

Abstract
Nuclear factor 90 (NF90), an RNA-binding protein implicated in the regulation of gene expression, exists as a heterodimeric complex with NF45. We previously reported that depletion of the NF90/NF45 complex results in a multinucleated phenotype. Time-lapse microscopy revealed that binucleated cells arise by incomplete abscission of progeny cells followed by fusion. Multinucleate cells arose through aberrant division of binucleated cells and displayed abnormal metaphase plates and anaphase chromatin bridges suggestive of DNA repair defects. NF90 and NF45 are known to interact with the DNA-dependent protein kinase (DNA-PK), which is involved in telomere maintenance and DNA repair by nonhomologous end joining (NHEJ). We hypothesized that NF90 modulates the activity of DNA-PK. In an in vitro NHEJ assay system, DNA end joining was reduced by NF90/NF45 immunodepletion or by RNA digestion to an extent similar to that for catalytic subunit DNA-PKcs immunodepletion. In vivo, NF90/NF45-depleted cells displayed increased γ-histone 2A.X foci, indicative of an accumulation of double-strand DNA breaks (DSBs), and increased sensitivity to ionizing radiation consistent with decreased DSB repair. Further, NF90/NF45 knockdown reduced end-joining activity in vivo. These results identify the NF90/NF45 complex as a regulator of DNA damage repair mediated by DNA-PK and suggest that structured RNA may modulate this process.
INTRODUCTION
Nuclear factor 90 (NF90) and nuclear factor 45 (NF45) form a heterodimeric core complex (18) that is abundant in many cells and tissues (36, 49, 55, 66). NF90 is a product of the interleukin enhancer-binding factor 3 gene, ILF3, and is also known as DRBP76, NFAR1, and TCP80. NF45, or ILF2, is encoded by the ILF2 gene.
NF90 and NF45 interact with numerous proteins and RNAs, generating higher-order complexes that participate in biological processes, including transcription, RNA transport, mRNA stability, and translation (2, 51). NF90 and NF45 were initially purified as DNA binding proteins (8), however, and evidence for their involvement in DNA metabolism is accumulating. Remarkably, they were found to copurify from human placenta together with all three subunits of the DNA-dependent protein kinase (DNA-PK) (63), which is involved in the repair of DNA double-strand breaks (DSBs) (29, 31). In addition, NF90/NF45 and DNA-PK have been identified as antigen receptor response element (ARRE) DNA-binding subunits and implicated in chromatin remodeling in T cells (58). Furthermore, a multiprotein complex containing these proteins was associated with a role in DNA replication, transcription, and repair (22).
Recently, we found that depletion of the NF90/NF45 core complex in HeLa cells, using RNA interference directed against either of its components, led to impaired DNA replication and cell proliferation. The cells displayed a mitotic defect resulting in the formation of giant multinucleated cells containing up to 11 interconnected nuclei (18). Such failure of proper mitotic progression is associated with disruption of genomic integrity. Multinucleated cells have been observed following DNA damage caused by X-irradiation of HeLa cells (14, 20, 57) or after depletion of proteins that play a part in chromosome segregation and DNA repair (6, 11, 13, 26, 27, 57), including DNA-PK (9, 11, 27, 57). Repair of DSBs, essential for the maintenance of genome integrity, can occur through homologous recombination or nonhomologous end joining (NHEJ). NHEJ is the predominant pathway used for repair of spontaneous and pathological DSBs during most of the cell cycle, and it plays a key role in the generation of antibodies by V(D)J and class switch recombination.
DNA-PK acts as a sensor for DSBs and an effector in the repair pathway. It is composed of two DNA-binding subunits, Ku70 and Ku80, and a catalytic subunit, DNA-PKcs, belonging to the phosphatidylinositol 3-kinase (PI3-kinase)-like kinase (PI3KK) family (29, 31). DNA-PKcs becomes autophosphorylated in the presence of DNA ends (10, 31, 50, 64), and it then phosphorylates a number of substrates, including the endonuclease Artemis (16, 31) and histone H2A.X (1, 23, 52), components of the NHEJ pathway (29, 31). Biochemical experiments indicated that DNA-PK forms a complex with NF90/NF45 in the presence of DNA and that recombinant NF90 specifically promotes the binding of DNA-PK to DNA (63). Moreover, NF90 and NF45 also serve as substrates of DNA-PK and become phosphorylated in a DNA-dependent manner (63). These findings are consistent with a functional relationship between NF90/NF45 and DNA-PK (63), although this proposition has not been explored further.
Taken together, these observations suggested that the phenotype of NF90/NF45-depleted HeLa cells could be due to a failure of NHEJ-mediated DSB repair. To examine this hypothesis, we first studied the events leading to genesis of multinucleated cells in NF45-depleted HeLa cells. As with DNA-PK insufficiency, time-lapse microscopy revealed that binucleated cells arise by incomplete abscission during mitosis due to the presence of DNA bridges, followed by cytoplasmic coalescence. Higher-order nucleated cells arise by karyokinesis without cytokinesis in a process of endoreduplication. Next, we confirmed that NF90/NF45 complexes interact with DNA-PK, and we established a novel in vitro assay to assess the involvement of NF90/NF45 in NHEJ. We found that NF90 depletion reduced end-joining activity similarly to depletion of DNA-PK. Finally, we showed that NF90/NF45-depleted cells display greatly increased numbers of γH2A.X foci, consistent with an accumulation of DSBs, and an increased sensitivity to ionizing radiation. Furthermore, NF90/NF45 depletion reduced end joining in vivo. These data identified a novel role for the NF90/NF45 complex in DNA metabolism as an important player in DSB repair mediated by DNA-PK.
MATERIALS AND METHODS
Cell culture.
Cell lines c, d3, and d5 were grown as described previously (18). Tetracycline-regulated HeLa stable cell lines carrying short hairpin RNA (shRNA) were generated using synthetic DNA sequences (d5-8, NF45 nucleotides [nt] 504 to 522; cms, mismatched control [18]) cloned into the pSingle-tTs-shRNA vector (Clontech) and selection for G418 (Invitrogen) resistance. Expression of shRNA was induced with 1 μg/ml doxycycline (Dox) (Sigma). For time-lapse microscopy, d5-8 cells were grown in T25 flasks in medium supplemented with 25 mM HEPES.
Time-lapse phase microscopy.
Flasks were equilibrated with 5% CO2 and placed in a 37°C chamber on an inverted Nikon microscope after the caps were tightened. Images were recorded with a 20× phase objective on a Scion CG7 grabber at 5-min intervals from 24 to 48 h postseeding.
Cell staining.
Cells were grown in chambered glass slides (Labtek) for 48 h, fixed in neutral buffered 4% paraformaldehyde for 2 to 4 h at room temperature (RT), and then washed with phosphate-buffered saline (PBS) (Cellgro). Cell membranes were stained with 25 μg/ml biotinylated wheat germ agglutinin (WGA) (Sigma) for 3 h at RT followed by streptavidin-Alexa 488 (Invitrogen) for 1 h. For DNA, cells were washed, fixed for 30 min, and then stained with 7-amino actinomycin D (7-AAD) (Invitrogen). For immunostaining, fixed cells were permeabilized with 0.2% Triton X-100 and blocked with bovine serum albumin (BSA). Nuclear membranes were detected with anti-lamin B2 antibody (Santa Cruz) overnight, followed by biotin-labeled goat anti-mouse antibody (Santa Cruz) for 2 h and streptavidin-conjugated Alexa 488 (Invitrogen) for 30 min. γ-H2A.X foci were detected with anti-phospho-histone H2A.X (Ser139) antibody (JBW301; Millipore) overnight and then Alexa 488-conjugated anti-mouse antibody (Invitrogen) for 1 h. Actin filaments were stained with Texas Red-X phalloidin (Invitrogen) for 30 min. Nuclei were stained with Hoechst 33342 (Molecular Probes) for 5 min. Stained cells were mounted in Vectashield (Vector Laboratories) or mounting medium (Sigma). Images were taken with a Zeiss LSM510 (confocal: 40× objective), Zeiss Axiovert 200 M (63× objective), or Nikon Eclipse E800 (10, 20, and 65× objective) microscope with the Axio Vision 4.6.3 or NIS elements BR3.0 software program.
In vitro NHEJ assay.
HeLa whole-cell extract (WCE) was prepared as described previously (65) and stored in aliquots at −80°C. DNA substrate with 5′-cohesive ends was prepared by digestion of pUC19 with SalI (New England BioLabs). DNA substrate with incompatible 3′-protruding ends was prepared by gel purification of the 5.7-kb fragment released by digestion of pSingle-tTS-shRNA with BstXI (New England BioLabs). Reaction mixtures containing 100 ng of pUC19/SalI DNA or 200 ng of the 5.7-kb DNA fragment were incubated for 60 or 180 min, respectively, with 20 μg WCE in reaction buffer (20 mM HEPES [pH 7.5], 10 mM MgCl2, 80 mM KCl, 1 mM dithiothreitol [DTT], 1 mM ATP, and 50 μM dATP, dGTP, dCTP, and dTTP) at 25°C. Where appropriate, the WCE was immunodepleted (see below) or preincubated with 1 μg antibody or RNase A for 1 h, or with drugs or vehicle for 15 min, before the addition of substrate. Reactions were terminated by addition of 50 mM EDTA and digestion with proteinase K (1 μg/μl; Roche) for 1 h at 37°C. Products were resolved in 0.7% agarose gels. DNA was stained with SYBR Gold (Molecular Probes) and detected by imaging with a Typhoon 8600 scanner (Molecular Dynamics) or Bio Image analyzer (Syngene). DNA bands were quantified with Image Quant 5.2 software (Molecular Dynamics) or ImageJ 1.43u software (NIH).
Amplification, cloning, and sequencing of end-joined products.
DNA products from the in vitro end-joining assay were purified by phenol-chloroform extraction and ethanol precipitation. The junction region was amplified using HotStarTaq DNA polymerase (Qiagen) and primers a (5′-ATGCAAGCTTCCTTTATTACCCAGAAGTCAGATGC-3′) and b (5′-ATGCTCTAGAGTAAACTCGCCCAGAAGCTAGG-3′), where substrate sequences are underlined and restriction endonuclease sites are in bold. PCR was carried out in a 50-μl reaction mixture for 35 cycles (denaturation, 94°C; annealing, 62°C; extension, 72°C). The amplified end-joined products were purified by ethanol precipitation, digested with XbaI and HindIII (New England BioLabs), repurified using Qiagen PCR purification columns, and cloned into the pUC19 vector digested with XbaI and HindIII using HB101 competent cells. The cloned DNA was examined by restriction enzyme digestion and sequenced using the M13F primer.
Immunoprecipitation, immunodepletion, and immunoblotting.
For immunoprecipitation, 300 μg of WCE protein was diluted in 300 μl of dialysis buffer (25 mM HEPES [pH 7.5], 100 mM KCl, 1 mM EDTA, 10% glycerol, 0.2 mM phenylmethylsulfonyl fluoride [PMSF], and 0.5 mM DTT), precleared by centrifugation at 10,000 × g for 5 min, and incubated with 2 μg of antibody conjugated to protein G beads (GE Healthcare). Antibodies were as follows: Omni (D5; Santa Cruz), Ku70 (N3H10; Santa Cruz), DNA-PKcs (18-2; Novus), NF90 (DRBP76), and NF45 (18). Immunoprecipitates were washed once with dialysis buffer and 4 times with IP lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and 1 mM DTT). For immunodepletion, 350 μg of WCE in 35 μl dialysis buffer was incubated with 2 μg antibody conjugated to protein G beads for 1 h at 4°C. The procedure was repeated twice more with fresh antibody-conjugated beads. Immunoprecipitated proteins and immunodepleted WCE were resolved in 7.5% polyacrylamide-SDS gels, electroblotted, and visualized using monoclonal antibodies against DNA-PKcs (G4; Santa Cruz), Ku80 (C20; Santa Cruz), NF45 (Everest Biotech), actin (C2; Santa Cruz), and other proteins as specified above. Where indicated, WCE was preincubated with Benzonase (Novagen) for 30 min at 37°C in the presence of 1 mM MgCl2 or with ethidium bromide at 4°C for 30 min, followed by centrifugation.
Clonogenic survival assay.
Stable cell lines were grown to 70% confluence in 6-well plates and irradiated with 6 Gy of 137Cs gamma rays (J.L. Shepherd Mark-I-type irradiator). Cells were seeded for colony formation within 10 min after irradiation. After an incubation of 10 days, the plates were rinsed with PBS, fixed in ethanol, stained with crystal violet, and colonies consisting of 50 cells or more were counted as survivors.
In vivo NHEJ assay.
HeLa cells (1.5 × 105) were transfected with 30 nM C, D3, and D5 short interfering RNA (siRNA) (18) using 5 μl of INTERFERin (Polyplus-transfection). At 48 h, 3 × 105 cells were transfected with 100 ng of pEGFP-Pem1-Ad2 reporter plasmid (56) digested with HindIII or I-SceI (New England Biologicals) and 25 ng of pDsRed-Express-C1 (Clontech) using jetPEI (Polyplus-transfection). The cells were harvested 24 h later, resuspended in 0.5 ml PBS, and assayed for the expression of enhanced green fluorescent protein (EGFP) and DsRed by flow cytometry (FACSCalibur; BD Biosciences).
RESULTS
Genesis of multinucleated cells.
NF90 and NF45 are coregulated at the level of protein stability, and their complexes play a role in cell division and DNA synthesis (18). Depletion of NF90/NF45 slows cell growth and leads to the formation of large, multinucleated cells in which the nuclei are joined by constrictions. These observations were made with HeLa cell lines stably expressing shRNA directed against NF90 or NF45 and were especially pronounced with the NF45-depleted d5 cell line (18). To obtain regulated expression of shRNA and increased numbers of multinucleated cells for microscopic analysis, we produced a stable cell line, d5-8, in which d5 shRNA is under the control of a conditional promoter. Although regulation was not completely tight, Dox-induced d5-8 cultures contained ∼20% multinucleated cells, compared to ∼8% in uninduced cultures (Fig. 1A and B). A cell line carrying control mismatched shRNA (cms) had <1% multinucleated cells irrespective of the presence of Dox. The multinucleated cells in induced d5-8 cultures were up to 8 times bigger than normal cells, contained up to 15 nuclei, and displayed gross disturbances of cytoskeletal architecture (Fig. 1C). These results corroborate the causal relationship between NF90/NF45 depletion and the multinucleate phenotype, and the high frequency of multinucleated cells in induced d5-8 cultures allowed us to observe their genesis by time-lapse microscopy.
Fig. 1.

Induction of multinucleated cells by NF45 shRNA. (A) Bright-field micrographs of HeLa stable cell lines without or after 72 h of induction (+ Dox) of control mismatch shRNA (cms) or d5 shRNA (d5-8). Arrows point to some of the multinucleated cells. (B) Quantitation of multinucleated cells from 2 independent experiments with standard deviations. (C) Morphology of multinucleated cells. Fluorescence microscopy images at low (×10) and high (×65) magnification of d5-8 and cms cell cultures grown in the presence of Dox. Nuclei are stained blue (Hoechst), and actin filaments are stained red (phalloidin). A representative multinucleated cell containing connected nuclei is depicted at magnification ×65.
The production of multinucleated cells began with the generation of binucleated cells. Individual mononucleated cells rounded and underwent mitosis to produce two daughter cells, some of which fused over time (Fig. 2A, red and blue arrows). The presence of two nuclei in single cells resulting from such a sequence of events is clearly visualized in an enlarged image (Fig. 2C). Occasionally, fusion occurred between cells after two consecutive mitoses. In these cases, a binucleated cell resulted from the fusion of a daughter cell with its “niece,” i.e., a daughter of its sister cell (Fig. 2A, black arrows). For cells that subsequently formed a binucleated cell, the timing of mitosis as a whole was variable and extended (see Table S1 in the supplemental material). Individual mitotic phases were extended by up to 2-fold in cells that subsequently underwent fusion, and fusion itself occurred at a highly variable and often prolonged time after mitosis appeared to be complete. In the case of the cells marked with red arrows (Fig. 2A), for example, ∼965 min elapsed from the time two daughter cells were first observed (460 min) until their fusion was complete (1,425 min).
Fig. 2.

Genesis of binucleated cells. (A) Images from phase-contrast microscopy time-lapse videos (magnification, ×20) of d5-8 cells treated with Dox, taken at 5-min intervals. Blue and red arrows show formation of binucleated cells from the incomplete division and fusion of two daughter cells. Black arrows show the genesis of a binucleated cell from a daughter and a granddaughter cell. (B) Enlargement of daughter cells, showing connection via a cytoplasmic bridge. (C) Enlargement of fused cells (black and red arrows). (D) Chromatin bridges between daughter cells of NF45-depleted cells. Confocal micrographs (×40) of cms and d5-8 cells treated with Dox and stained for cytoplasmic membranes (WGA) and for DNA (7-AAD). Arrows point to apparent persistent midbodies with a DNA bridge. (E) Anaphase chromatin bridges. Confocal micrographs (×40) showing high frequency of anaphase DNA bridges in d5-8, but not cms, cells treated with Dox.
Inspection of cells that subsequently fused revealed partial abscission failure. The presence of cytoplasmic bridges connecting these “daughter” cells after cytokinesis had apparently ceased is evident in several frames and is conspicuous in an enlarged image (Fig. 2B). The cytoplasmic bridges were examined by staining cells for DNA with 7-AAD, using fluorescent WGA to visualize the plasma membrane. Confocal microscopy revealed that the cytoplasmic bridges between daughter cells contained DNA and an apparent persistent midbody (Fig. 2D, lower panel). Remarkably, a high proportion of cells that underwent mitosis contained chromatin bridges during anaphase and telophase (Fig. 2E), suggestive of a defect in chromosome disjunction. These observations suggest that binucleated cells are generated by cytoplasmic merging of sibling cells that are connected by chromatin bridges at the midbody.
Some binucleated cells continued to undergo cell division. In one example, a binucleated cell (Fig. 3A, red arrow) produced three daughter cells that remained connected by cytoplasmic bridges (at 195 min). Two of the daughter cells subsequently fused (at 350 min), resulting in the formation of a trinucleated cell (Fig. 3A, blue arrow) and a singly nucleated cell that eventually died (not shown). Higher-order multinucleated cells continued to undergo mitosis with endoreduplication. Over a 23-h interval, one such large multinucleated cell, containing about 4 nuclei, rounded and underwent mitosis to produce a cell with about 8 nuclei and then rounded again (Fig. 3B). DNA staining revealed that multinucleated cells had abnormal organization of their chromosomes on the metaphase plates (Fig. 3C), impeding normal mitosis and resulting in partial karyokinesis without cytokinesis. As in the d3 and d5 cell lines (18), the multiple nuclei in Dox-induced d5-8 cultures were joined by DNA connections (Fig. 3D). The nuclei in these cells were surrounded by nuclear membranes, as shown by staining of lamin B (see Fig. S1 in the supplemental material).
Fig. 3.

Genesis of multinucleated cells. (A) Formation of trinucleated cells in d5-8 cultures treated with Dox. Images from time-lapse video, as in Fig. 2A, show a binucleated cell (red arrow) undergoing mitosis to produce a trinucleated cell (blue arrow) and a uninucleated cell. (B) Endoreduplication of multinucleated cells. Images from time-lapse video, as in Fig. 2A, show a multinucleated cell (black arrow) with at least four nuclei undergoing mitosis to produce a giant cell with at least 8 nuclei by karyokinesis without cytokinesis. (C) Metaphase plates in d5-8 multinucleated cells. Confocal (×40) micrographs show normal (left) and abnormal (middle and right) chromosome alignment. (D) Giant cells containing interconnected nuclei. Confocal images (×40) of DNA-stained multinucleated cells with incomplete karyokinesis.
Participation of NF90 in NHEJ in vitro.
The development of the multinucleated phenotype in NF90/NF45-depleted cells exhibited features associated with DNA damage and in particular resembled that of cells lacking DNA-PK. For example, DNA-PKcs deficiency leads to persistent anaphase bridges in mouse cells (17) and aberrant mitotic spindles and multinucleated human cells (57). Ku deficiency also results in enhanced basal levels of DSBs and chromosome aberrations and leads to the multinucleated phenotype in mouse and human cells (9, 11, 27). To determine whether NF90/NF45 interacts biochemically with DNA-PK in HeLa cell extracts, we conducted immunoprecipitation experiments (Fig. 4A). Immunoblotting demonstrated that NF90 coimmunoprecipitated with DNA-PKcs and Ku70, as well as with NF45 (Fig. 4A, top). Reciprocally, Ku70 coimmunoprecipitated with NF90 as well as DNA-PKcs (Fig. 4A, middle), and DNA-PKcs coimmunoprecipitated with NF90 as well as Ku70 (Fig. 4A, bottom). In all cases, a control antibody (Omni) gave little or no signal. Pretreatment with Benzonase to eliminate nucleic acids did not affect the coimmunoprecipitation of DNA-PK with NF90 (Fig. 4B, top). Similarly, the interactions were unaffected by the addition of ethidium bromide (24), providing evidence that they are DNA independent (Fig. 4B, bottom). These data confirm the existence and specificity of NF90/NF45 complexes with DNA-PK in these cells.
Fig. 4.

Inhibition of NHEJ by NF90 antibody. (A) NF90 is present in complexes with Ku70 and DNA-PKcs in HeLa cells. Immunoprecipitates from HeLa WCE with antibody against Omni (control), NF90, Ku70, NF45, and DNA-PKcs (DPKcs) were assayed by immunoblotting. (B) DNA independence of NF90–DNA-PK interactions. WCE was preincubated with Benzonase (100 U per 300 μg WCE protein) or ethidium bromide (EtBr) before immunoprecipitation with antibody against Omni or NF90 and immunoblotting. (C) Kinetics of in vitro NHEJ reaction using SalI-linearized pUC19 DNA substrate. Oligomeric end-joined species were resolved by agarose gel electrophoresis and quantified with the Image Quant software program. HM, higher multimers (pentamers and above). (D) Inhibition of NHEJ by drugs. Assays contained wortmannin (1, 5, 10, or 20 μM), KU55933 (0.025, 0.05, 0.5, or 1 μM), NU7026 (0.5, 1, 2.5, 5, or 10 μM), or solvent (dimethylsulfoxide [DMSO]; 0.25, 0.5, or 1%). EDTA, supplemented with 50 mM EDTA; −, no additions; M, DNA molecular markers. (E) WCE was treated with Omni, Ku70, DNA-PKcs (DPKcs), or NF90 antibody, left untreated (No Ab), heat inactivated at 90°C, or supplemented with 50 mM EDTA and then assayed for NHEJ activity. (F) Quantitation of residual monomeric substrate (Monomer) and higher multimeric products (higher multimer; pentamers and above) expressed relative to Omni control. Results are means of data from 3 independent experiments in duplicate with standard deviations.
Next, we examined the effect of adding antibodies to an established in vitro NHEJ assay using HeLa cell extract and SalI-linearized plasmid DNA as a substrate (65). Resolution of the reaction products in an agarose gel revealed a series of bands formed by ligation of the sticky ends. No ligation occurred in the presence of EDTA or with heat-treated HeLa cell extract. Quantitation showed that dimers formed first, followed by trimers and tetramers, and that each subsequently gave rise to pentamers and higher multimeric species (Fig. 4C). To determine whether the ligation is dependent on DNA-PK, we tested the effects of wortmannin, a general PI3-kinase inhibitor, and of NU7026 and KU55933, which selectively block DNA-PKcs and ATM, respectively (60, 65). Wortmannin and NU7026 strongly inhibited the formation of DNA multimers, whereas KU55933 did not, indicating that DNA-PKcs mediates DNA joining in this assay (Fig. 4D). Accordingly, addition of antibody against DNA-PKcs inhibited ligation, as seen most clearly for the multimeric products (Fig. 4E). Antibodies directed against NF90 or Ku70 elicited a qualitatively similar response, albeit lesser in magnitude, reducing the formation of multimers and increasing the level of the monomeric substrate (Fig. 4E and F). These findings suggested that NF90 plays a part in DNA end joining.
Although DNAs with cohesive ends have been used as substrates for in vitro NHEJ assays, such assays do not necessarily entail the action of the nuclease or polymerase characteristic of NHEJ (3, 4, 32, 65), and alternative substrates have been devised (5, 30, 42). We generated a 5.7-kb DNA substrate with incompatible protruding 3′ ends (Fig. 5A) that gave rise to ∼15% dimers when ligated in the in vitro NHEJ system (Fig. 5B). As with the cohesive-end substrate (Fig. 4), dimer formation was inhibited in a dose-dependent manner by wortmannin and NU7026 but not by KU55933 (Fig. 5B), verifying that the reaction is dependent on active DNA-PKcs. When analyzed by PCR amplification (Fig. 5C) and cloning (Fig. 5D) using specific primers (Fig. 5A), the expected products were generated. Sequence analysis of selected clones (Fig. 5E) revealed that the DNA had been processed by nuclease, polymerase, and ligase activities, as is characteristic of NHEJ (29).
Fig. 5.

In vitro NHEJ assay using DNA substrate with noncohesive ends. (A) Schematic showing the derivation of the 5.7-kb fragment with noncohesive BstXI termini, A and B. Corresponding PCR primers a and b contain HindIII and XbaI sites, respectively. Possible end-joining orientations are diagrammed. (B) Products of in vitro NHEJ reactions using the 5.7-kb substrate separated in an agarose gel (lanes 1 to 3). Inhibitors were added as for Fig. 4C. (C) PCR amplification products of uncut pSingle-tTs-shRNA plasmid (lane 1), unreacted 5.7-kb DNA substrate (lane 2), and total DNA isolated from NHEJ reactions (lane 3). (D) Cloned products of A-to-B end joining (orientation I; panel A) released by digestion with HindIII and XbaI and resolved in an agarose gel. (E) DNA sequences of cloned A-B end-joined products (Type 1 and Type 2) aligned with the A and B ends of the 5.7-kb substrate (Input). Dashes represent nucleotide deletions; arrow denotes nucleotide additions; rectangles represent microhomology sequence potentially used for end joining.
To determine whether NF90-containing complexes participate in NHEJ, we immunodepleted the HeLa cell extract with antibodies directed against NF90 or DNA-PKcs. Compared to mock-depleted extract or extract treated with anti-Omni antibody, NF90 immunodepletion inhibited dimer formation by ∼55% (Fig. 6A and B). Immunodepletion of DNA-PKcs inhibited dimer formation by ∼40%. These results corroborate the role of NF90 and DNA-PKcs in the in vitro NHEJ reaction. Immunoblotting demonstrated that NF90/NF45 was largely depleted by anti-NF90 antibody, whereas DNA-PKcs was incompletely removed from the extract by its cognate antibody (Fig. 6C). This protein remained refractory even after repeated treatment (data not shown), suggesting that the DNA-PKcs epitope is partially masked. Interestingly, the anti-NF90 antibody gave a small but reproducible reduction in DNA-PKcs, whereas antibody against DNA-PKcs achieved only a minor reduction in the levels of NF90 and NF45 (Fig. 6C). These findings imply that although a minor fraction of the cellular NF90/NF45 complexes contain DNA-PKcs, they account for about half of the in vitro NHEJ activity.
Fig. 6.
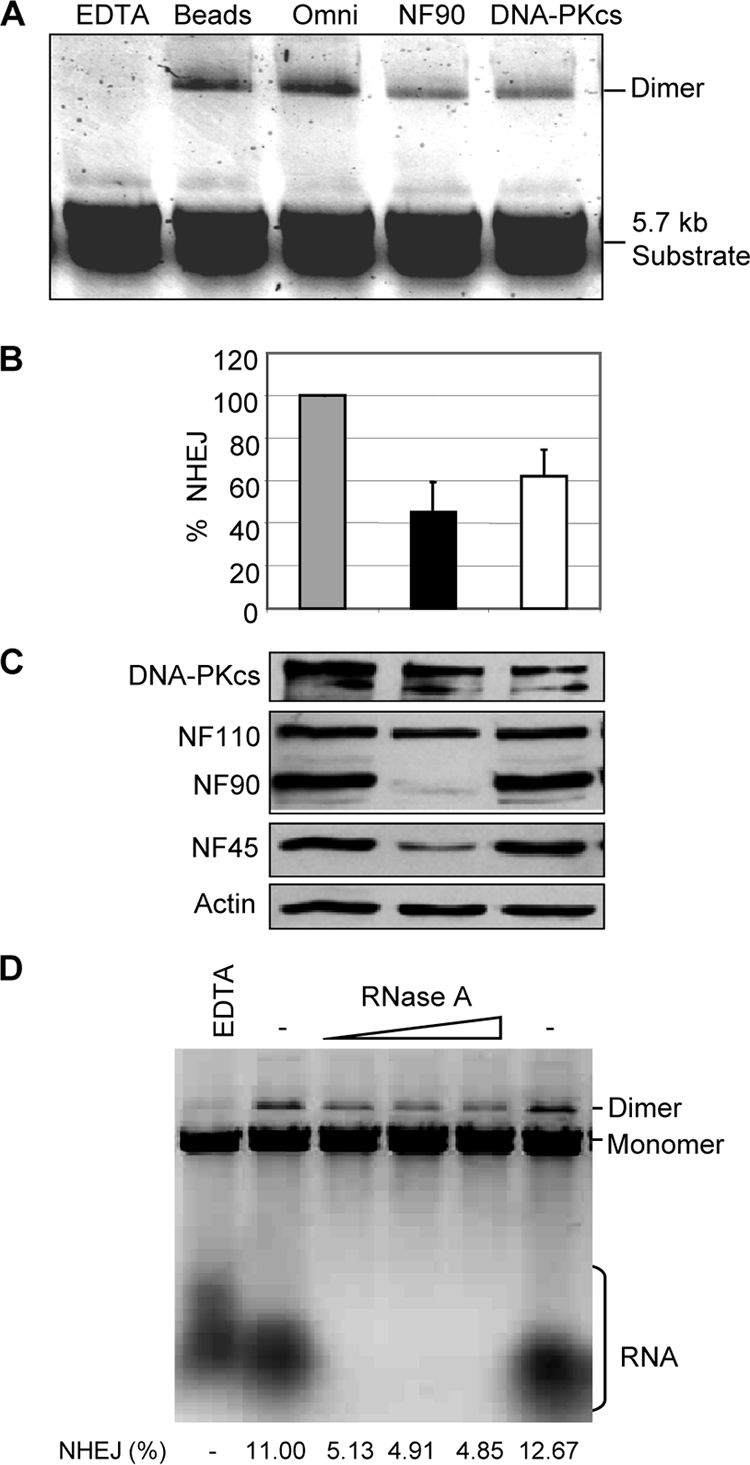
NF90 depletion inhibits NHEJ. (A) WCE was mock depleted (Beads) or immunodepleted with Omni, NF90, or DNA-PKcs antibody and then assayed for NHEJ with 5.7-kb substrate. (B) Quantitation of end joining after immunodepletion. Efficiency of dimer formation is expressed as a percentage of input substrate (n = 3, with standard deviations). (C) Immunoblot analysis of depleted extracts. (D) Effect of RNase treatment on NHEJ. Extract was preincubated with EDTA, diluent (−), or RNase A (0.5, 1.5, and 2.5 μg/ml), as indicated. The percent conversion of monomeric substrate to dimer products is shown.
Because NF90 is an RNA binding protein and is largely bound to RNA in HeLa cells (41), we examined the effect of RNase treatment on in vitro NHEJ. Pretreatment of HeLa cell extract with RNase A effectively destroyed its RNA and reduced the efficiency of end joining by up to 58% (Fig. 6D). As expected, RNase treatment had no effect on the levels of several proteins tested, including NF90, NF45, Ku70, Ku80, and DNA-PKcs (data not shown). Treatment with RNase T1 did not eliminate the RNA and failed to reduce end joining (data not shown). These findings suggest that RNA is involved in NHEJ, possibly acting as a regulator.
Decreased DNA damage repair in NF90/NF45-depleted cells.
These biochemical data, together with our cytological observations, suggested that NF90/NF45 is involved in DSB repair. Accordingly, NF90/NF45 depletion would be expected to increase the frequency of DSBs and the sensitivity of cells to DNA damage. To detect DSBs, we immunostained d3 and d5 cells with antibodies directed against Ser139-phosphorylated H2A.X (γ-H2A.X), a component of the DSB repair apparatus and a sensitive marker of DNA damage (21, 53). Stained foci marking the sites of DNA damage were abundant in d3 and d5 cells in comparison with results for control cells (Fig. 7A and B; see also Table S2 and Fig. S2 in the supplemental material). Most of the multinucleated cells (>80%) were positive for γ-H2A.X foci in both d3 and d5 cultures (Fig. 7B). The few multinucleated cells in control cultures were also largely γ-H2A.X positive (Fig. 7B; see also Table S2). Most notably, the frequency of γ-H2A.X foci was elevated in mononucleated d3 and d5 cells (>12% and 8%, respectively) but not in control cells (1.0%), indicating that DSBs arise in NF90/NF45-depleted cells prior to the formation of multinucleated cells.
Fig. 7.

NF90 and NF45 depletion increases susceptibility to DNA damage. (A) γ-H2A.X foci in cells depleted of NF90 and NF45. Fluorescence microscopy images of control (c), d3, and d5 cells stained for actin filaments (phalloidin), DNA (Hoechst), and γ-H2A.X (magnification, ×63). (B) Quantitation of γ-H2A.X-positive cells in control and depleted cultures (Total) and in their constituent mononucleated and multinucleated cells. (C) Decreased clonogenicity of depleted cultures after gamma irradiation. Data are presented as percentage survival relative to that of control cells (means of triplicates with standard deviations).
We used a clonogenic survival assay (48) to test the prediction that NF90/NF45-depleted cells would display increased susceptibility to DNA damage. Cells were exposed to gamma irradiation, decreasing their ability to form colonies by 5- to 6-fold. Irradiated d3 and d5 cell lines formed about half as many colonies as control cells (Fig. 7C; see also Table S3 in the supplemental material). Although several factors may account for the observed decrease in clonogenic survival, decreased DNA damage repair is likely a major mechanism.
NF90/NF45 depletion reduces joining of DNA breaks in vivo.
To evaluate the effect of NF90/NF45 depletion on end joining in cells, we used an in vivo plasmid repair assay (43, 56) based on a disrupted EGFP gene. The EGFP gene is present in a construct that contains an inserted stuffer sequence separating its 5′ and 3′ segments (Fig. 8A). The construct gives rise to EGFP in transfected cells when two conditions are met. First, a segment of the stuffer containing Ad2 sequence flanked by interfering splice signals must be removed. This is accomplished by digestion in vitro with either of two endonucleases, HindIII and I-SceI, which yield sticky and blunt ends, respectively. Second, the plasmid ends must be joined in the cell by a reaction that usually (HindIII) or necessarily (I-SceI) entails NHEJ (43, 56). The ligated DNA is transcribed to yield a precursor RNA that is then spliced to form uninterrupted EGFP mRNA. EGFP is detected by fluorescence-activated cell sorter (FACS) analysis, and data are normalized to red fluorescence generated from a control reporter (DsRed).
Fig. 8.

NF90/NF45 knockdown inhibits NHEJ in vivo. (A) Generation of end-joining substrate. The circular map of reporter plasmid pEGFP-Pem1-Ad2 diagrams the EGFP gene interrupted by a stuffer sequence containing an Ad2 intron. Endonuclease sites are marked. The linear diagram illustrates the positions of splice sites (solid circles) relative to the intron and endonuclease sites. Digestion with I-SceI or HindIII generates noncompatible and cohesive NHEJ substrates, respectively. (B) NHEJ in cells after NF90/NF45 knockdown. Cells were transfected with control, NF90, or NF45 siRNA (C, D3, and D5, respectively) and with the I-SceI or HindIII substrates and then analyzed by flow cytometry. Dot plots show red and green fluorescence (vertical and horizontal axes, respectively). Values inserted in individual plots enumerate EGFP-positive cells, percent of total). Immunoblots show the levels of NF90 and NF45 in siRNA-treated cells. (C) Quantitation of EGFP-positive cells normalized to DsRed-positive cells. P values: *, <0.05; **, <0.01; ***, <0.001 (based on five independent experiments with the I-SceI substrate and four independent experiments with the HindIII substrate).
The digested EGFP construct was introduced together with DsRed into HeLa cells that had been transfected with small interfering RNA (siRNA) directed against NF90 (D3) or NF45 (D5) or with control siRNA (C). In representative flow cytometry plots (Fig. 8B), ∼1.6 to 1.7% of the control cell population displayed EGFP fluorescence. Depletion of NF90 or NF45 reduced the EGFP-positive population to ∼0.5 to 0.7%. Normalized results of multiple experiments showed that end-joining efficiency was reduced by ∼20 to 40% (Fig. 8C). The inhibition was highly significant and was reproducible with both D3 and D5 and with both the HindIII and I-SceI substrates. This inhibition was somewhat less than that observed in vitro (Fig. 5 and 6), a difference that may be attributable to the relative efficacy of depletion by antibody versus siRNA (compare Fig. 6C and 8B). Nevertheless, the results obtained with the in vivo NHEJ assay strongly support those obtained with the in vitro assays. We conclude that the NF90/NF45 complex, like DNA-PK, is involved in DNA repair.
DISCUSSION
The results presented here indicate that the NF90/NF45 complex plays a positive role in the DNA damage repair pathway in vivo and implicate the complex in DSB repair mediated by DNA-PK via NHEJ both in vivo and in vitro. This role is compatible with reports identifying NF90/NF45 as a component of DNA- and RNA-binding multiprotein complexes that participate in DNA replication, chromatin integrity, DNA repair, and transcription (18, 22, 58). We propose that NF90/NF45, by virtue of its ability to bind many nucleic acids and proteins, functions as an adaptor complex bridging these processes.
Our findings provide an explanation for the generation of multinucleated cells in NF90/NF45-depleted HeLa cell cultures (18). An almost identical phenotype was observed in HeLa cells that were X-irradiated (14, 20, 57) or that failed to repair their DNA efficiently as a consequence of deficiency of DNA-PKcs or Ku80 (9, 11, 27, 57). As with NF45-depleted cells, the X-irradiated cells remained connected at the midbody via chromatin bridges, and some cells fused and formed multinucleated cells. DNA damage was correlated with increased γ-H2A.X foci and DNA bridges, reflecting a lack of proper DNA repair. The infrequent appearance of multinucleated cells in untreated cultures following cell fusion and endoreduplication was also noted in the early days of HeLa cell culture (37, 38). Evidently HeLa cells can override the G2 damage checkpoint and continue endoreduplication, leading to the formation of giant multinucleated cells (14, 20, 57).
Outside the S/G2 phases of the mammalian cell cycle, repair of damaged DNA occurs chiefly by NHEJ. Inactivation of the human genes encoding the Ku proteins or DNA-PKcs results in cells deficient for DSB repair, although some other mammalian species, including the mouse, are less severely debilitated (35, 54). DNA-PK is found at the ends of chromosomes, consistent with a role in telomere maintenance and the prevention of chromosomal end fusion (54). The observation of DNA bridges and cell fusion are indicative of the involvement of NF90/NF45 in these processes, potentially mediated via their interactions with DNA-PK. DNA-PKcs is also required for DNA rearrangements involved in generating the antigen-binding sites of T-cell receptors and immunoglobulin molecules. In view of the role of NF90/NF45 in NHEJ, knockout of either of its components may also have deleterious effects on the immune system. It is notable that DNA-PKcs deficiency leads to severe combined immunodeficiency in the mouse (61), whereas knockout of the ILF3 gene, encoding NF90, is lethal (46, 59), consistent with the broad role of this protein in multiple pathways. Interestingly, regulation of the DNA-PK–NF90/NF45 complex during T-cell activation has been documented (58).
The precise action of NF90/NF45 in NHEJ remains to be elucidated, but two lines of evidence suggest that the complex acts at an early stage. First, although dispensable for NHEJ in a reconstituted in vitro system (30), NF90 and NF45 facilitate the binding of DNA-PK to DNA in vitro (63). On their own, NF90 and NF45 did not appear to form complexes with DNA (63), and their binding to DNA-PK is not DNA dependent (Fig. 4B). This implies that NF90/NF45 interacts with DNA-PK and contributes to its recruitment to DSBs or to the stability of the DNA-PK interaction with damaged DNA. Second, foci containing γ-H2A.X, believed to help in the binding and retention of essential repair proteins, are not attenuated in NF90/NF45-depleted cells. On the contrary, the number of γ-H2A.X foci increases markedly, suggesting that phosphorylation of this histone is enhanced or that its dephosphorylation is slowed when NF90/NF45 is limiting. H2A.X phosphorylation can be catalyzed by two other PI3KKs, ataxia-telangiectasia mutated (ATM) and ATM-Rad3 related (ATR), as well as by DNA-PKcs. It is possible that NF90/NF45 affects cross talk between DNA-PK and the other PI3KKs (15, 33). Alternatively, reduced binding of DNA-PKcs to DSBs in the absence of NF90/NF45 might hamper DNA repair by reducing the phosphorylation of a substrate such as Artemis. DNA-PKcs also interacts with the catalytic subunits of the protein phosphatases PP2A and PP6 and with regulatory subunits of PP6 (7, 12). These interactions are thought to help in the recruitment of PP6 to γ-H2A.X foci and the dephosphorylation of H2A.X (12), which occur after completion of DSB repair (23). Inhibition of DNA-PKcs kinase activity results in the inhibition of both in vitro end-joining activity and in vivo dephosphorylation of γ-H2A.X (28). Thus, NF90/NF45 might modulate DNA-PKcs activity at multiple levels.
NF90 and NF45 are phosphoproteins in vivo (19, 33, 41, 45, 67) and they can be phosphorylated by DNA-PK in vitro in a DNA-dependent fashion (63). Although it has not been established whether DNA-PK subserves this activity in vivo, we surmise that the phosphorylation of NF90/NF45 could regulate their interactions with the DNA repair machinery with consequences for NHEJ. Furthermore, as shown here, destruction of RNA reduces DNA end joining in vitro, implying a role for RNA in NHEJ. It is notable that NF90 has nucleic acid binding domains, including tandem double-stranded RNA binding motifs (dsRBMs) and an RGG domain, and a DZF (dsRBM- and zinc finger-associated) protein interaction motif (51, 62) that is shared with NF45 and is important for development in flies and mice (34). In addition to NF90 and NF45, DNA-PK copurified from human placenta with the translation initiation factor eIF2 (63), also an RNA-binding protein. NF90 binds to the α subunit of eIF2 (40), and both NF90 and NF45 interact with the eIF2α kinase PKR (19, 25, 39, 44, 55). DNA-PKcs plays a part in regulating protein synthesis in UV-irradiated cells, and translational inhibition in these cells correlated with increased eIF2α phosphorylation mediated by the GCN2 kinase (47). Taken together, these findings raise the possibility that the large DNA-PK–NF90/NF45–eIF2 complex serves as a means of communication between nuclear and cytoplasmic processes, possibly modulated by RNA ligands.
Supplementary Material
ACKNOWLEDGMENTS
We thank Vera Gorbunova for providing the EGFP reporter plasmid and Prasad Neti for assistance in cell irradiation.
This work was supported in part by NIH R01 grant AI034552 to M.B.M.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
Published ahead of print on 3 October 2011.
REFERENCES
- 1. An J., et al. 2010. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol. Biol. 11: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barber G. N. 2009. The NFAR's (nuclear factors associated with dsRNA): evolutionarily conserved members of the dsRNA binding protein family. RNA Biol. 6: 35–39 [DOI] [PubMed] [Google Scholar]
- 3. Baumann P., West S. C. 1998. DNA end-joining catalyzed by human cell-free extracts. Proc. Natl. Acad. Sci. U. S. A. 95: 14066–14070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Budman J., Chu G. 2005. Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J. 24: 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S., et al. 2001. Accurate in vitro end joining of a DNA double strand break with partially cohesive 3′-overhangs and 3′-phosphoglycolate termini: effect of Ku on repair fidelity. J. Biol. Chem. 276: 24323–24330 [DOI] [PubMed] [Google Scholar]
- 6. Chesnokov I. N., Chesnokova O. N., Botchan M. 2003. A cytokinetic function of Drosophila ORC6 protein resides in a domain distinct from its replication activity. Proc. Natl. Acad. Sci. U. S. A. 100: 9150–9155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chowdhury D., et al. 2005. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 20: 801–809 [DOI] [PubMed] [Google Scholar]
- 8. Corthésy B., Kao P. N. 1994. Purification by DNA affinity chromatography of two polypeptides that contact the NF-AT DNA binding site in the interleukin 2 promoter. J. Biol. Chem. 269: 20682–20690 [PubMed] [Google Scholar]
- 9. Difilippantonio M. J., et al. 2000. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404: 510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding Q., et al. 2003. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 23: 5836–5848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dmitrieva N. I., Celeste A., Nussenzweig A., Burg M. B. 2005. Ku86 preserves chromatin integrity in cells adapted to high NaCl. Proc. Natl. Acad. Sci. U. S. A. 102: 10730–10735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Douglas P., et al. 2010. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol. Cell. Biol. 30: 1368–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eldridge A. G., et al. 2006. The evi5 oncogene regulates cyclin accumulation by stabilizing the anaphase-promoting complex inhibitor emi1. Cell 124: 367–380 [DOI] [PubMed] [Google Scholar]
- 14. Erenpreisa J., et al. 2005. Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol. Int. 29: 1005–1011 [DOI] [PubMed] [Google Scholar]
- 15. Giunta S., Belotserkovskaya R., Jackson S. P. 2010. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 190: 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodarzi A. A., et al. 2006. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 25: 3880–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goytisolo F. A., Samper E., Edmonson S., Taccioli G. E., Blasco M. A. 2001. The absence of the DNA-dependent protein kinase catalytic subunit in mice results in anaphase bridges and in increased telomeric fusions with normal telomere length and G-strand overhang. Mol. Cell. Biol. 21: 3642–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guan D., et al. 2008. Nuclear factor 45 (NF45) is a regulatory subunit of complexes with NF90/110 involved in mitotic control. Mol. Cell. Biol. 28: 4629–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harashima A., Guettouche T., Barber G. N. 2010. Phosphorylation of the NFAR proteins by the dsRNA-dependent protein kinase PKR constitutes a novel mechanism of translational regulation and cellular defense. Genes Dev. 24: 2640–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang H., et al. 2008. Abnormal cytokinesis after X-irradiation in tumor cells that override the G2 DNA damage checkpoint. Cancer Res. 68: 3724–3732 [DOI] [PubMed] [Google Scholar]
- 21. Kantidze O. L., Iarovaia O. V., Razin S. V. 2006. Assembly of nuclear matrix-bound protein complexes involved in non-homologous end joining is induced by inhibition of DNA topoisomerase II. J. Cell Physiol. 207: 660–667 [DOI] [PubMed] [Google Scholar]
- 22. Karmakar S., Mahajan M. C., Schulz V., Boyapaty G., Weissman S. M. 2010. A multiprotein complex necessary for both transcription and DNA replication at the beta-globin locus. EMBO J. 29: 3260–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kinner A., Wu W., Staudt C., Iliakis G. 2008. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 36: 5678–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lai J. S., Herr W. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U. S. A. 89: 6958–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Langland J. O., Kao P. N., Jacobs B. L. 1999. Nuclear factor-90 of activated T-cells: a double-stranded RNA-binding protein and substrate for the double-stranded RNA-dependent protein kinase, PKR. Biochemistry 38: 6361–6368 [DOI] [PubMed] [Google Scholar]
- 26. Li F., et al. 1999. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1: 461–466 [DOI] [PubMed] [Google Scholar]
- 27. Li G., Nelsen C., Hendrickson E. A. 2002. Ku86 is essential in human somatic cells. Proc. Natl. Acad. Sci. U. S. A. 99: 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S., et al. 2003. Modification of the ionizing radiation response in living cells by an scFv against the DNA-dependent protein kinase. Nucleic Acids Res. 31: 5848–5857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lieber M. R. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79: 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma Y., et al. 2004. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 16: 701–713 [DOI] [PubMed] [Google Scholar]
- 31. Mahaney B. L., Meek K., Lees-Miller S. P. 2009. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 417: 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mason R. M., Thacker J., Fairman M. P. 1996. The joining of non-complementary DNA double-strand breaks by mammalian extracts. Nucleic Acids Res. 24: 4946–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuoka S., et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- 34. Meagher M. J., et al. 1999. Identification of ZFR, an ancient and highly conserved murine chromosome-associated zinc finger protein. Gene 228: 197–211 [DOI] [PubMed] [Google Scholar]
- 35. Meek K., Gupta S., Ramsden D. A., Lees-Miller S. P. 2004. The DNA-dependent protein kinase: the director at the end. Immunol. Rev. 200: 132–141 [DOI] [PubMed] [Google Scholar]
- 36. Neplioueva V., Dobrikova E. Y., Mukherjee N., Keene J. D., Gromeier M. 2010. Tissue type-specific expression of the dsRNA-binding protein 76 and genome-wide elucidation of its target mRNAs. PLoS One 5: e11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oftebro R., Wolf I. 1967. Mitosis of bi- and multinucleate HeLa cells. Exp. Cell Res. 48: 39–52 [DOI] [PubMed] [Google Scholar]
- 38. Palyi I. 1976. Mechanisms of spontaneous and induced heteroploidization and polyploidization. Acta Morphol. Acad. Sci. Hung. 24: 307–315 [PubMed] [Google Scholar]
- 39. Parker L. M., Fierro-Monti I., Mathews M. B. 2001. Nuclear factor 90 is a substrate and regulator of the eukaryotic initiation factor 2 kinase double-stranded RNA-activated protein kinase. J. Biol. Chem. 276: 32522–32530 [DOI] [PubMed] [Google Scholar]
- 40. Parker L. M., Fierro-Monti I., Reichman T. W., Gunnery S., Mathews M. B. 2001. Double-stranded RNA-binding proteins and the control of protein synthesis and cell growth. Cold Spring Harb. Symp. Quant. Biol. 66: 485–497 [DOI] [PubMed] [Google Scholar]
- 41. Parrott A. M., Walsh M. R., Reichman T. W., Mathews M. B. 2005. RNA binding and phosphorylation determine the intracellular distribution of nuclear factors 90 and 110. J. Mol. Biol. 348: 281–293 [DOI] [PubMed] [Google Scholar]
- 42. Pastwa E., Somiari R. I., Malinowski M., Somiari S. B., Winters T. A. 2009. In vitro non-homologous DNA end joining assays—the 20th anniversary. Int. J. Biochem. Cell Biol. 41: 1254–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patel A. G., Sarkaria J. N., Kaufmann S. H. 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U. S. A. 108: 3406–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patel R. C., et al. 1999. DRBP76, a double-stranded RNA-binding nuclear protein, is phosphorylated by the interferon-induced protein kinase, PKR. J. Biol. Chem. 274: 20432–20437 [DOI] [PubMed] [Google Scholar]
- 45. Pei Y., et al. 2008. Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by AKT-dependent phosphorylation at Ser647 in response to CD28 costimulation. J. Immunol. 180: 222–229 [DOI] [PubMed] [Google Scholar]
- 46. Pfeifer I., et al. 2008. NFAR-1 and -2 modulate translation and are required for efficient host defense. Proc. Natl. Acad. Sci. U. S. A. 105: 4173–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Powley I. R., et al. 2009. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev. 23: 1207–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puck T. T., Marcus P. I. 1956. Action of x-rays on mammalian cells. J. Exp. Med. 103: 653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ranpura S. A., Deshmukh U. S., Reddi P. P. 2007. NF45 and NF90 in murine seminiferous epithelium: potential role in SP-10 gene transcription. J. Androl. 29: 186–197 [DOI] [PubMed] [Google Scholar]
- 50. Reddy Y. V., Ding Q., Lees-Miller S. P., Meek K., Ramsden D. A. 2004. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J. Biol. Chem. 279: 39408–39413 [DOI] [PubMed] [Google Scholar]
- 51. Reichman T. W., Mathews M. B. 2003. The NF90 family of double-stranded RNA binding proteins: regulators of viral and cellular function, p. 335–342. In Bradshaw R., Dennis E. (ed.), Cell signaling handbook. Academic Press, San Diego, CA [Google Scholar]
- 52. Reitsema T., Klokov D., Banath J. P., Olive P. L. 2005. DNA-PK is responsible for enhanced phosphorylation of histone H2AX under hypertonic conditions. DNA Repair (Amst.) 4: 1172–1181 [DOI] [PubMed] [Google Scholar]
- 53. Rogakou E. P., Boon C., Redon C., Bonner W. M. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruis B. L., Fattah K. R., Hendrickson E. A. 2008. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 28: 6182–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saunders L. R., et al. 2001. Characterization of two evolutionarily conserved, alternatively spliced nuclear phosphoproteins, NFAR-1 and -2, that function in mRNA processing and interact with the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 276: 32300–32312 [DOI] [PubMed] [Google Scholar]
- 56. Seluanov A., Mittelman D., Pereira-Smith O. M., Wilson J. H., Gorbunova V. 2004. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. U. S. A. 101: 7624–7629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shang Z. F., et al. 2010. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 phosphorylation in response to DNA damage. Cancer Res. 70: 3657–3666 [DOI] [PubMed] [Google Scholar]
- 58. Shi L., et al. 2007. Dynamic binding of Ku80, Ku70 and NF90 to the IL-2 promoter in vivo in activated T-cells. Nucleic Acids Res. 35: 2302–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi L., et al. 2005. NF90 regulates cell cycle exit and terminal myogenic differentiation by direct binding to the 3′-untranslated region of MyoD and p21WAF1/CIP1 mRNAs. J. Biol. Chem. 280: 18981–18989 [DOI] [PubMed] [Google Scholar]
- 60. Soutoglou E., Misteli T. 2008. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320: 1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stavnezer J., Guikema J. E., Schrader C. E. 2008. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 26: 261–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tian B., Bevilacqua P. C., Diegelman-Parente A., Mathews M. B. 2004. The double-stranded-RNA-binding motif: interference and much more. Nat. Rev. Mol. Cell. Biol. 5: 1013–1023 [DOI] [PubMed] [Google Scholar]
- 63. Ting N. S., Kao P. N., Chan D. W., Lintott L. G., Lees-Miller S. P. 1998. DNA-dependent protein kinase interacts with antigen receptor response element binding proteins NF90 and NF45. J. Biol. Chem. 273: 2136–2145 [DOI] [PubMed] [Google Scholar]
- 64. Uematsu N., et al. 2007. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177: 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang H., Perrault A. R., Takeda Y., Qin W., Iliakis G. 2003. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 31: 5377–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao G., Shi L., Qiu D., Hu H., Kao P. N. 2005. NF45/ILF2 tissue expression, promoter analysis, and interleukin-2 transactivating function. Exp. Cell Res. 305: 312–323 [DOI] [PubMed] [Google Scholar]
- 67. Zhu P., et al. 2010. IL-2 mRNA stabilization upon PMA stimulation is dependent on NF90-Ser647 phosphorylation by protein kinase CbetaI. J. Immunol. 185: 5140–5149 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.