

Abstract
A fundamental objective in molecular biology is to understand how DNA is organized in concert with various proteins, RNA, and biological membranes. Mitochondria maintain and express their own DNA (mtDNA), which is arranged within structures called nucleoids. Their functions, dimensions, composition, and precise locations relative to other mitochondrial structures are poorly defined. Superresolution fluorescence microscopy techniques that exceed the previous limits of imaging within the small and highly compartmentalized mitochondria have been recently developed. We have improved and employed both two- and three-dimensional applications of photoactivated localization microscopy (PALM and iPALM, respectively) to visualize the core dimensions and relative locations of mitochondrial nucleoids at an unprecedented resolution. PALM reveals that nucleoids differ greatly in size and shape. Three-dimensional volumetric analysis indicates that, on average, the mtDNA within ellipsoidal nucleoids is extraordinarily condensed. Two-color PALM shows that the freely diffusible mitochondrial matrix protein is largely excluded from the nucleoid. In contrast, nucleoids are closely associated with the inner membrane and often appear to be wrapped around cristae or crista-like inner membrane invaginations. Determinations revealing high packing density, separation from the matrix, and tight association with the inner membrane underscore the role of mechanisms that regulate access to mtDNA and that remain largely unknown.
INTRODUCTION
Mitochondria are multifunctional cellular organelles that maintain and express their own DNA (mtDNA). Of the several thousand different proteins comprising the mitochondria, only 13 are encoded by mtDNA, which also contains genes for ribosomal and transfer RNAs necessary for mitochondrial mRNA translation. Transcription initiates from a minimal control region, resulting in strand-specific polycistronic RNAs. The 13 mtDNA-encoded proteins are essential subunits of the mitochondrial complexes responsible for ATP production through oxidative phosphorylation. The function of these complexes is coupled to a multitude of processes involved in intersecting metabolic pathways, reactive oxygen generation, calcium homeostasis, growth and differentiation, and apoptosis. The importance of these processes is emphasized by the numerous human genetic disorders associated with nuclear and mtDNA mutations leading to defects in mtDNA inheritance, maintenance, and expression (63). The small circular mitochondrial genome is unique both in its simplicity of organization and in that its expression is coordinated with that of nuclear-encoded proteins in response to cellular demands and mitochondrial capacity. mtDNA is organized into structures called nucleoids (34, 59). There are ∼300 to 800 nucleoids in a cell, each containing ∼1 to 10 mtDNAs, RNA, and proteins. Although the basic enzymology of mtDNA replication, expression, and repair is reasonably well understood, challenges remain in determining how these processes are integrated within a cell. As with other organisms, the packaging of mtDNA and the remodeling of nucleic acid-protein complexes are thought to regulate mitochondrial genetic events and their signaling pathways. Nucleoids are associated with proteins that are involved with mtDNA maintenance and expression, along with others that have no obvious relationship with mtDNA. The latter include those involved in protein stability and folding, mitochondrial import, RNA helicases, amino acid, carbon, and lipid metabolism and others with inner mitochondrial membrane locations (8, 19). In Saccharomyces cerevisiae, the nucleoid is thought to have a dynamic protein composition that responds to various cellular conditions while enrolling alternative functions of metabolic proteins (31). Although the yeast paradigm might generally apply to mammalian mtDNA, the evolutionary patterns with respect to their mitochondrial genomes, metabolic needs, and nucleoid compositions are dissimilar (30).
Mitochondrial transcription factor A (TFAM; also known as mtTFA) is regarded as the predominant core nucleoid protein. In mammals, this HMG box protein binds to mtDNA with high affinity at mtDNA promoters, where it functions as a transcription factor. It also binds DNA less specifically while functioning as an mtDNA packaging factor and is one of the few proteins with a clear structural role within the nucleoid. At maximum capacity, there are potentially ∼450 TFAM binding sites on the circular ∼16-kb mammalian mtDNA (27). TFAM localizes almost exclusively to nucleoids and is unstable in mitochondria without mtDNA (32, 33). The high DNA binding capacity and nucleoid-specific localization are properties of TFAM that have been useful in this work.
High-resolution imaging of submitochondrial structures has been achieved by electron microscopy (EM), which is mandated by spatial constraints within the organelle. Mammalian mitochondria are compartmentalized by outer and inner membranes, which form intermembrane spaces (IMS) that are typically only 20 to 50 nm wide (16). mtDNA was initially imaged using EM and was described as embodying elongated fibrils or rods residing within electron-lucent regions of the mitochondrial matrix (39). Unfortunately, the appearance of mtDNA was very much dependent upon the sample processing method, leaving uncertainty about the native organization of mtDNA. Iborra et al. later used immunogold EM analysis and reported that mammalian nucleoids are spheres 65 to 70 nm in diameter (24). Although that range represents a reasonable approximation, immunogold EM-labeled structures lack dimensional resolution. Nucleoids may have been directly imaged by EM according to a more recent report (47), but that study lacked any experimental evidence that the putative nucleoid structures contained mtDNA. Fluorescence microcopy has also been useful for producing images that reveal nucleoid location and frequency and for observing the dynamics of fusion and fission (6, 19, 54). However, in conventional fluorescence microscopy, the diffraction of light restricts the image resolution of objects to those that are more than 200 to 350 nm apart. This limit of diffraction has been surpassed using both two-dimensional (2D) (7, 22, 28, 52) and three-dimensional (23, 25, 55, 61) approaches. Other novel strategies continue to advance the field of superresolution microscopy at a rapid pace (56).
Two-dimensional photoactivated localization microscopy and three-dimensional photoactivated localization microscopy (PALM and iPALM, respectively) are two of the fluorescence techniques capable of locating objects at resolutions of 20 to 50 nm. This is accomplished through the use of photoactivatable fluorescent proteins (PA-FPs), which are activated stochastically and at low frequency rates in order to achieve spatial separation of their diffraction-limited fluorescent images. The location of each PA-FP molecule is determined with high precision by fitting the images to 2D Gaussian profiles (7). In iPALM, interferometry is added to locate molecular positions in the z dimension (58). Here we describe how the use of hyperbolic mirrors facilitates discrimination of multiple interferometric fringes and extends the measurement range along the z axis from ∼250 nm to ∼750 nm. We used this extended-range iPALM method to visualize mitochondrial nucleoids in 500-nm-thick sections. We also describe how imaging of gold nanoparticles in both channels allows two-color PALM image registration and accurate estimation of colocalization errors. Two-color 2D PALM is applied to determine the relative locations of mitochondrial nucleoids, matrix proteins, and cristae with unprecedented spatial resolution.
MATERIALS AND METHODS
Plasmid construction and mtDNA analysis.
For inducible gene expression, we used the GeneSwitch system (Invitrogen). A commercial pGeneV5/His plasmid was modified by inserting an AscI site into the multicloning sequence between the EcoRI and BamHI sites. The AscI linker was created by hybridizing oligonucleotides 5′-GATCCACTAGTGGCGCGCCAG-3′ and 5′-AATTCTGGCGCGCCACTAGTG-3′ to yield single-stranded overhangs suitable for ligation. The AscI-bearing plasmid (pGS) was then used to subclone mEos2 and Dronpa fragments into the NotI and AgeI sites, resulting in the plasmids pGS-mEos2 and pGS-Dronpa. The mEos2 cloning fragment was amplified by PCR using primers 5′-AACTGCGGCCGCATGAGTGCGATTAAGCCAG-3′ and 5′-GCAGACCGGTTATCGTCTGGCATTGTC-3′. The Dronpa cloning fragment was amplified by PCR using primers 5′-AACTGCGGCCGCATGGTGAGTGTGATTAAACCAG-3′ and 5′-GCAGACCGGTTACTTGGCCTGCCTCG-3′. TFAM and LACTB sequences were ligated upstream and in-frame using the AscI and NotI sites of pGS-mEos2 and pGS-Dronpa, respectively. The TFAM fragment was PCR amplified using primers 5′-CATTACGGCGCGCCAGTAATGGCGCTGTTCCGGG-3′ and 5′-TAGATGCGGCCGCAATGCTCAGAGATGTCTCC-3′. The LACTB fragment was amplified by PCR using primers 5′-CATTACGGCGCGCCAGTCATGTACCGGCTCCT-3′ and 5′-TAGATGCGGCCGCTGGACTGAATGGGGACGGC-3′. All constructs were analyzed by sequencing. TFAM has a T-to-C transition at nucleotide position 643, resulting in a lysine-to-glutamate substitution at amino acid position 215. The cytomegalovirus (CMV) promoter-driven CoxVIII1–29-Dronpa and CoxVIII1–29-Eos plasmids were previously described (7, 61). To determine the average number of mtDNA molecules per cell, 3T3sw cells were harvested during the late log phase of growth and counted using a Vi-CELL XR analyzer (Beckman Coulter). Total DNA was prepared from 100,000 cells by the use of 10 independent samples and the method of Legros et al. (33). To account for the efficiency of mtDNA recovery, half of the cell samples were spiked with a known amount of reference plasmid DNA containing the entire mouse mtDNA genome (p501-1). Whole-cell DNA preparations were then diluted, and mtDNA was quantified against a standard curve using the p501-1 plasmid in a quantitative PCR (qPCR) assay as previously described (9).
Cell culture.
The mifepristone-inducible 3T3 Switch (3T3sw) mouse fibroblast cell line was purchased from Invitrogen. These cells express a transcriptional activator protein consisting of the yeast GAL4 binding domain, a progesterone ligand binding domain, and a p65 activation domain from NF-κB. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM)–10% NCS–1 mM sodium pyruvate–2 mM l-alanyl-l-glutamine (GlutaMax)–50 μg/ml hygromycin B (Gibco/Invitrogen). The inducible expression plasmids carrying TFAM-mEos2 and LACTB(1–68)-Dronpa were transfected into 3T3sw cells by the use of Amaxa Nucleofector as previously described (10). Stably transfected cell lines were maintained under conditions of selection using Zeocin (200 μg/ml). Gene expression was induced with mifepristone (150 to 200 pM) for 7 to 8 h. For two-color PALM, the stable cell lines were transiently transfected with the appropriate second expression plasmid. The regulated genes were induced with mifepristone 20 h after transient transfection was performed as outlined above.
Confocal microscopy and analysis.
Cells were grown in Lab-Tek II chambers with no. 1.5 borosilicate coverglass bottoms (Nunc) coated with human fibronectin (Millipore) (15 μg/ml) overnight at 4°C. For mitochondrial imaging, cells were stained with 0.050 mM MitoTracker Red CMXRos (Invitrogen) in complete media for 15 min at 37°C, rinsed once with dye-free media, and imaged live. For mtDNA staining, cells were stained with PicoGreen (Invitrogen) diluted 1:500 in complete media for 30 min at 37°C and then incubated in dye-free media for 1 h. Colocalization by immunofluorescence was accomplished by fixing cells with 2% paraformaldehyde in 60 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES)–25 mM HEPES–10 mM EGTA–2 mM MgCl2, pH 7.0, for 15 min at 37°C, blocking in 5% goat serum, and incubating with anti-single-stranded DNA (anti-ssDNA) monoclonal antibody (clone BV16-19 [Millipore]) diluted 1:4,000 in 0.25% Triton X–100-phosphate-buffered saline (PBS) overnight at 4°C. Cells were then reacted with an AlexaFluor 568 goat anti-mouse IgG secondary antibody (Invitrogen) diluted 1:3,000 in 0.25% Triton X-100–PBS for 1 h at room temperature. Cells were imaged using a Zeiss LSM 510 META microscope equipped with a 100× 1.4-numerical aperture (NA) Plan-Apochromat objective. A 15-mW DPSS 561-nm diode laser was used for excitation of MitoTracker Red and Alexa-Fluor 568. The 488 line of a 30-mW multiline gas argon laser was used for excitation of TFAM-mEos2, PicoGreen, and LACTB1–68-Dronpa. All images represent 1,024-by-1,024, 12-bit z-stack acquisitions created using sequential scanning and 0.8-μm steps. Volocity software (Perkin Elmer) was used to identify and count nucleoids by the following protocol: objects are found using intensity levels and clipped to region of interest, and touching objects are separated by 0.2 μm3. Microsoft Excel was used for basic statistical analysis.
Cell fixation and cryosectioning.
Cells in T flasks (75 cm2) were washed with PBS and fixed in monolayers with 10 ml of freshly prepared 4% paraformaldehyde-0.2% glutaraldehyde-100 mM sodium phosphate (pH 7.4). Initial fixation for 15 min at 37°C was followed by an additional hour with fresh fixative at room temperature. Cells were washed with PBS and incubated with 0.05 M glycine-PBS for 15 min and then scraped from the flask surface into PBS with 1% bovine serum albumin (BSA), centrifuged at 2,000 × g for 5 min, harvested, and repelleted in a 1.5-ml microcentrifuge tube. For embedding and cryosectioning, cell pellets were suspended in 75 μl of warm 10% gelatin. Cooled gelatin fragments were infiltrated overnight at 4°C with 2.3 M sucrose-100 mM sodium phosphate (pH 7.4). Samples were mounted onto specimen pins, immersed in liquid nitrogen, and transferred to a precooled cryochamber affixed to a microtome. Interferometric PALM data were also collected from samples embedded in LR White resin. This modified Tokuyasu method performed using LR White resin embedding was previously described (10).
Two-dimensional superresolution microscopy.
Basic PALM methods were described previously (57). For typical TFAM-mEos2 data collection, images were taken at intervals of 50 ms, and 15,000 to 25,000 frames were collected at a 561-nm laser power of approximately 1,000 W/cm2. For typical Dronpa PA-FP data collection, images were taken at 100 ms, and 4,000 to 8,000 frames were collected at a 488-nm laser power of approximately 500 W/cm2. Molecule localization accuracy is represented here as the standard deviation of the position assignment value (σ). In these experiments, the average σ for Eos was 3.0 nm and for Dronpa was 3.1 nm. To determine whether nucleoid dimensions are altered by expression of TFAM-mEos2, we compared molecular maps from PALM to those obtained using antibody-based dSTORM imaging (21). For dSTORM imaging, nucleoids were labeled with a monoclonal antibody (clone BV16-19) as described above. A 1:8,000 dilution of an Alexa Fluor 568-labeled goat anti-mouse IgG antibody (Invitrogen) was used for the secondary labeling. dSTORM imaging of 150-nm-thick cryosections was done using PBS containing 100 mM mercaptoethylamine and a 488-nm laser at ∼3 kW/cm2. Approximately 6,000 images were obtained, with frames acquired every 20 ms. Half-maximum histogram peak widths determined from two perpendicular axes were used to determine nucleoid diameters for dSTORM and PALM analyses as shown here (see Fig. 2).
Fig. 2.

PALM data representation and measurement of a single nucleoid defined by TFAM-mEos2 fluorescence localization. (A) Fluorescence localization data displayed as a two-dimensional distribution of points, each representing the centroid position of the diffraction-limited image of the fluorescent protein (FP) molecule TFAM-mEos2. (B) Histogram distribution of the FP molecules shown in panel A in the x dimension. The relative position of each bin is displayed on the x axis, with marks every 10 nm. Measurement of the nucleoid x axis width is obtained from the full-width half-maximum of the histogram peak height (FWHM), shown here as a red line with asterisks. (C) The same data displayed as a color-coded probability map of FP molecule locations. The color intensity scale (bottom left) indicates the FP molecule probability per square nanometer. The FWHM positions are also shown in panels A and C with white hash marks. For this molecule, the FWHM value for the x dimension is about 125 nm. (A and C) Bars, 50 nm.
Measurement, calibration, and validation of z-coordinate positions in iPALM.
Basic iPALM methods were described previously (58). For typical TFAM-mEos2 data collection, images were collected at 12.5 frames/s with an excitation time of 50 mS, collecting 20,000 to 50,000 frames at a 561-nm laser power of approximately 2,000 W/cm2. Molecule localization accuracy is reported here as the standard deviation of the position assignment value (σ). In these experiments, the average accuracy values for the x, y, and z dimensions were 11.9 nm, 10.1 nm, and 10.5 nm, respectively. The z determinations of the iPALM technique in this work have been improved by extending the range to ∼750 nm from a previous limit of ∼250 nm. Hyperbolic mirror parameters were adjusted in order to ensure that the ellipticity point-spread functions (PSF) depended on the emitter z coordinate. Modeling suggested that the mirror curvature radius required to ensure this dependence of PSF ellipticity on z-coordinate values is on the order of 105 cm. The hyperbolic mirrors impart weak saddle-shaped wavefront distortions to infinity-corrected beams at the back focal planes of the microscope objectives in the iPALM setup. As a result, the diffraction-limited images of single fluorescent molecules become elliptical, with the degree and angle of ellipticity being functions of the z coordinate. This allows discrimination of adjacent interferometric fringes and extension of the measurement range from ∼250 nm to ∼750 nm. Astigmatic point-spread functions (PSF) can be approximated by determinations performed using 2D Gaussian profiles with different x and y widths. The x-y ellipticity of the PSF can be defined as follows:
 |
where σx and σy represent Gaussian widths along the x and y axes. In a typical calibration run, we translated the sample with gold nanoparticles over a vertical range of ∼800 nm and recorded the fluorescent images every 8 nm. We then performed 2D Gaussian fitting and extracted x-y ellipticity values as defined in equation 1. The z coordinate could then be extracted in the following manner. During the iPALM calibration, the dependence of x-y ellipticity on the sample z coordinate (see Fig. 3) is recorded along with the standard iPALM interferometric calibration (58). The polynomial fitting is performed according to the following formula:
where ε is the x-y ellipticity of PSF as defined in equation 1 and m1, m2, and m3 are the fitting parameters determined from the calibration step. Then, during the iPALM measurements, we extract two values: zinterf (which is the z coordinate determined from the recorded interferometric patterns as described in reference 58), and Zε (which is the z coordinate determined using equation 2 from the recorded value of ε and m1, m2, and m3 determined during calibration).
Fig. 3.

Point-spread function ellipticity and interferometric z-coordinate display. (A) Three examples of PSF images of gold nanoparticles at different vertical positions are shown. x-y ellipticity is plotted relative to the z position (shown in red). The black line represents the polynomial fit corresponding to equation 2. (B) Interferometric coordinates are displayed as black dots relative to collected frame number. (C) The full z coordinate is displayed relative to frame number as the sample is displaced along the z axis. Sample positions from panels B and C are shown in red.
The z coordinate is then calculated using the following formula:
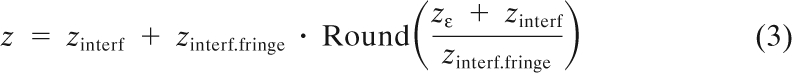 |
where the constant zinterf.fringe represents a single interferometric fringe value of ∼250 nm.
The assignment of the z coordinate was validated using biotinylated Eos fluorescent protein (FP) molecules bound to the surface of a coverslip. No. 1.5 coverglasses (Werner Instruments) were cleaned and coated with 0.1% poly-l-lysine for 30 min, rinsed, and dried. Gold nanorod particles (NanoPartz) were then applied by application of a 0.5% solution for 5 min. After being rinsed, the glass was coated with biotin (1 mg/ml)-BSA-PBS (Sigma Chemical A8549) followed by streptavidin–PBS (Sigma Chemical S462) (1 mg/ml solution). Biotin-conjugated mEos (gift of George Patterson) was then bound to the streptavidin at 3.2 μg/ml for 5 min. We then took a wide-field (20 μm by 20 μm) image of the molecules while translating the sample along the z axis in 8.75-nm steps (pausing for 99 frames at each step). The entire sample excursion along the z axis was ∼750 nm (or 3 interferometric fringes). We then extracted first interferometric and then full z coordinates for each detected FP molecule (total count, ∼85,000) by the use of equation 3 and the procedure described above. Data from biological samples were obtained using 500-nm-thick sections and both the modified Tokuyasu method and the LR White resin-embedded samples as described above.
Alignment of two-color PALM images and evaluation of alignment errors.
Two-color superresolution imaging requires a way to differentiate and align two fluorescent labels. The colocalization of fluorescent labels in 2 channels is determined by the following equation (44):
where σloc1 and σloc2 represent the localization accuracies in each color channel and σreg represents the accuracy with which positions between 2 channels can be registered. Assuming the simplest case with no chromatic aberrations, the relationship between the images collected in the 2 color channels should be revealed by a similarity transformation involving shift, rotation, and slightly different magnifications:
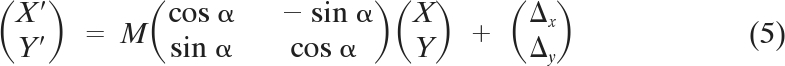 |
where M represents magnification.
In order to determine the transformation parameters in equation 5, we used 100-nm-diameter gold nanoparticles (Microspheres-Nanospheres, Cold Spring Harbor, NY), which exhibit fluorescence in both channels. These particles exhibit surface plasmon resonance-enhanced photoluminescence with wide spectra in the visible region (14, 62). The spectra of individual nanoparticles deposited on glass coverslips with excitation at 488 nm and 561 nm were determined using a SPEX 500 spectrometer and averaged over a range of at least 5 nanoparticles in each case. In order to evaluate the two-color registration accuracy (σreg), we deposited gold nanoparticles on a standard coverslip and imaged them in both channels, collecting 5,000 frames from each channel. We used Semrock RU-568/FF01-593-40 and RU-488/FF01-520-35 long-pass and band-pass filter pairs (the same filters used in PALM measurements for fluorescent protein localization) to reject the excitation signal and limit the emission spectra to relevant wavelength ranges. We then performed the standard localization procedure for all fiducials within a field of 25 by 25 μm in both channels. For each nanoparticle, we then determined the x and y coordinates in each channel, determining the averages of the distributions over 5,000 frames. The pairs of coordinates determined for each nanoparticle in the 2 channels should satisfy equation 5, so we can use the linear regression procedure to extract the transformation coefficients.
First, it is convenient to use complex variable Z = X + jY (where j represents complex unity), so we can rewrite equation 5 as follows:
where Mc = Mejα, Δ = Δx + jΔy, and Mc represents complex magnification. We can derive a least-square optimization (linear regression) procedure for finding Mc and Δ:
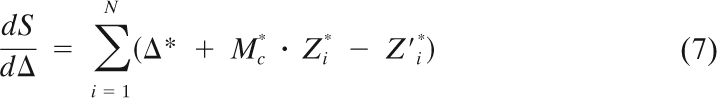 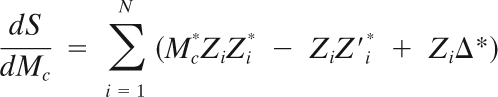
|
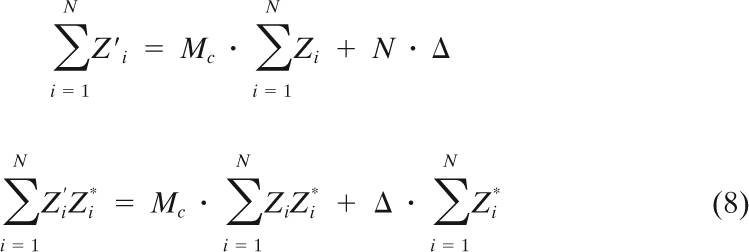 |
One can then solve the system shown above by linear decomposition to get the values of Mc and Δ.
In order to evaluate the 2-channel registration accuracy (σreg), we again determined the averaged x and y coordinates of each nanoparticle in each channel after transformation. Then we calculated the differences between the averaged x and y coordinates in 2 channels for each nanoparticle.
EM analysis of mitochondrial matrix dimensions.
To determine the average dimensions of the mitochondrial matrix, 3T3sw and 3T3sw cells expressing inducible LACTB1–68-Dronpa were harvested and fixed as outlined above. Cell pellets were washed with 1% BSA–0.1 M phosphate buffer (PB) (pH 7.4) and resuspended in 0.1 M sodium cacodylate buffer prior to immobilization of pelleted cells in 1.2% agar–PB at 44°C. Agar pellet fragments were secondarily fixed overnight in 2% paraformaldehyde, washed three times with cacodylate buffer, and postfixed with reduced 1% OsO4 for 1 h. Samples were again washed with 0.1 M sodium cacodylate, stained for 1 h in 1% uranyl acetate, dehydrated stepwise in ethanol, and embedded in Epon. EM images were obtained using an FEU Tecnai G2 20 Twin transmission EM (TEM) at magnifications of ×5,000 to ×11,500. The average matrix space between cristae and the outer edges was determined using multiple measurements from 80 different mitochondria and SigmaScan Pro (Systat). Measurements of distances between various membranes were taken at 2 to 6 perpendicular locations for each.
Immunogold particle electron microscopy.
A stable 3T3sw cell line carrying an inducible LACTB1–68-Dronpa gene was induced, fixed, and frozen as described above. Cryosections (60 nm thick) were cut and placed on Pioloform-coated, carbon-stabilized 200-mesh nickel grids. The sections were washed three times on drops of PBS (pH 7.4) for 5 min with shaking. The sections were then incubated in 0.05 M glycine–PBS for 15 min followed by a 20-min incubation in a blocking solution of 1% BSA, 0.1% Tween 20, 0.1% Triton X-100, and PBS (pH 7.4). The primary anti-Dronpa-green antibody (MBL, Nagoya, Japan) was diluted 1:20 in blocking buffer and incubated on sections for 2 h, followed by three 10-min washes in 0.1× blocking buffer. This was followed with 60 min of incubation with a goat anti-mouse secondary antibody conjugated to a 10-nm-diameter gold particle (Ted Pella, Redding, CA), three 10-min washes with 0.1× blocking buffer, and two 5-min washes with PBS. Sections were fixed with 1% glutaraldehyde–PBS for 15 min, followed by two 5-min washes in PBS and five 3-min washes in distilled water. Lastly, the cryosections were stained with 0.5% uranyl acetate–2% methylcellulose for 10 min on ice.
Immunogold labeling was analyzed as described by Rabouille (49). Images of labeled cells were taken at a magnification of ×29,000 for a field of view of 3.26 ×106/nm2 with a Tecnai T20 TEM (FEI) operated at 80 kV. The surface areas of mitochondria, nucleus, and cytoplasm were determined for each electron micrograph using the point-hit method and ImageJ software (http://imagej.nib.gov/ij/) to overlay the micrograph with line spacing at 100 nm, representing a surface area of 10,000 nm2. For submitochondrial localization, we used a finer-mesh grid such that each point represented 5,000 nm2. The relative labeling density for each organelle and compartment was determined by dividing the number of gold particles located on the organelle and compartment by the surface area as determined by the point-hit method. Nuclear labeling was used as the background signal, and specific gold labeling was determined by subtracting the background labeling from the relative labeling density.
RESULTS
TFAM-mEos2 is incorporated into nucleoids without apparent mitochondrial alteration.
To label nucleoids, TFAM (NM_009360.4) was fused into the mEos2 translational reading frame (FJ707374.1) encoding PA-FPs with green to red fluorescence (36). To limit effects of overexpression, we used the GeneSwitch system (Invitrogen), which employs an inducible synthetic promoter. We have also established mouse 3T3 Switch (3T3sw) fibroblasts that stably express the inducible TFAM-mEos2 gene in order to limit the copy number effects inherent in transfection. The cells retain the native TFAM gene. It is difficult to unequivocally state that heterologous gene expression is without consequence with respect to unintended effects. However, we have made efforts to determine whether the TFAM-mEos2 fusion protein significantly alters the relevant properties of the mitochondria.
Figure 1 displays confocal microscopy images showing that TFAM-mEos2 accumulates at punctate foci within the mitochondria. The pattern of these foci is consistent with nucleoid images obtained by others (19). Mitotracker red staining showed that the foci are limited to the mitochondria and that the reticular morphology of the mitochondria was retained (Fig. 1B to D). This indicates that TFAM-mEos2 expression is well tolerated, as fragmentation of mitochondria is often associated with distressed mitochondrial function and apoptosis (29). TFAM-mEos2-labeled nucleoids were counted and compared to the number of nucleoids in control cells stained with the DNA-binding dye PicoGreen (4). The number of TFAM-mEos2 foci per cell (mean = 291) was similar to the nucleoid count in PicoGreen-stained 3T3sw cells not expressing the fluorescent fusion protein (mean = 275 [Table 1]). Anti-DNA antibody immunostaining showed extensive colocalization with TFAM-mEos2 (Fig. 1E to H). In addition, induction of TFAM-mEos2 expression did not alter either the mtDNA amount determined using qPCR or the mitochondrial morphology, as seen in electron micrographs of EPON-embedded cells (data not shown).
Fig. 1.

Genetically expressed TFAM-mEos2 localizes to mtDNA. (A and E) Confocal images of cells displaying the green-state fluorescence of TFAM-mEos2. (B) Mitochondrial staining with Mitotracker Red. (C) Merged images from panels A and B, with box outlining the magnified region displayed in panel D. Arrows in panel D indicate yellow nucleoids within mitochondria. (F) Alexa-Fluor 568 immunofluorescence staining with an anti-DNA antibody. (G) Merged images from panels E and F, with box outlining the magnified region displayed in panel H. Left and right arrows in panel H point to nucleoids with lesser and greater TFAM-mEos2 signals, respectively. Bars, 10 μm (A and E), 5 μm (D and H).
Table 1.
Properties of 3T3sw mitochondrial nucleoids, mitochondria, and relative DNA packing density
| Property | Value | Basis or reference |
|---|---|---|
| Mean no. of nucleoids/3T3sw cell (±SD)d | ||
| PicoGreen DNA staining | 275 (±77) | |
| TFAM-mEos2 (green state) labeling | 291 (±92) | |
| Mean no. of mtDNA copies/3T3sw cellb (±SD) | 821 (±163) | |
| mtDNA/nucleoidsc | 2.98 | 821/275 |
| Mean nucleoid vol (nm3) | 830,000 | Fig. 4 |
| Mean matrix space between cristae (nm)d (±SD) | 68 (±24) | |
| Mean matrix diam (nm) (±SD)e | 238 (±75) | |
| ρpackf | ||
| Bacteriophage T7 | 0.49 | 48 |
| ECV304 (human) mt nucleoid | 0.110 | 24 |
| P. polycephalum mt nucleoid | 0.069 | 5 |
| 3T3 (mouse) mt nucleoid | 0.063g | |
| Nuclear DNA | 0.031 | 3.3 × 109 bp/113 μm3 |
| E. coli | 0.005 | 4.64 × 106 bp/0.9 μm3 |
| Nucleoid diam (nm) | ||
| 2D PALM (TFAM-mEos2) | 110 (±59) | |
| dSTORM (DNA antibody) | 110 (±46) |
Data were determined by whole-cell confocal microscopy.
Data were determined by qPCR.
Average mtDNA genomes/cell divided by average nucleoids/cell.
Data were determined by measurements from EM images using the formula (mitochondrial length/number of cristae) − average crista width (21.5 nm); n = 80 mitochondria.
Data were determined from 60 width measurements taken between inner boundary membranes; sample range, 115 to 430 nm.
Data were determined using the equation
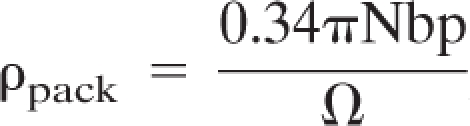 , where Nbp represents the number of base pairs and Ω represents the package volume in cubic nanometers. mt, mitochondrial.
, where Nbp represents the number of base pairs and Ω represents the package volume in cubic nanometers. mt, mitochondrial.
Data were determined using averages of 3 genomes/nucleoid and the equation shown in footnote f.
To determine whether TFAM-mEos2 expression affects nucleoid structure, we compared these nucleoid dimensions to those seen in the absence of TFAM-mEos2 expression. This was done using dSTORM (21) imaging of nucleoids labeled with an antibody to DNA. TFAM-mEos2- and anti-DNA antibody-labeled nucleoids are similar in size (Table 1). Thus, it does not appear that expression of TFAM-mEos2 protein leads to abnormal packaging of nucleoids. Under the conditions used, dSTORM imaging suffered from a comparatively low labeling density. Therefore, the resolution of nucleoid structure determinations performed using dSTORM is less than that seen with PALM.
PALM and iPALM image display and dimension measurement.
The distribution of TFAM-mEos2 molecules within a nucleoid obtained using PALM can be displayed in several ways, as shown in Fig. 2. Final PALM data consist of a distribution of points representing assigned locations of molecules of a fluorescent protein (7). Panel A in Fig. 2 represents the simplest molecule distribution using point assignments. PALM images are most often displayed as a space- and color-filled distribution that expresses the statistical certainty of the assignment of each molecular location. An example of such a probability map is shown in Fig. 2C. Each of the probability density values represents the probability of an observed molecular label occurring in a given area. These values are derived from the Gaussian profile determined for each molecular event imaged and are reported as the probability divided by the area in square nanometers. For example, a point of light with dimensions of 20 by 20 nm would be binned over a grid with dimensions of 20 by 20 nm, with each grid cell value calculated at 1/400 per nm2, or 0.0025/nm2, as the maximum probability value for this event. A broader Gaussian function would yield a lesser value, reflecting the broader area of each grid space. In the rendered image, each space is given a color on a gradient that reflects the probability value for each event. These are then overlaid on a single image. The utility of this image is that it supplies further information about the quality of the centroid point assignments. This probability map display is used in the remaining figures of this work, and a detailed description of the rendering algorithm appears in earlier work (7) (see section 6 of the online supporting information associated with that work). In images that are densely labeled, the data representing the localization probability of a given individual molecule are lost in the summed projection. However, this positional information is preserved and used in the histogram analysis. Figure 2B displays a histogram that plots the frequency of the same molecules at a given position along a single axis. Importantly, the width of the frequency histogram at half the maximum peak value (full-width half-maximum of the histogram peak height; FWHM) represents a statistically robust and conservative measurement of the width of the nucleoid in this dimension. The use of FWHM measurements applied to PALM has previously been validated using cytoskeletal fibers with known dimensions (58).
Extension of iPALM measurement range to 750 nm by the use of hyperbolic mirrors.
The original iPALM method was able to achieve highly accurate lateral (x and y) localization of fluorescent particles and a vertical z localization range of ∼250 nm. Extension of this vertical z range is based on the principle that point sources of light correspond to point-spread functions (PSF) that can be made elliptical using hyperbolic mirrors. Figure 3A shows that the ellipticity of the resulting x-y PSF is dependent on the location of the light source in the z dimension. Thus, the ellipticity can be used to determine the z coordinate of a particular fluorescent molecule. Figures 3B and C show that the procedure works well over a range that is greater than ∼750 nm, which is close to the depth of field of the high-NA objective used in the experiment. There was a small fraction of molecules for which the interferometric fringe was determined incorrectly (faint traces 250 nm above and below the center trace in Fig. 3C). The total fraction of these ghosts, or the error ratio, was 0.75%, and it did not exceed 5% for any z-position.
Dimensional analysis of ellipsoid-shaped nucleoids determined using 3-dimensional iPALM.
Three-dimensional iPALM of TFAM-mEos2-defined nucleoids reveals that about 65% of nucleoids have roughly ellipsoidal shapes. Examples of three ellipsoidal nucleoids are shown in Fig. 4, where the TFAM-mEos2 distributions are displayed in two-dimensional projections, along with a three-dimensional drawing of each nucleoid with FWHM measurements of each dimension. The nucleoid shown in Fig. 4D to F has dimensions that are close to the average within the ellipsoid subset. The short, middle, and long nucleoid axis measurements are shown as distributions in Fig. 5A. Ellipsoidal nucleoid dimensions differ over a 10-fold range from shortest to longest dimension (31 to 318 nm). The shapes of the ellipsoids also differed. However, most nucleoids have either three unequal dimensions or one dimension that is larger than the other two. The mean values of the three major axes are 85 by 108 by 146 nm, as shown in Fig. 5A. The ellipsoidal subset was also used to estimate nucleoid volume, and that distribution is shown in Fig. 5B. A histogram plotting the frequency of binned nucleoid volumes reveals that the distribution is positively skewed. Individual nucleoid volumes, along with the mean value of 830,000 nm3, are plotted in Fig. 5C. The standard deviation of the mean encompasses a 25-fold range from about 60,000 to 1,600,000 nm3. Giant nucleoids above this range exist but are infrequent. A relative estimation of the number of TFAM-mEos2 molecules per nucleoid can be approximated using PALM. There is no correlation between TFAM-mEos2 molecular counts and nucleoid volume. It is therefore unlikely that the fluorescent molecules influenced the size of the nucleoids.
Fig. 4.

Ellipsoidal nucleoid projections and their graphical dimensions obtained from iPALM imaging of TFAM-mEos2. Panels A to C, panels D to F, and panels G to I represent three different nucleoids. In panels A, D, and G, each nucleoid image is projected in x (horizontal) and y (vertical) dimensions. In panels B, E, and H, the z dimension replaces y on the vertical axis. In panels C, F, and I, each nucleoid is graphically displayed along with measurements of full width at half-maximum (FWHM) in three dimensions. Bar, 50 nm (the scale bar in panel A applies to all panels). The fluorescent molecule scale maximum values (representing probability per square nanometer) differ among the panels and range from 0.019 to 0.034.
Fig. 5.

Ellipsoidal nucleoid axial dimension and volume distribution. (A) For each nucleoid (n = 98), a short-, middle-, and long-axis length is plotted to display the distribution. Bars representing means (± standard deviations [SD]) are plotted above each data set; the long-axis mean is 146 (±47) nm, the middle-axis mean is 108 (±30) nm, and the short-axis mean is 85 (±27) nm. (B) A 24-bin histogram displaying the frequency distribution of calculated nucleoid volumes. (C) The individual nucleoid volumes (n = 98) are plotted as points along the x axis shared with panel B. The mean (8.5 × 105 nm3) ± SD (7.77 × 105 nm3) is displayed above the raw distribution.
Analysis of nucleoid size, relative spatial limits, and estimated mtDNA density.
We were interested in learning whether nucleoid size measurements represented any spatial constraints within the mitochondria. Dimensions of mitochondrial compartments were obtained from EM images of 3T3sw cells (Table 1). The average diameter of the mitochondrial matrix between inner boundary membranes measured 238 (±75) nm, which is sufficient to accommodate most nucleoids. However, the mean available matrix space between cristae measured only 68 (± 24) nm, which is inadequate for most nucleoids. The relative dimensions of the nucleoid and the crista-limited matrix indicate either an intimate association between cristae and nucleoids or the existence of specialized, crista-free nucleoid domains within the matrix.
We have also estimated the packing density of mtDNA in mouse fibroblasts by the use of cell population averages. Table 1 contains a summary of the data used in these calculations. Quantitative PCR yielded an average value of 821 mtDNA genomes per cell. Whole-cell nucleoid counts obtained from confocal microscopy indicated an average of 275 nucleoids per cell. Therefore, there are, on average, approximately 3 mtDNA molecules per nucleoid in 3T3sw cells. Although carcinoma cells typically yield higher estimates, our determined value is consistent with a previous estimation of 2.4 genomes per nucleoid in fibroblasts (33). The DNA packing density (ρpack) is determined by dividing the volume of a DNA cylinder of known length by the volume of space that it occupies (48). Calculated using this method, the mammalian nucleoid has an average ρpack value of 0.063, which is similar to the Physarum polycephalum mtDNA nucleoid ρpack value of 0.069 (Table 1) but less than that calculated from measurements made by Iborra et al. (24). By comparison, Escherichia coli and mammalian nuclear genomes, with ρpack values of 0.031 and 0.005, are less compact. This level of mtDNA condensation has implications with respect to mechanisms of packaging and mtDNA accessibility.
iPALM and PALM imaging analysis of variously shaped nonellipsoidal nucleoids.
Although the majority of nucleoids were roughly ellipsoidal, as described above, the remaining 35% displayed an assortment of shapes. We can loosely categorize these nucleoids into three equally represented subsets based on their predominant features. The first category comprises nucleoids with either a crescent shape or one side that is largely concave. Examples of these are shown in Fig. 6A and D. The second category includes nucleoids that are amorphous or branched, as displayed in Fig. 6C. The third category consists of nucleoids that have either an internal gap or are split, as shown in Fig. 6B and E. It is important that the heterogeneity in size and shape also applies to the elliptical subset and that these categories, while not fully discrete, have descriptive value. The crescent or concave shapes demonstrate that nucleoids are not restricted to a globular form and may indicate structural constraints imposed by mitochondrial membranes. The amorphous nucleoids show that the TFAM-mEos2 population can be unevenly distributed into discrete core domains with branches that are less condensed. The images of the split nucleoids are consistent with previous observations of nucleoid fusion and fission (19). More subtly gapped nucleoids may encompass protrusions of cristae. Alternatively, uneven distributions of TFAM-mEos2 could represent core TFAM binding sites, thus resolving discrete mtDNA molecules within a more loosely organized nucleoid.
Fig. 6.

iPALM imaging reveals that nucleoids are not restricted to ellipsoidal boundaries. TFAM-mEos2-labeled nonelliptical nucleoids exhibit concave, split, and amorphous forms. The fluorescent molecule scale maximum values (representing probability per square nanometer) differ among the panels and range from 0.14 to 0.19. Collectively, these examples represent about 35% of the nucleoid population. Bar, 100 nm (the scale bar in panel A applies to all panels).
Alignment of two-color PALM images and colocalization accuracy.
In this study, we sought to determine the location of nucleoids relative to those of other mitochondrial compartments. This required the imaging of two fluorescent proteins within the same sample. Superresolution imaging requires an accurate and robust method to determine the relative locations of different fluorescent proteins within the same field. This can be done by constructing labels with different excitation spectra and identical emission spectra (5) or by using fluorescent labels with different excitation spectra (57). In the latter case, PALM images in 2 channels with different fluorescent wavelengths are typically not aligned “as acquired,” and images may be shifted and tilted due to different filter sets used in acquisition and slight magnification differences of optics at different wavelengths. We have used gold nanoparticles as fiducial markers for dual-label alignment. To characterize the utility of gold particles for such alignments, we first measured their photoluminescence spectra. As can be seen from Fig. 7A, the emission spectrum of 100-nm gold nanoparticles is fairly broad and covers the wavelength range of interest. This allows flexibility in the choice of the fluorescent proteins used. It should be pointed out that, when one of the fluorescent labels, such as Alexa 647, produces emissions that are further toward the red end of the spectrum, the gold particles cannot be used, since their emission at 650 nm is weak. However, particles with a properly chosen size/aspect ratio work well (62). An example of gold particle alignment is displayed in Fig. 7B. This alignment results in a low localization error, i.e., the distance between averaged coordinates in the 520-nm and 590-nm channels after alignment. The distributions of the x and y localization errors are shown in Fig. 7C. The standard deviations of the x and y localization errors shown in Fig. 7C are 5.1 nm and 6.7 nm, respectively, and thus we can estimate
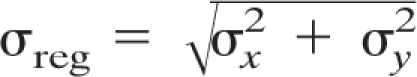 (in this case, σreg = ∼8.4 nm). For the two-color PALM measurement, we used enough fiducials to follow the same procedure, which allowed us both to perform image alignment and to estimate dual-label registration accuracy (σreg), which is typically below 10 nm. This high degree of accuracy of two-color image alignment is crucial for interpreting colocalization studies at high resolution, and we have applied this method to the colocalization of mitochondrial nucleoids within the organelle. Prior to this work, methods for determining two-color localization accuracy were lacking.
(in this case, σreg = ∼8.4 nm). For the two-color PALM measurement, we used enough fiducials to follow the same procedure, which allowed us both to perform image alignment and to estimate dual-label registration accuracy (σreg), which is typically below 10 nm. This high degree of accuracy of two-color image alignment is crucial for interpreting colocalization studies at high resolution, and we have applied this method to the colocalization of mitochondrial nucleoids within the organelle. Prior to this work, methods for determining two-color localization accuracy were lacking.
Fig. 7.

Alignment of dual-label PALM images and colocalization accuracy. Panel A displays emission spectra of 100-nm-diameter gold nanoparticles. Data representing the emission filters used for registration in the 520-nm channel (Semrock FF01-520-35; green dashes) and 590-nm channel (Semrock FF01-593-40; orange dashes) are also shown. (B and C) Two-color PALM image alignment. Localizations of gold nanoparticles at 520 nm (green dots) and 590 nm (red dots) before (B) and after (C) alignment. Bars, 20 nm. Data points were collected from 5,000 frames and peak coordinates extracted for each channel. (D) Two-color localization error distributions. Distances plotted are between averaged coordinates in the 520-nm and 590-nm channels after alignment. Localization differences within the given error range are shown in red for the x axis and in blue for the y axis. The standard deviation for X (σx) is 5.1 nm and for Y (σy) is 6.7 nm.
TFAM-mEos2 nucleoid location and mitochondrial matrix-targeted CoxVIII1-29-Dronpa are largely mutually exclusive.
The N-terminal mitochondrial targeting sequence from CoxVIII (NM_004074.2) is commonly used to deliver fluorescent proteins to the mitochondrial matrix and has also been applied in PALM (7, 51). We used a previously characterized CoxVIII1–29-Dronpa expression plasmid (61) to deliver the PA-FP Dronpa to the mitochondrial matrix. Dronpa (AB180726.1) encodes a green photoactivatable fluorescent protein used as a second sequential marker in PALM that is paired with the green-to-red-fluorescence PA-FP mEos (18, 57). Figure 8 displays examples of 2D PALM imaging data obtained from cells coexpressing the nucleoid-specific TFAM-mEos2 and the matrix-specific CoxVIII1-29-Dronpa. The large majority (84%) of the cells show that the nucleoid and matrix proteins have discrete distributions (Fig. 8A to E). Gaps between the matrix and nucleoid proteins are not uncommon. The remaining 16% show various degrees of overlapping expression of the nucleoid and matrix proteins (Fig. 8F). This could represent a true mixing of the proteins. However, it is also possible that the two signals were emitted from different planes within the 150-nm-thick section. We have been unable to obtain two-color iPALM data to answer the question of how frequently these proteins truly colocalize. Regardless, the predominantly discrete localization shows that the nucleoid most often excludes freely diffusing matrix proteins.
Fig. 8.

TFAM-mEos2 nucleoid location relative to the mitochondrial matrix-targeted CoxVIII1–29-Dronpa. TFAM-mEos2 nucleoids are displayed in red. The mitochondrial matrix protein CoxVIII1-29-Dronpa is shown in green. The fluorescent molecule scale maximum values (probability per square nanometer) differ per panel and range from 0.005 to 0.01 for Dronpa and 0.01 to 0.02 for mEos2. The nucleoid and matrix proteins have discrete boundaries in most (84%) images, as displayed in panels A to E. These edges are less discrete in the remaining 16%, as represented by panel F. Bar, 100 nm (the bar in panel A applies to all panels).
Amino-terminal peptides from LACTB faithfully target Dronpa to the mitochondrial IMS.
Prior data indicate that the protein encoded by LACTB (NM_030717.1) localizes to the mitochondrial intermembrane space (IMS) within cristae. To create a marker for the mitochondrial IMS and cristae, we fused Dronpa to the nucleotide sequence encoding the N-terminal 68 amino acids of LACTB that were previously identified as the mitochondrial targeting sequence (45). LACTB1–68-Dronpa is seen exclusively in the mitochondria, according to the results of confocal microscopy (Fig. 9A, B, and C). As shown by the use of PALM at higher resolutions, the majority of LACTB1–68-Dronpa appears on the inside of the mitochondria (Fig. 9D, E, and F). The outside edge of the mitochondria is not labeled, indicating that neither the outer membrane nor the boundary IMS exhibits significant fluorescence. Without further reference structures, this internal labeling is consistent with a matrix or crista location.
Fig. 9.

Genetically expressed LACTB1–68-Dronpa localizes to the mitochondrial intermembrane space. (A to C) Confocal images of a single cell expressing LACTB1–68-Dronpa (A) and mitochondria stained with Mitotracker Red (B); (C) a merged image representing the two labels. (A) Bar, 10 μm. (D to E) Two-color PALM images expressing LACTB1–68-Dronpa (green) and the mitochondrial matrix protein encoded by CoxVIII1-29-mEos2 (red). (D) PALM image showing multiple mitochondria in a cell cross-section; bar, 2 μm. Five gold fiducial particles are circled in white. (E and F) High-resolution two-color PALM images of sectioned mitochondria; bars, 200 nm. The fluorescent molecule scale maximum values (probability per square nanometer) in panels E and F are about 0.0023 for Dronpa and 0.025 for mEos2. (G) Immunogold EM labeling of cryosections of cells with anti-Dronpa antibody, showing LACTB1–68-Dronpa localization to the mitochondrial inner membrane space. M, matrix; C, cristae; BM, boundary membrane. Arrows highlight several gold particles localized to cristae.
To further specify the location of LACTB1–68-Dronpa, we performed immunogold-labeling EM with an antibody against Dronpa (Fig. 9G). The results of immunogold labeling indicated that nearly the entire specific LACTB1–68-Dronpa signal localized to mitochondria (86% ± 3.4% standard error of the mean [SEM] [n = 2,129]). Cytoplasmic labeling, which presumably included LACTB1–68-Dronpa being translated at and/or trafficked into the cytoplasm, represented the remaining 14% of specific labeling. The cytoplasmic signal may also represent some form of labeling in mitochondria that is not clearly visible in the low-contrast cryosections. Because we wanted to use LACTB1–68-Dronpa protein as an IMS-crista marker, we analyzed its submitochondrial localization. The primary and secondary antibody-gold complex used for immunogold-labeling EM may span a distance of 20 nm; therefore, a gold particle that is located 20 nm away from a compartment may still represent detection of an antigen within that compartment. Since mitochondrial cristae have an average cross-sectional diameter of ∼30 nm, we assigned a gold particle to a compartment only when its center was in that compartment and used a point-hit method to determine specific labeling densities by adjusting for compartment surface area values (see Materials and Methods). Using nuclear labeling as background and subtracting the associated value to determine specific labeling, we found that 77% ± 3.6% (SEM) (n = 1,245) of gold particles labeled the mitochondrial IMS. The crista IMS was labeled more frequently than the boundary IMS (48% and 29% of total specific labeling).
Nucleoid TFAM-mEos2 and IMS-targeted LACTB1–68-Dronpa are closely associated.
To determine the location of nucleoids relative to the inner mitochondrial membrane, we took advantage of the IMS localization of LACTB1–68-Dronpa by the use of two-color PALM. Figure 10 shows a close association between the nucleoid TFAM-mEos2 and the LACTB1–68-Dronpa. Two types of relative positioning are seen. The first is displayed in Fig. 10A to C, where the nucleoid is immediately adjacent to the IMS, with various degrees of overlap at their margins. The second is displayed in Fig. 10D to F, in which the nucleoid appears to surround a region of IMS. This close proximity exceeds that most often seen between the matrix and the nucleoid (Fig. 8). Thus, it appears that nucleoids are often either adjacent to the boundary of crista IMS or partially or fully wrapped around a crista-like structure. As with the matrix (CoxVIII1-29-Dronpa) colocalization described above, some overlapping nucleoid and IMS signals cannot be completely resolved in the z dimension of the 150-nm-thick sections. In such cases, the nucleoid and the membrane may reside in different z planes. In the x and y dimensions, we have determined that the dual-label image alignment accuracy is high, with σ values ranging from 22 to 26 nm. The alignment was quantified using multiple gold fiducials within each field as described above.
Fig. 10.

TFAM-mEos2 nucleoid location relative to the IMS targeted LACTB1–68-Dronpa. Two-dimensional, dual-label PALM images of cells expressing inducible TFAM-mEos2 (red) and LACTB1–68-Dronpa (green). The fluorescent molecule scale maximum values (probability per square nanometer) differ per panel and range from 0.002 to 0.01 for Dronpa and from 0.011 to 0.019 for mEos2. TFAM-labeled nucleoids either were located adjacent to the cristae-IMS as shown in panels A to C or surrounded the cristae-IMS as shown in panels D to F). Bars, 100 nm.
DISCUSSION
Preliminary iPALM measurements of mitochondrial nucleoids revealed that the structures were larger than anticipated. Nucleoids were frequently truncated at the boundaries of 200-nm-thick sections. To facilitate data collection and achieve full-length measurements of nucleoids that are larger than 200 nm, we developed and employed a version of the iPALM with an expanded vertical z-coordinate range. The original iPALM method allows both high lateral x and y localization and high vertical z localization of a fluorescent particle over a vertical range of ∼250 nm. Beyond that range, the high precision can be maintained with the next interference fringe; however, one cannot discriminate between adjacent interference fringes. This results in a position ambiguity of n × zinterf.fringe, where n represents the integer number and zinterf.fringe (∼250 nm) represents the interferometric fringe.
The problem that the position ambiguity presents may be overcome by establishing a property that varies with the z coordinate at a low rate, which would allow discrimination of adjacent interferometric fringes. This could be done by analyzing image moments (3), but that approach does not have a high enough signal-to-noise ratio to work well with relatively dim fluorescent proteins. Here we replaced the original turning 22.5° mirrors in iPALM with mildly hyperbolic mirrors that added a saddle-shaped phase shift across the pupil plane. As a result, the PSF of images of point sources became elliptical, and, as proposed by Kao and Verkman (26), this ellipticity should vary with the axial coordinate. The hyperbolic shapes of the two turning mirrors were matched such that the phase differences between the two interfering beams remained constant across the pupil plane; thus, the multiphase interferometry critical to the original iPALM method was preserved. The ellipticity is dependent on the sample z coordinate and can be used to extract the vertical location of a molecule (Fig. 3). This method has much lower accuracy than the iPALM technique but is not limited to a single interferometric fringe. The procedure can be used to determine the fringe order, which, in turn, allows accurate determination of the z coordinate over a wider range.
Mitochondrial nucleoid dimensions were previously estimated by using anti-DNA immunogold EM to measure the diameters of clustered particles (24). Those data indicate that the nucleoids from a human carcinoma are roughly spherical, with a diameter that averages 65 to 70 nm and ranges from 31 to 132 nm. In contrast, our data indicate that most (65%) nucleoids appear ellipsoidal and have a much broader size range of 31 to 318 nm. The difference in these two measurements is notably at the upper end of the distributions. It is possible that there are real differences in the nucleoid sizes between human carcinoma cells and the mouse fibroblasts used in this study. However, the larger nucleoid sizes detected with iPALM are perhaps better explained by the 3-dimensional capacity of iPALM. Immunogold EM is a surface-limited technique and is sensitive only to those epitopes that are exposed on a thin section. It is less likely that longer nucleoids would be exposed on a thin sectional plane that reveals the entire nucleoid length. In contrast, iPALM is a 3-dimensional technique, revealing fluorescent protein locations within all dimensions of our 500-nm-thick samples. Therefore, we believe that the larger and more variously shaped nucleoids revealed by iPALM better represent their true dimensions. This has led us to reconsider both the relative external and internal organizations of nucleoids.
We used the dimensions of the examples in the ellipsoidal subset to estimate nucleoid volumes and found them to vary over a large range. Previous DNA fluorescence intensity data indicate that nucleoid sizes reflect their mtDNA content (6). Although we have not determined numbers of mtDNAs within individual nucleoids, the distribution of our volumetric data is consistent with this conclusion. The pattern of volume per nucleoid distribution shown in Fig. 5A is remarkably similar to typical distributions of mtDNAs per nucleoid described by others (11, 42, 54). This correlation is substantiated by the conversion of volumes to genomes. The average nucleoid volume in 3T3sw cells was determined to be 830,000 nm3 (Fig. 4A). By counting nucleoids and measuring mtDNA content per cell, we calculate that these cells have an average of 3 genomes per nucleoid (Table 1). If nucleoid volumes scale primarily with mtDNA content, then a single-genome nucleoid would have an approximate volume of 276,650 nm3. This conversion yields a range of 1 to 6 mtDNAs per nucleoid within the standard deviation shown in Fig. 5 and is congruent with multiple prior estimates of numbers of genomes per nucleoid (34). We hypothesize that nucleoid volumes might also be affected by changes in the packaging of mtDNA. However, in this analysis of steady-state populations, genome content appears to have a dominant role in ellipsoidal nucleoid volume variation.
These data also allowed us to determine the degree to which mtDNA is packaged. Surprisingly, we found that it is more condensed than both E. coli and mammalian nuclear DNA. Previous studies have led to the popular view that mtDNA is loosely packaged. EM imaging and psoralen cross-linking experiments indicated that only the noncoding control region of mtDNA is densely bound by protein (2, 13, 40, 46). More recently, the packaging of mtDNA has been reconsidered. TFAM clearly has mtDNA maintenance functions separate from transcription and exhibits the capacity to condense DNA in vitro (15, 27). However, its contribution to packaging is a subject of current controversy. One argument in this debate is that TFAM is abundant enough to saturate the mtDNA and effectively packages DNA at high protein/DNA ratios (1). The counterargument is that intracellular TFAM levels are, in fact, insufficient to cover mtDNA and that full binding is incompatible with transcription (12, 35). Considering the very high mtDNA ρpack value calculated from our data, the most straightforward conclusion is that, within the ellipsoidal nucleoids, mtDNA is maximally bound by TFAM. This may in fact be the case, and this condensed structure would exclude the possibility that the core nucleoid is sparsely bound at the steady state. However, our data also led to a caveat. The high packing density of mouse fibroblast mtDNA is similar to that found in the mitochondrial nucleoid of the slime mold P. polycephalum. This was unexpected, as there are major differences in the mtDNA packaging proteins used by these organisms. Mammalian TFAM condenses DNA via two HMG box domains. In addition to these domains, the P. polycephalum TFAM homolog (Glom) contains additional lysine-rich and proline-rich domains that are responsible for maintaining this highly compact yet functional mtDNA (53). Without these domains in TFAM, it is unclear how mammalian nucleoids would achieve the same mtDNA packing density. Furthermore, although yeast Abf2 and mammalian TFAM have the ability to condense DNA in vitro, mathematical modeling and volume estimates of numbers of fully bound DNA in vitro appear to be insufficient to account for this large ρpack value (17, 27). More recently, methylation protection analyses concurrent with altered TFAM expression indicate that mtDNA is unlikely to be fully bound by TFAM (50). If this is the case, then an alternative mechanism of mtDNA condensation is required to reconcile these data. It is possible that there are additional strategies for packaging mtDNA within the nucleoid. If other packaging mechanisms are present and their relative contributions are unknown, then full TFAM binding is not an obligatory feature. mtDNA condensation resulting from the presence of other proteins, macromolecular crowding, or DNA supercoiling has not been investigated.
The high mtDNA packing density finding is also supported by two-color PALM data obtained using matrix-targeted CoxVIII1-29-Dronpa (Fig. 8). A similar fluorescent fusion protein (CoxVIII-green fluorescent protein) has been shown to be freely diffusible within the mitochondrial matrix (43). If the nucleoid were loosely packed, we should see widespread colocalization of matrix Dronpa with TFAM-mEos2. However, Dronpa protein within the mitochondrial matrix is largely excluded from the nucleoid. Interestingly, this nucleoid exclusion was previously seen by Iborra and coworkers in a study using matrix-targeted yellow fluorescent protein. They also noted that areas surrounding the nucleoids were devoid of fluorescence (24). The latter observation was partially replicated here and may indicate an additional barrier between the nucleoid and the matrix. A compressed and isolated structure is also consistent with recent data indicating that nucleoids are genetically autonomous (20). The high packing density and limited diffusional access by matrix proteins are also directly relevant in defining how the nucleoid receives signals and alters its organization. These are physical impediments for an unmediated exchange with the matrix that have likely been underappreciated. Mechanisms for achieving the exchange in bacteria and in nuclei involve strategies for remodeling of DNA binding and packaging proteins. The regulation of mtDNA topology within the mitochondria is only beginning to be explored (50). It has recently been shown in studies using Drosophila cells that the mitochondrial Lon protease can regulate TFAM levels, which in turn alter the rate of transcription of mtDNA (35). Thus, it may be that local turnover of TFAM, other nucleoid proteins, or RNA is a general strategy for altering the local topology.
Nucleoid remodeling proteins and intercompartmental communication might also function through interactions with the inner membrane. Therefore, we have considered nucleoid position relative to cristae. There is a large amount of heterogeneity among nucleoids with respect to both size and shape. Among examples in the ellipsoidal subset of nucleoids, there is a 25-fold range in volumes just within the standard deviation. It is clear that nucleoid size is loosely restricted. One constraint affecting nucleoid size could be the physical space available within the mitochondria. The mitochondrial matrix space is defined by both the width of the space between the inner boundary membranes and the frequency and size of the cristae that partition that width. In two dimensions, 3T3sw mitochondria show an average width of 238 (± 75) nm between the inner boundary membranes. The mean dimensions of a nucleoid are 85 by 108 by 146 nm. Therefore, the mitochondrial matrix is wide enough to easily accommodate the majority of nucleoids if the cristae are excluded. However, the spaces between cristae may be more restrictive for the nucleoid. The average matrix space between cross-sectioned cristae is only 68 (± 24) nm. Although these are average dimensions, they encourage speculation that either the cristae membrane influences nucleoid structure or the inner membrane is arranged to accommodate the nucleoid.
We present two additional results that point toward a close relationship between nucleoids and the inner membrane. First, analyses of nucleoid shape yielded circumstantial evidence that the inner membrane might influence nucleoid contour. A total of 65% of nucleoids are ellipsoidal and have no obvious structural influence from the inner membrane. The remaining 35% have concave edges, splits, or branches that are consistent with membrane interactions. In direct support of this proposition, LACTB1–68-Dronpa localization indicates a very close relationship with nucleoids (Fig. 10). EM imaging has shown that mtDNA (24, 41) and TFAM (19) are located in the mitochondrial matrix. Therefore, the 5- to 10-nm-wide inner membrane lies between the nucleoid and the IMS, although this was not resolved in our PALM analysis. Essentially all of the nucleoids seen in two-color PALM were very close to or overlap the LACTB1–68-Dronpa signal. There are two common relative positions of the nucleoid and IMS marker (Fig. 10). The IMS (and therefore the inner membrane) either penetrates the nucleoid boundaries or is tangentially positioned. Those nucleoids with tangentially oriented IMS might be located next to either the boundary membrane or cristae. Those that are more fully engaged with the IMS are likely to be wrapped around an inner membrane cristae tubule. Our current imaging results do not always distinguish between the IMS of the boundary membrane and the IMS of cristae, and either location is plausible. However, prior localization of LACTB and our current immunolocalization of LACTB1–68-Dronpa reveal the novel prospect that there is an intimate association of the nucleoid with specialized cristae. Conventional microscopy has suggested that, in yeast, a subpopulation of nucleoids is associated with a protein complex that spans both the inner and outer mitochondrial membranes (37). As we cannot rule out boundary membrane interactions, it remains possible that mammalian nucleoids interact with analogous structures. The primary evidence for membrane association in mammals came from early EM imaging, which revealed that partially purified mtDNA contains short regions that are bound to detergent-soluble fragments (2, 38). The current PALM data bear more directly and fully on this interaction.
Although two-color PALM is able to detect fluorescent proteins residing within the cryosections, it is a two-dimensional method and lacks the capacity to assign molecular locations in the z dimension. We were therefore unable to confidently determine the exact frequency of membrane-associated nucleoids. However, we propose an initial conservative estimate that at least 35% of nucleoids are in contact with the inner membrane, as inferred from that fraction of nucleoids that are not ellipsoidal. However, an ellipsoidal shape does not necessarily preclude membrane association or even membrane penetration. It is worth noting that our nucleoid volume calculations do not account for space that may be occupied by cristae in some of the ellipsoidal nucleoids. Therefore, the volume occupied by the mtDNA in this space may be lower than shown in Table 1. Thus, the nucleoid density value reported here might also represent a low estimate.
The list of nucleoid-associated proteins that have been identified is long and increasing (8, 59). A future challenge is to spatially and temporally categorize members of this list. Some nucleoid-associated proteins are located in the inner membrane. Others are located in the matrix, the intermembrane space, and even the outer membrane. Many of these have primary functions that appear unrelated to mtDNA and largely reside outside the nucleoid. Others that have known functions within the nucleoid, such as mtDNA repair enzymes, are principally located within the membrane, independently of mtDNA (60). From a functional standpoint, it is important to make a distinction between core proteins and those that are transient residents.
The available data allow us to propose a model for the organization of mammalian nucleoids that has testable features and emphasizes current questions (Fig. 11). The core of the nucleoid is displayed in Fig. 11 in dark orange. Size appears to primarily reflect genome content, and shape may indicate “active” nucleoids. A very high mtDNA packing density commonly limits the protein composition within the core to those that have a direct role in mtDNA packaging. Diffusible matrix proteins may not freely infiltrate most nucleoids. Other nucleoid-associated proteins may gain fuller or more local access to mtDNA through regulated modifications of nucleoid structures. This remodeling could be mediated by proteins located in the matrix or at the inner membrane or both. The nucleoid-membrane interface remains poorly defined. However, the exchange of transiently associated nucleoid proteins could be mediated by cristae and inner membrane dynamics. The local availability of the inner membrane proteins and the intermembrane space lends additional routes for intercompartmental communication. Additional analysis of this membrane interaction awaits further development of two-color iPALM or other high-resolution microscopy approaches.
Fig. 11.
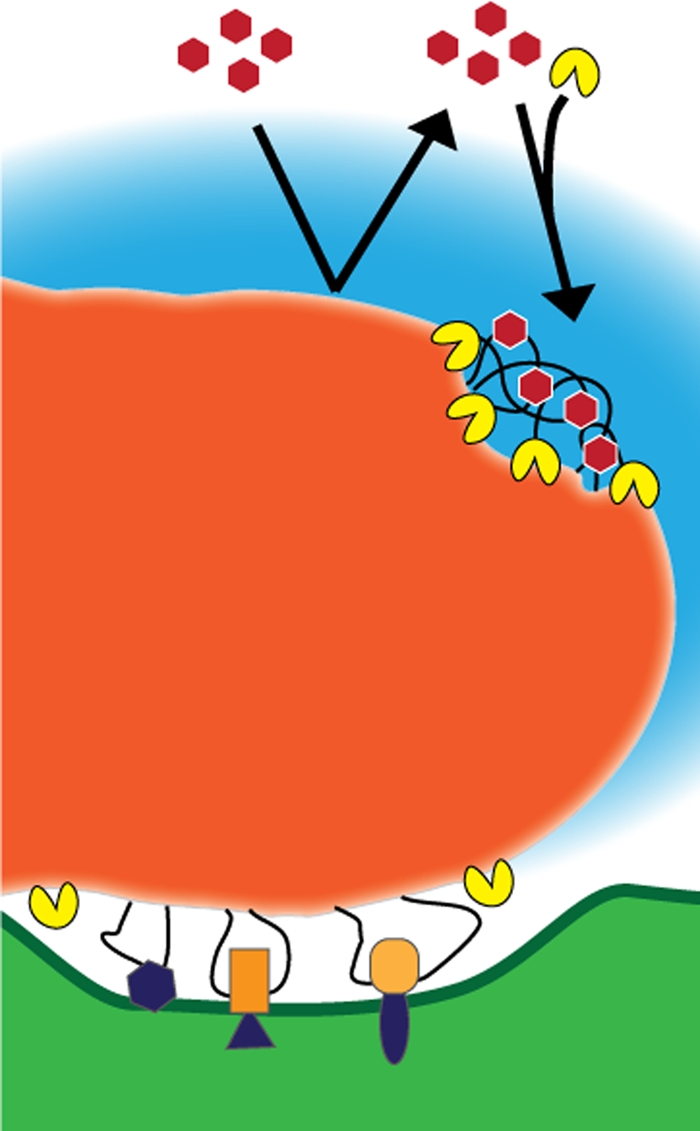
Model illustration of the mammalian mitochondrial nucleoid. The nucleoid core is shown in dark orange. An RNA cloud adjacent to the nucleoid is depicted in blue. Inner membrane connections are displayed here as being mediated by DNA loops that are associated with proteins that are either directly (purple) or indirectly (orange) bound to the membrane. Matrix proteins (red hexagons) are normally excluded from the nucleoid by the highly condensed mtDNA. Matrix and membrane nucleoid-associated proteins gain access to the mtDNA through the regulation of remodeling proteins (yellow).
ACKNOWLEDGMENTS
We thank Yalin Wang for contributions in optimizing and providing cryosections for PALM, Wei-Ping Li for providing TEM images of 3T3sw cells used for mitochondrial dimension analysis, and Rick Fetter for LR White resin sample processing. Eric Betzig, Hari Shroff, Jim Galbraith, and Cathy Galbraith provided invaluable instruction and advice with PALM.
This work was supported by the Howard Hughes Medical Institute (T.A.B., A.N.T., G.S., B.G.K., H.F.H., D.A.C.) and grant AGSS237009 from the Ellison Medical Foundation Foundation (D.F.B.).
We disclose that Harald Hess has licensed PALM technology to Carl Zeiss, Inc.
Footnotes
Published ahead of print on 17 October 2011.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Alam T. I., et al. 2003. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 31:1640–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albring M., Griffith J., Attardi G. 1977. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. U. S. A. 74:1348–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aquino D., et al. 2011. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat. Methods 8:353–359 [DOI] [PubMed] [Google Scholar]
- 4. Ashley N., Harris D., Poulton J. 2005. Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp. Cell Res. 303:432–446 [DOI] [PubMed] [Google Scholar]
- 5. Bates M., Huang B., Dempsey G. T., Zhuang X. 2007. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 317:1749–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bereiter-Hahn J., Vöth M. 1996. Distribution and dynamics of mitochondrial nucleoids in animal cells in culture. Exp. Biol. Online 1:1–17 [Google Scholar]
- 7. Betzig E., et al. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–1645 [DOI] [PubMed] [Google Scholar]
- 8. Bogenhagen D. F., Rousseau D., Burke S. 2008. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 283:3665–3675 [DOI] [PubMed] [Google Scholar]
- 9. Brown T. A., Clayton D. A. 2002. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′-dideoxycytidine. Nucleic Acids Res. 30:2004–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown T. A., Fetter R. D., Tkachuk A. N., Clayton D. A. 2010. Approaches toward super-resolution fluorescence imaging of mitochondrial proteins using PALM. Methods 51:458–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavelier L., Johannisson A., Gyllensten U. 2000. Analysis of mtDNA copy number and composition of single mitochondrial particles using flow cytometry and PCR. Exp. Cell Res. 259:79–85 [DOI] [PubMed] [Google Scholar]
- 12. Cotney J., Wang Z., Shadel G. S. 2007. Relative abundance of the human mitochondrial transcription system and distinct roles for h-mtTFB1 and h-mtTFB2 in mitochondrial biogenesis and gene expression. Nucleic Acids Res. 35:4042–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeFrancesco L., Attardi G. 1981. In situ photochemical crosslinking of Hela cell mitochondrial DNA by a psoralen derivative reveals a protected region near the origin of replication. Nucleic Acids Res. 9:6017–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dulkeith E., et al. 2004. Plasmon emission in photoexcited gold nanoparticles. Phys. Rev. B. 70:205424 [Google Scholar]
- 15. Ekstrand M. I., et al. 2004. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genetics 13:935–944 [DOI] [PubMed] [Google Scholar]
- 16. Frey T. G., Mannella C. A. 2000. The internal structure of mitochondria. Trends Biochem. Sci. 25:319–324 [DOI] [PubMed] [Google Scholar]
- 17. Friddle R. W., et al. 2004. Mechanism of DNA compaction by yeast mitochondrial protein Abf2p. Biophys. J. 86:1632–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frost N. A., Shroff H., Kong H. H., Betzig E., Blanpied T. A. 2010. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron 67:86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garrido N., et al. 2003. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 14:1583–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gilkerson R. W., Schon E. A., Hernandez E., Davidson M. M. 2008. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 181:1117–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heilemann M., van de Linde S., Mukherjee A., Sauer M. 2009. Super-resolution imaging with small organic fluorophores. Angew. Chem. Int. Ed. Engl. 48:6903–6908 [DOI] [PubMed] [Google Scholar]
- 22. Hess S. T., Girirajan T. P. K., Mason M. D. 2006. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91:4258–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang B., Wang W. Q., Bates M., Zhuang X. W. 2008. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319:810–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iborra F. J., Kimura H., Cook P. R. 2004. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Juette M. F., et al. 2008. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5:527–529 [DOI] [PubMed] [Google Scholar]
- 26. Kao H. P., Verkman A. S. 1994. Tracking of single fluorescent particles in three dimensions: use of cylindrical optics to encode particle position. Biophys. J. 67:1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaufman B. A., et al. 2007. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell 18:3225–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klar T. A., Dyba M., Hell S. W. 2001. Stimulated emission depletion microscopy with an offset depleting beam. App. Phys. Lett. 78:393–395 [Google Scholar]
- 29. Knott A. B., Perkins G., Schwarzenbacher R., Bossy-Wetzel E. 2008. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9:505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kucej M., Butow R. A. 2007. Evolutionary tinkering with mitochondrial nucleoids. Trends Cell Biol. 17:586–592 [DOI] [PubMed] [Google Scholar]
- 31. Kucej M., Kucejova B., Subramanian R., Chen X. J., Butow R. A. 2008. Mitochondrial nucleoids undergo remodeling in response to metabolic cues. J. Cell Sci. 121:1861–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larsson N. G., Oldfors A., Holme E., Clayton D. A. 1994. Low levels of mitochondrial transcription factor A in mitochondrial DNA depletion. Biochem. Biophys. Res. Commun. 200:1374–1381 [DOI] [PubMed] [Google Scholar]
- 33. Legros F., Malka F., Frachon P., Lombès A., Rojo M. 2004. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 117:2653–2662 [DOI] [PubMed] [Google Scholar]
- 34. Malka F., Lombès A., Rojo M. 2006. Organization, dynamics and transmission of mitochondrial DNA: focus on vertebrate nucleoids. Biochim. Biophys. Acta 1763:463–472 [DOI] [PubMed] [Google Scholar]
- 35. Matsushima Y., Goto Y., Kaguni L. S. 2010. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc. Natl. Acad. Sci. U. S. A. 107:18410–18415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKinney S. A., Murphy C. S., Hazelwood K. L., Davidson M. W., Looger L. L. 2009. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 6:131–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meeusen S., Nunnari J. 2003. Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J. Cell Biol. 163:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nass M. M. K. 1969. Mitochondrial DNA. I. Intramitochondrial distribution and structural relations of single- and double-length circular DNA. J. Mol. Biol. 42:521–528 [DOI] [PubMed] [Google Scholar]
- 39. Nass M. M. K., Nass S. 1963. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J. Cell Biol. 19:593–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nass M. M. K. 1966. Circularity of mitochondrial DNA. Proc. Natl. Acad. Sci. U. S. A. 56:1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nass S., Nass M. M. K. 1963. Intramitochondrial fibers with DNA characteristics. II. Enzymatic and other hydrolytic treatments. J. Cell Biol. 19:613–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Navratil M., Poe B. G., Arriaga E. A. 2007. Quantitation of DNA copy number in individual mitochondrial particles by capillary electrophoresis. Anal. Chem. 79:7691–7699 [DOI] [PubMed] [Google Scholar]
- 43. Partikian A., Olveczky B., Swaminathan R., Li Y. X., Verkman A. S. 1998. Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J. Cell Biol. 140:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pertsinidis A., Zhang Y., Chu S. 2010. Subnanometre single-molecule localization, registration and distance measurements. Nature 466:647–651 [DOI] [PubMed] [Google Scholar]
- 45. Polianskyte Z., et al. 2009. LACTB is a filament-forming protein localized in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 106:18960–18965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Potter D. A., Fostel J. M., Berninger M., Pardue M. L., Cech T. R. 1980. DNA-protein interactions in the Drosophila melanogaster mitochondrial genome as deduced from trimethylpsoralen crosslinking patterns. Proc. Natl. Acad. Sci. U. S. A. 77:4118–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prachar J. 2010. Mouse and human mitochondrial nucleoid—detailed structure in relation to function. Gen. Physiol. Biophys. 29:160–174 [PubMed] [Google Scholar]
- 48. Purohit P. K., et al. 2005. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 88:851–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rabouille C. 1999. Quantitative aspects of immunogold labeling in embedded and nonembedded sections. Meth. Mol. Biol. 117:125–144 [DOI] [PubMed] [Google Scholar]
- 50. Rebelo A. P., Williams S. L., Moraes C. T. 2009. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37:6701–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rizzuto R., Simpson A. W., Brini M., Pozzan T. 1992. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358:325–327 [DOI] [PubMed] [Google Scholar]
- 52. Rust M. J., Bates M., Zhuang X. W. 2006. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3:793–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sasaki N., et al. 2003. Glom is a novel mitochondrial DNA packaging protein in Physarum polycephalum and causes intense chromatin condensation without suppressing DNA functions. Mol. Biol. Cell 14:4758–4769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Satoh M., Kuroiwa T. 1991. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 196:137–140 [DOI] [PubMed] [Google Scholar]
- 55. Schermelleh L., et al. 2008. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 320:1332–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schermelleh L., Heintzmann R., Leonhardt H. 2010. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 190:165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shroff H., et al. 2007. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. U. S. A. 104:20308–20313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shtengel G., et al. 2009. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl. Acad. Sci. U. S. A. 106:3125–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spelbrink J. N. 2010. Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life 62:19–32 [DOI] [PubMed] [Google Scholar]
- 60. Stuart J. A., Mayard S., Hashiguchi K., Souza-Pinto N. C., Bohr V. A. 2005. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 33:3722–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vaziri A., Tang J., Shroff H., Shank C. V. 2008. Multilayer three-dimensional super resolution imaging of thick biological samples. Proc. Natl. Acad. Sci. U. S. A. 105:20221–20226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu X., et al. 2010. High-photoluminescence-yield gold nanocubes: for cell imaging and photothermal therapy. ACS Nano. 4:113–120 [DOI] [PubMed] [Google Scholar]
- 63. Zeviani M., Di Donato S. 2004. Mitochondrial disorders. Brain 127:2153–2172 [DOI] [PubMed] [Google Scholar]