrite produced during growth on aerobic plates for strain 2.4.3 and the various nap mutants. Nitrite levels are given by absorbance at 540 nm (see Materials and Methods for an explanation).
To determine if the oxidation state of the carbon source impacted Nap-α activity, 2.4.3 was grown on different carbon sources on medium containing nitrate. Growth on pyruvate and lactate resulted in nitrite production levels similar to the level produced during growth on succinate (not shown). Strain 2.4.3 grew very slowly on butyrate, regardless of the presence or absence of nitrate. The amount of nitrite produced was similar to the amount produced during growth on succinate. Unexpectedly, strain 2.4.3 was unable to grow on malate-containing medium if 10 mM nitrate was present. Growth was restored if nitrate was reduced to 5 mM, and under this condition no nitrite was produced.
Control of nap-β expression.
The observed pattern of nap-β expression suggests that oxygen is the major environmental factor influencing expression. Expression of nap-β did not change in mutant strains lacking two of the global oxygen-responsive regulators, PrrA and FnrL (Fig. 1) (12, 53). Wild-type expression in the fnrL mutant was surprising since the nap-β promoter region contains an Fnr-type binding motif (5′-TTGATccatATCAA-3′ [lowercase letters indicate variable residues that are not involved in binding]) centered −81.5 bp upstream of the predicted translation start (22). Since nap-β was not in the FnrL regulon, it was postulated that the Fnr responsible for controlling nap-β expression would be present only in those strains with nap-β, namely, strains 2.4.3 and 2.4.1. As predicted, nap-β expression in 2.4.1 was similar to that in strain 2.4.3, while expression in R. sphaeroides 2.4.9 was not strongly induced by decreasing oxygen (Fig. 1). The R. sphaeroides 2.4.3 genome contains 10 genes encoding putative Fnr-type regulators. Of this set, 2.4.3, 2.4.1, and 2.4.9 share a core set of five conserved FNR proteins including FnrL, NnrR, and FixK (22). Of the remaining putative regulators, four are not highly conserved across the three strains. The final putative regulator, Rsph17025_3486, is highly conserved in 2.4.1 (88% identity) but is only 68% identical to its closest relative in 2.4.9; this was considered a candidate for regulation of nap-β. However, inactivation of Rsph17025_3486 did not change nap-β expression (data not shown).
Control of nap-α expression.
Microarray studies of the PrrA regulon in strain 2.4.1 suggest that nap-α is repressed by PrrA (12). To test for PrrA-dependent regulation of nap-α in 2.4.3, expression was assessed in a prrA mutant background. In cells experiencing an oxic to microoxic transition in liquid culture, there was no significant difference between nap-α expression levels in the wild-type and the prrA strains (not shown). Transition from anoxic to oxic conditions could not be carried out since prrA mutants will not grow photosynthetically. Using an alternative strategy, nap-α expression was assessed in a ΔccoN mutant background. This mutant, which lacks the cbb3-cytochrome oxidase, has been demonstrated to have higher levels of phosphorylated PrrA (PrrA∼P) under oxic conditions (36). The enhanced levels of PrrA∼P under oxic conditions did not impact nap-α expression since no change in final levels of expression were observed after a transition from photosynthetic growth to oxic conditions (Fig. 3). Expression of nap-α in the ccoN and prrA mutants was also measured on solid medium (Fig. 4). Under oxic conditions maximal nap-α expression in the ΔccoN mutant was 65% of the wild-type level, but there was no difference between expression in the wild-type and prrA mutant strains (Fig. 4). Under low oxygen there was 3-fold higher expression of nap-α in the prrA mutant than in wild type while expression could not be tested in the ccoN mutant since it will not grow under microoxic conditions (Fig. 4).
A previous report has linked the quorum-sensing regulator, QsrR, to expression of a number of redox proteins in R. sphaeroides 2.4.1, including nitrate reductase (17). Density-dependent expression might explain the observed increase in nap-α expression on solid medium although the physiological rationale for this would be unclear. To test this, a strain was constructed in which the qsrR ortholog in 2.4.3 was insertionally inactivated. Expression of nap-α in this strain on solid medium was identical to that of the wild type with expression high under atmospheric oxygen and repressed under low oxygen (data not shown). Expression in the mutant was also indistinguishable from wild-type under photosynthetic, anoxic conditions and in aerobic liquid culture (not shown). The qsrR mutant also had similar Nap activity to wild-type under all conditions tested. These results indicate that QsrR is not an important regulator of nap-α expression in 2.4.3.
DISCUSSION
While it is not uncommon for bacteria to have multiple nitrate reductases, it is rare for a bacterium to have multiple nitrate reductases of the same type. Among the alphaproteobacteria, genome analysis shows that only Pseudovibrio sp. JE062 and R. sphaeroides 2.4.3 have two nap clusters. Among other proteobacteria, two nap clusters are also found in a notable number of the Shewanella strains (43, 7). In Shewanella piezotolerans WP3, both nap gene clusters are involved in nitrate respiration, seemingly redundant in function (7). However, in mutant studies, one of the nap gene clusters provides a growth benefit over the other (7). In strain 2.4.3 the different gene order and gene content make it unlikely nap-α and nap-β are paralogous. Given the limited distribution of nap-β among the alphaproteobacteria, it may have been acquired via lateral gene transfer. However, the GC content of the cluster provides little clue as to the origin. Other than napC-β, which has a GC content of 63.4%, the rest of the nap-β cluster has a GC content that is near or slightly above the 68.2% GC content of the genome. The nap-β genes all show a significant preference for G or C in the third codon position. These data suggest either that nap-β has been in the genome long enough to be nearly fully adapted to the GC usage of R. sphaeroides or that it was acquired from a bacterium with a similar GC content.
Nap-α activity was detected only in cells growing on solid medium (Fig. 5). Nitrite production was not reliant on reduced substrate but occurred even with substrates at oxidation levels similar to the oxidation level of cell biomass (27). However, growth on malate, a substrate more oxidized than biomass, did not lead to nitrite production on solid medium. This result strongly supports the hypothesis that nitrite production on solid medium is a result of imbalances in the availability of electron donors and acceptors. As a colony forms on the surface of the plate, cells close to the agar will become oxygen limited as the colony gets larger. However, due to the diffusion of substrates within the agar, the cells near the agar surface will continue to have ready access to carbon. These gradients will likely produce imbalances in availability of the electron donors and acceptors. There are some similarities between plate culture and a natural mode of bacterial growth, the biofilm. In both instances, cell accumulation results in steep gradients of nutrients and oxygen (44). Under these dynamic conditions, Nap-α would be useful for redox homeostasis. Nap-α activity on plates was not specific to strain 2.4.3 since 2.4.1 also produced nitrite when it was grown on solid medium (data not shown). The absence of nitrite production in liquid culture likely reflects the fact that under these conditions the cells do not experience the electron donor and acceptor gradients that occur during growth on a solid surface.
While growth on agar increased nap-α expression, the regulatory mechanism leading to this increase in expression is not clear. It seems likely that nap-α expression requires multiple regulators, and there may be a regulatory system that responds to redox imbalances in the cell. This could explain why nap-α expression is reduced in the presence of nitrate on solid medium since nitrate reduction could be used to mitigate redox imbalances in the cell. While there is enhanced nap-α expression on plates, expression in this milieu still responds to decreases in external oxygen concentrations, likely by PrrBA-dependent repression. Cells growing under low-oxygen conditions on solid medium had levels of nap-α expression similar to the levels found in liquid medium under the same conditions. There is also a measurable decrease in nap-α expression in the ΔccoN mutant, which has higher levels of PrrA∼P (30, 31). Loss of the cbb3 oxidase, the high-affinity terminal oxidase, would likely lead to overall higher oxygen levels in the colony (16, 35). A higher level of oxygen is not consistent with the observed decrease in nap-α expression, suggesting that increased levels of PrrA∼P are blocking the increased expression occurring on agar (1). Of note, the observed Nap-α expression pattern and activity are distinct from those of Nap-α of the related organism Rhodobacter sphaeroides f. sp. denitrificans, where this enzyme is dedicated to denitrification and expressed under anoxic conditions (48). Instead, the Nap-α data are more consistent with the Nap studies from Paracoccus denitrificans, where the enzyme plays a role in redox homeostasis while a respiratory nitrate reductase (Nar) is dedicated to denitrification (10, 11, 39).
nap-β expression is inversely proportional to oxygen levels. nap-β is somewhat unusual in that it is not part of either the FnrL or PrrBA regulon in spite of its products being important bioenergetic components under microoxic growth conditions (12). The lack of nitrate control was also unexpected. In some organisms, the nap-α cluster is not regulated in response to nitrate but is, instead, regulated in response to redox signals, consistent with a physiological role for Nap-α as an overflow outlet (6, 10, 11, 13). Nap-β plays a respiratory role, and most genes encoding enzymes of this type require the presence of substrate for maximal expression. In R. sphaeroides this occurs for dimethyl sulfoxide (DMSO) reductase in 2.4.1 and nitrite and NO reductase in 2.4.3 (23, 28). The nap-β orthologs in Shewanella and E. coli require nitrate for expression (2, 46). It is not known if the nap-β orthologs in related alphaproteobacteria require nitrate for maximal expression. In E. coli NapGH has specificity for ubiquinol, perhaps constraining Nap function to quinone type (3). Ubiquinol is the major quinone used by R. sphaeroides, suggesting that NapGH may have a similar functional role in both bacteria.
The multiple types and functional roles of nitrate reductases demonstrate how each enzyme can be tailored to fit physiological needs, which will be governed by the lifestyle of each bacterial species (14, 37). Among the Nap enzymes, the functional specificity may be conferred by the accessory genes found in different operons or by differences in the core NapABC subunits. This study has demonstrated distinct physiological roles for Nap-α and Nap-β, making strain 2.4.3 an excellent system for further study of the enzymological features possibly constraining Nap function.
ACKNOWLEDGMENTS
We thank Peter Choi and Iichiro Shiratsuchi for preliminary aspects of this work.
Portions of this work were supported by Department of Energy grant 95ER20206.
Footnotes
Published ahead of print on 23 September 2011.
REFERENCES
- 1. Arai H., Roh J. H., Kaplan S. 2008. Transcriptome dynamics during the transition from anaerobic photosynthesis to aerobic respiration in Rhodobacter sphaeroides 2.4. 1. J. Bacteriol. 190:286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beliaev A. S., et al. 2005. Global transcriptome analysis of Shewanella oneidensis MR-1 exposed to different terminal electron acceptors. J. Bacteriol. 187:7138–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brondijk T. H. C., Nilavongse A., Filenko N., Richardson D. J., Cole J. A. 2004. NapGH components of the periplasmic nitrate reductase of Escherichia coli K-12: location, topology and physiological roles in quinol oxidation and redox balancing. Biochem. J. 379:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bru D., Sarr A., Philippot L. 2007. Relative abundances of proteobacterial membrane-bound and periplasmic nitrate reductases in selected environments. Appl. Environ. Microbiol. 73:5971–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Canfield D. E., Glazer A. N., Falkowski P. G. 2010. The evolution and future of earth's nitrogen cycle. Science 330:192–196 [DOI] [PubMed] [Google Scholar]
- 6. Castillo F., et al. 1996. Molecular and regulatory properties of the nitrate reducing systems of Rhodobacter. Curr. Microbiol. 33:341–346 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y., Wang F., Xu J., Mehmood M. A., Xiao X. 2011. Physiological and evolutionary studies of NAP systems in Shewanella piezotolerans WP3. ISME J. 5:843–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung C. T., Niemela S. L., Miller R. H. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. U. S. A. 86:2172–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donohue T., Kaplan S. 1991. Genetic techniques in Rhodospirillaceae. Methods Enzymol. 204:459–485 [DOI] [PubMed] [Google Scholar]
- 10. Ellington M. J., Fosdike W. L., Sawers R. G., Richardson D. J., Ferguson S. J. 2006. Regulation of the nap operon encoding the periplasmic nitrate reductase of Paracoccus pantotrophus: delineation of DNA sequences required for redox control. Arch. Microbiol. 184:298–304 [DOI] [PubMed] [Google Scholar]
- 11. Ellington M. J., Bhakoo K. K., Sawers G., Richardson D. J., Ferguson S. J. 2002. Hierarchy of carbon source selection in Paracoccus pantotrophus: strict correlation between reduction state of the carbon substrate and aerobic expression of the nap operon. J. Bacteriol. 184:4767–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eraso J., et al. 2008. Role of the global transcriptional regulator PrrA in Rhodobacter sphaeroides 2.4.1: combined transcriptome and proteome analysis. J. Bacteriol. 190:4831–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gavira M., Roldan M., Castillo F., Moreno-Vivian C. 2002. Regulation of nap gene expression and periplasmic nitrate reductase activity in the phototrophic bacterium Rhodobacter sphaeroides DSM158. J. Bacteriol. 184:1693–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gonzalez P. J., Correia C., Moura I., Brondino C. D., Moura J. J. 2006. Bacterial nitrate reductases: molecular and biological aspects of nitrate reduction. J. Inorg. Biochem. 100:1015–1023 [DOI] [PubMed] [Google Scholar]
- 15. Hanahan D. 1985. Techniques for transformation of E. coli, p. 109–135 In Glover D. M. (ed.), DNA cloning: a practical approach, vol. 1 IRL Press, Oxford, United Kingdom [Google Scholar]
- 16. Hartsock A., Shapleigh J. P. 2010. Mechanisms of oxygen inhibition of nirK expression in Rhodobacter sphaeroides. Microbiology 156:3158–3165 [DOI] [PubMed] [Google Scholar]
- 17. Hwang W., Lee K. E., Lee J. K., Park B. C., Kim K. S. 2008. Genes of Rhodobacter sphaeroides 2.4.1 regulated by innate quorum-sensing signal, 7,8-cis-N-(tetradecenoyl) homoserine lactone. J. Microbiol. Biotechnol. 18:219–227 [PubMed] [Google Scholar]
- 18. Jain R., Shapleigh J. 2001. Characterization of nirV and a gene encoding a novel pseudoazurin in Rhodobacter sphaeroides 2.4.3. Microbiology 147:2505–2515 [DOI] [PubMed] [Google Scholar]
- 19. Keen N. T., Tamaki S., Kobayashi D., Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in Gram-negative bacteria. Gene 70:191–197 [DOI] [PubMed] [Google Scholar]
- 20. Kern M., Mager A. M., Simon J. 2007. Role of individual nap gene cluster products in NapC-independent nitrate respiration of Wolinella succinogenes. Microbiology 153:3739–3747 [DOI] [PubMed] [Google Scholar]
- 21. Kokotek W., Lotz W. 1989. Construction of a lacZ-kanamycin-resistance cassette, useful for site-directed mutagenesis and as a promoter probe. Gene 84:467–471 [DOI] [PubMed] [Google Scholar]
- 22. Korner H., Sofia H., Zumft W. 2003. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol. Rev. 27:559–592 [DOI] [PubMed] [Google Scholar]
- 23. Kwiatkowski A., Shapleigh J. 1996. Requirement of nitric oxide for induction of genes whose products are involved in nitric oxide metabolism in Rhodobacter sphaeroides 2.4.3. J. Biol. Chem. 271:24382–24388 [DOI] [PubMed] [Google Scholar]
- 24. Laratta W., Choi P., Tosques I., Shapleigh J. 2002. Involvement of the PrrB/PrrA two-component system in nitrite respiration in Rhodobacter sphaeroides 2.4.3: evidence for transcriptional regulation. J. Bacteriol. 184:3521–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lueking D. R., Fraley R. T., Kaplan S. 1978. Intracytoplasmic membrane synthesis in synchronous cell populations of Rhodopseudomonas sphaeroides. J. Biol. Chem. 253:451–457 [PubMed] [Google Scholar]
- 26. Maniatis T., Fritsch E. F., Sambrook J. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. McKinlay J. B., Harwood C. S. 2010. Carbon dioxide fixation as a central redox cofactor recycling mechanism in bacteria. Proc. Natl. Acad. Sci. U. S. A. 107:11669–11675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mouncey N. J., Kaplan S. 1998. Cascade regulation of dimethyl sulfoxide reductase (dor) gene expression in the facultative phototroph Rhodobacter sphaeroides 2.4.1. J. Bacteriol. 180:2924–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nicholas D., Nason A. 1957. Determination of nitrate and nitrite. Methods Enzymol. 3:981–984 [Google Scholar]
- 30. O'Gara J., Eraso J., Kaplan S. 1998. A redox-responsive pathway for aerobic regulation of photosynthesis gene expression in Rhodobacter sphaeroides 2.4.1. J. Bacteriol. 180:4044–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oh J., Ko I., Kaplan S. 2004. Reconstitution of the Rhodobacter sphaeroides cbb3-PrrBA signal transduction pathway in vitro. Biochemistry 43:7915–7923 [DOI] [PubMed] [Google Scholar]
- 32. Olesen K., et al. 1998. Spectroscopic, kinetic, and electrochemical characterization of heterologously expressed wild-type and mutant forms of copper-containing nitrite reductase from Rhodobacter sphaeroides 2.4.3. Biochemistry 37:6086–6094 [DOI] [PubMed] [Google Scholar]
- 33. Penfold R. J., Pemberton J. M. 1992. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene 118:145–146 [DOI] [PubMed] [Google Scholar]
- 34. Peters A. C., Wimpenny J. W., Coombs J. P. 1987. Oxygen profiles in, and in the agar beneath, colonies of Bacillus cereus, Staphylococcus albus and Escherichia coli. J. Gen. Microbiol. 133:1257–1263 [DOI] [PubMed] [Google Scholar]
- 35. Preisig O., Zufferey R., Thony-Meyer L., Appleby C. A., Hennecke H. 1996. A high-affinity cbb3-type cytochrome oxidase terminates the symbiosis-specific respiratory chain of Bradyrhizobium japonicum. J. Bacteriol. 178:1532–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ranson-Olson B., Zeilstra-Ryalls J. 2008. Regulation of the Rhodobacter sphaeroides 2.4.1 hemA gene by PrrA and FnrL. J. Bacteriol. 190:6769–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richardson D. J., Berks B. C., Russell D. A., Spiro S., Taylor C. J. 2001. Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cell Mol. Life. Sci. 58:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Richardson D. J., et al. 1988. The role of auxiliary oxidants in maintaining redox balance during phototrophic growth of Rhodobacter capsulatus on propionate or butyrate. Arch. Microbiol. 150:131–137 [Google Scholar]
- 39. Sears H. J., Sawers G., Berks B. C., Ferguson S. J., Richardson D. J. 2000. Control of periplasmic nitrate reductase gene expression (napEDABC) from Paracoccus pantotrophus in response to oxygen and carbon substrates. Microbiology 146:2977–2985 [DOI] [PubMed] [Google Scholar]
- 40. Shapleigh J. P. 2011. Oxygen control of nitrogen oxide respiration, focusing on α-proteobacteria. Biochem. Soc. Trans. 39:179–183 [DOI] [PubMed] [Google Scholar]
- 41. Simon J., van Spanning R. J. M., Richardson D. J. 2008. The organisation of proton motive and non-proton motive redox loops in prokaryotic respiratory systems. Biochim. Biophys. Acta 1777:1480–1490 [DOI] [PubMed] [Google Scholar]
- 42. Simon R., Priefer U., Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology (NY) 1:784–791 [Google Scholar]
- 43. Simpson P. J. L., Richardson D. J., Codd R. 2010. The periplasmic nitrate reductase in Shewanella: the resolution, distribution and functional implications of two NAP isoforms, NapEDABC and NapDAGHB. Microbiology 156:302–312 [DOI] [PubMed] [Google Scholar]
- 44. Stewart P. S. 2003. Diffusion in biofilms. J. Bacteriol. 185:1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stewart V., Parales J. 1988. Identification and expression of genes narL and narX of the nar (nitrate reductase) locus in Escherichia coli K-12. J. Bacteriol. 170:1589–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stewart V. 1993. Nitrate regulation of anaerobic respiratory gene expression in Escherichia coli. Mol. Microbiol. 9:425–434 [DOI] [PubMed] [Google Scholar]
- 47. Sussman I., Erecińska M., Wilson D. F. 1980. Regulation of cellular energy metabolism. The Crabtree effect. Biochim. Biophys. Acta 591:209–223 [DOI] [PubMed] [Google Scholar]
- 48. Tabata A., Yamamoto I., Matsuzaki M., Satoh T. 2005. Differential regulation of periplasmic nitrate reductase gene (napKEFDABC) expression between aerobiosis and anaerobiosis with nitrate in a denitrifying phototroph Rhodobacter sphaeroides f. sp. denitrificans. Arch. Microbiol. 184:108–116 [DOI] [PubMed] [Google Scholar]
- 49. Tosques I., Shi J., Shapleigh J. 1996. Cloning and characterization of nnrR, whose product is required for the expression of proteins involved in nitric oxide metabolism in Rhodobacter sphaeroides 2.4.3. J. Bacteriol. 178:4958–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tosques I., Kwiatkowski A., Shi J., Shapleigh J. 1997. Characterization and regulation of the gene encoding nitrite reductase in Rhodobacter sphaeroides 2.4.3. J. Bacteriol. 179:1090–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vemuri G. N., Eiteman M. A., McEwen J. E., Olsson L., Nielsen J. 2007. Increasing NADH oxidation reduces overflow metabolism in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 104:2402–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yanisch-Perron C., Vieira J., Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 53. Zeilstra-Ryalls J., Kaplan S. 1995. Aerobic and anaerobic regulation in Rhodobacter sphaeroides 2.4.1: the role of the fnrL gene. J. Bacteriol. 177:6422–6431 [DOI] [PMC free article] [PubMed] [Google Scholar]