

Abstract
Diverse malfunctions in the expression and regulation of matrix metalloproteinases (MMPs) are often the cause of severe human diseases, bringing the identification of specific MMP inhibitors into major focus, particularly in anticancer treatment. Here, we describe a novel bioassay based on recombinant yeast cells (Pichia pastoris) that express, deliver, and incorporate biologically active human MMP-2 and MMP-9 at the yeast cell surface. Using Sed1p for cell wall targeting and covalent anchorage, a highly efficient bioassay was established that allows high-throughput screening and subsequent validation of novel MMP inhibitors as potential anticancer drugs. In addition, we developed a straightforward synthesis of a new aspartate-derived MMP inhibitor active in the nM range and bearing an amino functionality that should allow the introduction of a wide range of side chains to modify the properties of these compounds.
INTRODUCTION
Diverse malfunctions in the regulation of human matrix metalloproteinases (MMPs), especially MMP-2 and MMP-9, play a key role in the development of a wide range of diseases, including diabetes mellitus, arthritis, cardiovascular diseases, and, most of all, cancer (5, 16). Recent oncology research is therefore focusing on the specific inhibition of distinct MMPs as potential therapeutic targets (3). Most of these inhibitors are peptidic succinates with a hydroxamate functionality, binding to the zinc ion at the active site of the proteolytic enzyme (1, 7, 10). For example, hydroxamate 1 (Fig. 1, structure 1) shows significant selectivity toward MMP-2, -8, and -9 relative to other MMPs (17), which can be explained by the interaction of the nonpolar phenylpropyl side chain with the deep, tunnel-like binding pockets of these enzymes (20). The introduction of polar substituents (e.g., R = OH) onto the α-position of the hydroxamate group in general results in higher solubility and oral bioavailability (23, 27).
Fig. 1.
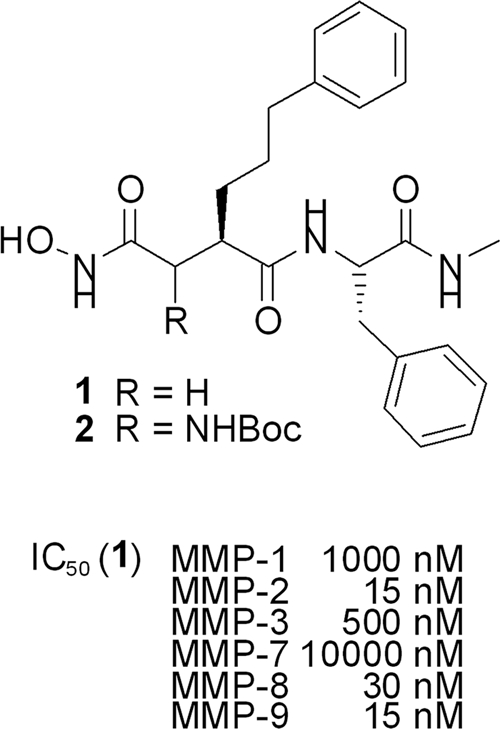
Hydroxamate based-selective MMP inhibitors. IC50, 50% inhibitory concentration.
Nevertheless, early generations of MMP inhibitors (MMPIs) did not meet the high expectations for them in clinical trials, as poor inhibitor specificities caused massive side effects due to the inhibition of non-MMP targets (5, 14, 16, 24). Thus, identification of more specific inhibitors is currently a major focus in MMPI development. Until now, the design and validation of novel inhibitors have often been hampered by costly and time-consuming MMP purification from human tumor cell lines or primary fibroblasts (9), as well as by the lack of a suitable high-throughput bioassay for comprehensive MMP inhibitor screening. To bypass such a limitation, the major objective of the present study was to immobilize biologically active human MMPs on the surface of yeast (Pichia pastoris) and to establish a cell-based bioassay for MMP inhibitor screening. In addition, we developed a straightforward synthesis of a potential inhibitor of these MMPs based on structure 1 (Fig. 1), bearing an amino functionality at the α position, which should allow the introduction of a wide range of side chains to modify the properties of these compounds.
MATERIALS AND METHODS
Cell surface expression of human MMP-2 and MMP-9 on P. pastoris.
Expression vectors were established using an Invitrogen Pichia pastoris Expression Kit Version F. The SED1 cell wall anchor sequence was amplified by PCR using Saccharomyces cerevisiae genomic DNA as a template and subsequently cloned into the pPIC9 expression vector using AvrII and NotI restriction enzymes, yielding pPIC9-SED1. The gene sequences of human MMP-2 and MMP-9, respectively, were amplified by PCR from human cDNA lacking their native endoplasmic reticulum (ER) import signal sequence. These proMMP genes were cloned into pPIC9-SED1 via EcoRI and AvrII/XbaI restriction sites, resulting in the MMP expression plasmids pPIC9-MMP-2-SED1 and pPIC9-MMP-9-SED1. All amplified genes had been sequenced prior to further use (for the primers, see Table S1 in the supplemental material). The resulting gene fusions contained the yeast ER signal sequence of the S. cerevisiae pheromone α mating factor (α-MF) at the 5′ end, followed by a signal peptidase cleavage site, the respective proMMP, and the SED1 gene at the 3′ end. The expression plasmids were linearized by 4 h of digestion with StuI and subsequently used for transformation of P. pastoris KM71 by electroporation. Homologous recombination into the his4 locus of the Pichia genome yielded histidine prototrophic yeast clones that were selected on agar plates lacking histidine. All yeast strains were cultivated in shaking flasks at 220 rpm and 30°C. Preprecultures were grown overnight in synthetic medium lacking histidine and precultures in “buffered medium glycerol” (BMG) (100 mM potassium phosphate buffer, pH 6.0, 1% [wt/vol] NH4SO4, 3.4% [wt/vol] yeast nitrogen base [YNB], 0.00004% [wt/vol] biotin, 2% [vol/vol] glycerol) until the cultures reached an optical density at 600 nm (OD600) of at least 30; inducing conditions were achieved by shifting into “buffered medium methanol” (BMM) (100 mM potassium phosphate buffer, pH 6.0, 1% [wt/vol] NH4SO4, 3.4% [wt/vol] YNB, 0.00004% [wt/vol] biotin, 1% [vol/vol] methanol) at a feeding rate of 1% (vol/vol) methanol twice a day. After 96 h of methanol induction, cells were harvested by centrifugation, washed with sterile H2O, and lyophilized.
Cell-based MMP gelatinase assay.
The gelatinase assay was based on the Invitrogen EnzCheck Gelatinase/Collagenase Assay, which was adapted to match the requirements of a cell-based test system. To perform the assay, lyophilized cells were reconstituted in assay reaction buffer (50 mM Tris, 150 mM NaCl, 5 mM CaCl2) at a cell density (OD600) of 5 (solution A). Inhibitors were likewise diluted in assay reaction buffer at 4-fold the concentration that was supposed to be tested (solution B). Solution C contained 100 μg ml−1 of fluorescein-conjugated DQ gelatin substrate (as provided by the manufacturer) in assay reaction buffer. Fluorescence detection was carried out in black 96-well microtiter plates (Nunc) with a composition of 100 μl solution A, 50 μl solution B, and 50 μl solution C in each well (yielding final concentrations [OD600 = 2.5] of 25 μg ml−1 DQ gelatin substrate and 1-fold inhibitor). Data were recorded at 37°C for 20 h at 20-min intervals with 19.5 min of shaking in between (700 rpm; 1-mm diameter) by a Labsystems Fluoroskan Ascent CF microtiter plate reader (excitation wavelength [λA] = 485 nm; emission wavelength [λE] = 527 nm). Subsequently, the slopes of the resulting graphs (see Fig. S2 in the supplemental material) were determined and depicted as a function of the inhibitor concentration by using the data from serial inhibitor dilutions. Mathematical regression of the data resulted in functions that allowed calculation of the inhibitor concentration that reduced enzyme activity by 50% (Km) as a measure of inhibitor function.
Inhibitor synthesis.
A key step in this synthesis (see Fig. 4) was a chelate-enolate Claisen rearrangement (11, 13, 15) of the chiral allylic ester (see Fig. 4, structure 3), which proceeded with an excellent yield of 93%, chirality transfer, and diastereoselectivity, giving rise to the unsaturated amino acid, which was directly converted into the corresponding benzyl ester (see Fig. 4, structure 4). Ozonolysis of the double bond and subsequent oxidation of the aldehyde obtained provided substituted aspartate (see Fig. 4, structure 5) (12), which was coupled with (S)-phenylalanine methyl-amide leading to structure 6 (see Fig. 4). The synthesis was finalized by hydrogenolytic removal of the benzyl ester, coupling with O-benzylated hydroxyl-amine, and cleavage of the benzyl-protecting group.
Fig. 4.

Synthesis and evaluation of MMP inhibitor 2. The reagents and conditions were as follows. (a) 1.2 eq ZnCl2, 2.5 eq lithium diisopropylamide (LDA), tetrahydrofurane (THF), −78°C to room temperature (RT), 20 h. (b) 1.1 eq Cs2CO3, 3.5 eq benzyl bromide (BnBr), dimethylformamide (DMF), 0°C to RT, 20 h. (c) 1, O3, CH2Cl2, −78°C, 5 min; 2, PPh3, −78°C, 30 min. (d) 10 eq NaClO2, 7 eq NaH2PO4 · 2H2O, 20 eq 2-methyl-2-butene, t-butanol/H2O, RT, 16 h. (e) 1.0 eq 2-(1H-benzotriazole-1-yl-1,1,3,3-tetramethylaminum tetrafluoroborate (TBTU), 1.0 eq NEt3, 1.2 eq (S)-PhNHMe, CH2Cl2, 0°C to RT, 18 h. (f) Pd/C (10%), H2, THF (90%). (g) 1.0 eq ClCOOiBu, 1.0 eq NEt3, 2.0 eq O-benzylhydroxylamine, THF, −20°C to RT, 18 h (90%). (h) Pd/C (10%), H2, DMF (83%). ee, enantiomeric excess; ds, diastereoselectivity.
RESULTS AND DISCUSSION
To analyze the inhibitory potential of the newly synthesized MMPIs, we developed a cell-based bioassay containing biologically active human MMPs expressed at high density on the yeast cell surface. In this respect, recombinant MMP production is usually unsuccessful in a prokaryotic host, since MMPs, as zinc-dependent endopeptidases, are naturally secreted as immature zymogens containing an N-terminal prodomain that keeps the enzyme inactive during passage through the eukaryotic secretory pathway (21, 26). Subsequent activation requires processing of a so-called cysteine switch, which is responsible for complexing the catalytic zinc ion (6, 18, 22). In an initial attempt to validate eukaryotic yeasts as hosts for the expression of human MMPs, we screened various biotechnologically relevant species, such as Kluyveromyces lactis, P. pastoris, S. cerevisiae, Schizosaccharomyces pombe, and Zygosaccharomyces bailii, for efficient processing and secretion of human proMMP-2 and proMMP-9. In each case, a species-specific N-terminal signal peptide was used to ensure proMMP import into the yeast secretory pathway (2, 4). Successful secretion of human MMPs into the culture supernatant was verified by Western analysis and zymography, indicating that P. pastoris was the only yeast capable of producing and secreting human MMPs in a biologically active form and at a constant and sufficiently high quality (unpublished results; see Fig. S1 in the supplemental material). Subsequent MMP immobilization on the P. pastoris cell surface was achieved by using the C-terminal glycosylphosphatidylinositol (GPI) anchor sequence of the S. cerevisiae SED1 gene (25), resulting in an in-frame protein fusion containing Sed1p at the C terminus of either proMMP-2 or proMMP-9.
MMP bioassay.
A schematic outline of the experimental setup for MMP inhibitor screening through yeast cell surface display of biologically active MMP-2 and MMP-9 is illustrated in Fig. 2a to d. MMP expression was driven from the methanol-inducible alcohol oxidase promoter AOX1, and proMMP import into the lumen of the endoplasmic reticulum was ensured by an N-terminal signal peptide derived from the S. cerevisiae α-MF (Fig. 2a). In each case, the resulting expression cassette was inserted into the his4 locus of the P. pastoris genome, yielding recombinant strains in which MMP-2 or MMP-9 was targeted to the yeast cell surface (Fig. 2b). Immunofluorescence microscopy of the resulting yeast transformants confirmed successful cell surface localization of both MMPs, which were fully accessible to the corresponding anti-MMP antibody, resulting in a bright fluorescent signal at the cell periphery; as expected, no cell surface fluorescence was detectable in negative-control cells (Fig. 3).
Fig. 2.

Experimental setup and schematic outline for MMP inhibitor screening through cell surface display of biologically active MMP-2 and MMP-9 in P. pastoris. (a) Expression cassette containing an N-terminal signal sequence (α-MF) to ensure proMMP import into the secretory pathway and signal peptidase cleavage, releasing proMMP into the ER lumen. The C-terminal anchoring domain of Sed1p mediates covalent MMP immobilization on the yeast cell wall via a GPI anchor-mediated process. (b) Recombinant proteins are delivered to the P. pastoris cell surface through the secretory pathway and are subsequently activated by autocatalytic removal of the MMP prodomain. (c) Activated MMPs are supplied with the desired inhibitor (red triangles) and fluorescein-labeled gelatin as a substrate (spirals). Subsequent cleavage of the substrate liberates fluorescing fragments that are detectable in a microplate fluorescence reader. (d) Model kinetics of remaining MMP activity at a given MMP inhibitor concentration (slopes in different colors) and subsequent calculation of a dose-effect diagram allowing quantitative comparison of different MMPIs. RFU, relative fluorescence units.
Fig. 3.
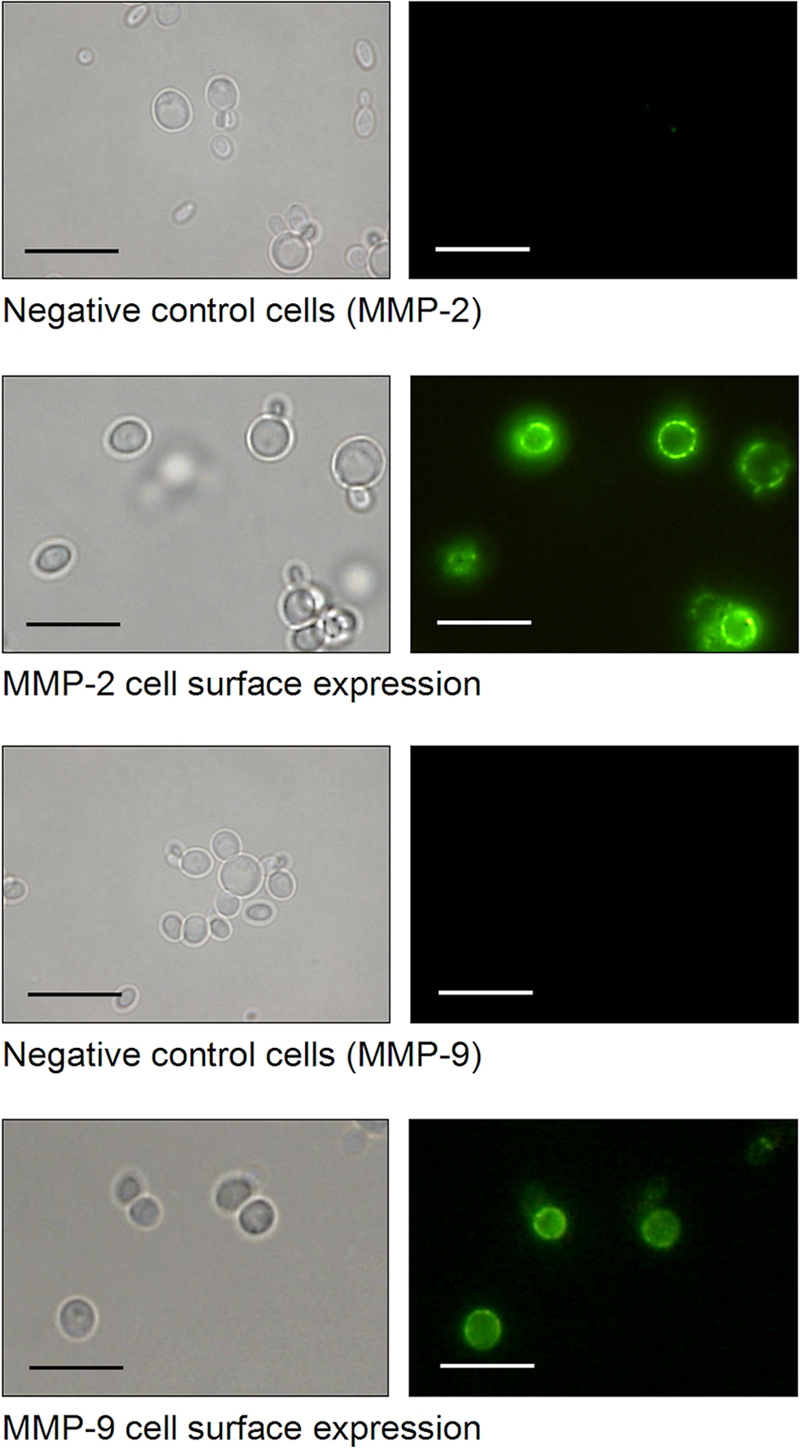
P. pastoris cell surface display of human MMP-2 and MMP-9 verified by indirect immunofluorescence microscopy. Cells of P. pastoris KM71 expressing the indicated MMP were cultivated for 72 h under inducing conditions in the presence of methanol. The MMPs were labeled with anti-MMP antibodies and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG and analyzed by fluorescence microscopy (Keyence BZ-8000; λA = 480 nm; λE > 510 nm). Scale bars = 10 μm.
MMP activity was quantified using fluorescein-labeled gelatin as a substrate. Intense fluorescein labeling of the MMP substrate leads to mutual quenching, which prevents fluorescence of intact gelatin. After cleavage by gelatinolytic MMPs, quenching is reversed, and liberated fluorescent fragments can be detected and semiquantified photometrically (Fig. 2c). Within each reaction well, MMP activity is directly proportional to the increase in fluorescence intensity over time within the linear part of the resulting graph (Fig. 2d). Surprisingly, cell surface display of latent proMMP genes led to fully active enzymes without the requirement for any preactivation step. An additional increase in MMP activity by 80% (MMP-2) and 150% (MMP-9) could be achieved by incubating the cells in TTC buffer (50 mM Tris-HCl, pH 7.5, 0.05% [vol/vol] Triton X-100, 5 mM CaCl2) or by lyophilization. TTC buffer contains Triton X-100, which (at higher concentrations) is conventionally used for in vitro MMP activation in zymography-based assays (8). In our case, the applied concentrations seemed to promote subsequent MMP activation. In contrast, treatment with the commonly used in vitro MMP-activating compound APMA (4-aminophenylmercuric acid) resulted in a total loss of MMP activity, although as shown by immunofluorescence microscopy, the enzymes had not been released from the surface (data not shown). APMA is known to cause conformational destabilization and subsequent autocatalytic cleavage of the MMP proregion (19), which in the case of cell surface-expressed MMPs might also cause MMP inactivation, perhaps due to the prevention of proper refolding of cell wall-anchored MMPs. Interestingly, expression of N-terminally truncated MMP variants resulted in a significant loss of MMP activity, indicating that the MMP proregion functions as an intramolecular chaperone required for proper MMP folding.
Assay optimization.
Adaptation of the in vitro assay to the requirements of a cell-based bioassay demanded several optimizations and modulations, such as defining the optimum cell density, substrate concentration, and culture growth conditions. Increasing the sample cell density resulted in a linear increase in MMP gelatinolytic activity up to an OD600 of 2.5. Higher cell densities negatively affected the light permeability of the suspension and resulted in a significant decrease in MMP activity. In an attempt to further optimize the cost efficiency of the bioassay, the MMP substrate could be reduced to a minimal concentration of 25 μg ml−1, thereby ensuring an adequate balance between applicability and cost reduction (see Fig. S2 in the supplemental material).
Supplementing yeast cultures with increasing concentrations of methanol likewise influenced MMP activity severely: cell feedings with 1% (vol/vol) methanol twice a day enhanced MMP-2 and MMP-9 activity by 82% and 14%, respectively, compared to 0.5% (vol/vol) methanol, while higher methanol concentrations (>2% [vol/vol]) exceeded the metabolic load and capacity of the cells, leading to a significant decrease in both cell viability and MMP activity. Methanol induction times longer than 80 to 90 h did not result in a further increase in MMP activity, as the MMP incorporation capacity of the yeast cell wall became limited. Scaling up induction times from 48 h to 96 h resulted in an increment of 130% for MMP-2 and 70% for MMP-9 cells; further elongation resulted in only a minor increase in MMP activity (see Fig. S2 in the supplemental material). To determine the total number of MMP molecules on a single cell, we compared yeast MMP activities with the activities of commercial MMPs extracted from human cell lines. Calculation by linear regression yielded >5 mgl−1 immobilized MMPs for yeast cultures at a cell density (OD600) of 100, demonstrating high-density MMP expression corresponding to about 5 × 104 molecules of MMP-2 and/or MMP-9 per cell (see Fig. S3 in the supplemental material).
To finally evaluate the power of the yeast-based bioassay for MMP inhibitor screening, we tested serial dilutions of MMPIs using the optimized parameters identified previously. We determined the slope of the linear graph sections and created a dose-effect diagram (Fig. 2d). Exponential regression permitted a calculation of the dose-effect relation for each inhibitor-MMP combination. Based on the enzyme activity in the absence of any inhibitor (Vmax), we determined the inhibitor concentration that reduced enzyme activity by 50% (Km). This yielded an objective and comparable value for classifying the capacity of the inhibitor (Fig. 2d).
Initial screenings were carried out with our new hydroxamate (Fig. 4, structure 2), with a hydrophobic side chain addressing the deep S1′ pockets of MMP-2 and MMP-9. Indeed, both MMPs were inhibited in the nM range (MMP-2, 390 nM; MMP-9, 150 nM), and the ease of the test underlines the potential of this innovative screening system (see Fig. S4 in the supplemental material). Although the activity of this new inhibitor is 10-fold lower than that of the known inhibitor (Fig. 4, structure 1), it is an excellent lead structure for further modifications to the amino functionality, an option that is not available with most of the common structures. By these modifications, critical issues, such as solubility or oral bioavailability, can be addressed easily.
Conclusions.
Heterologous expression of human MMPs in yeast is accompanied by significant advantages with respect to safety, handling, and cost efficiency. Imai and Okada recently described effective but time-consuming chromatographic procedures to separate MMPs from mammalian cell cultures (9). MMP-expressing yeast cells contain only the respective recombinant gelatinase in an immobilized form on the cell surface, thus enabling efficient MMP purification in a single centrifugation step, thereby reducing the time scale from weeks to minutes. Furthermore, activation of proMMP genes by simple lyophilization and/or incubation in an uncritical buffer bypasses the usual need to use a toxic organomercuric compound (such as APMA) for activation. In addition, MMP production in easy-to-work-with yeast cells provides constant availability at low cost: a 1-liter yeast culture provides sufficient cells to perform approximately 2,000 assays, thereby reducing total costs by 85% compared to conventionally obtained MMPs. Finally, MMP-expressing yeast cells can be provided and transported in a freeze-dried form.
In conclusion, we established a safe, rapid, standardized, and highly efficient yeast-based test system allowing high-throughput screening of prospective inhibitors of cancer-promoting human matrix metalloproteinases. This bioassay should hasten the progression of inhibitor development, as it enables quantitative comparison of MMP inhibitor capacity, which might contribute to novel approaches in cancer therapy in the near future. Although the metalloproteinases MMP-2 and MMP-9 featured above play a key role in tumor formation, malfunctions in the expression of almost every other human MMP were found to be important promoters of tumor invasion and metastasis. Besides gelatinases, MMP-1, MMP-3, MMP-7, and MMP-13 represent the metalloproteinases that are most frequently involved in cancer development (3, 18). For each of these MMPs, several fluorogenic substrates are available that could be easily adapted to the yeast bioassay described in this paper, thus enabling comprehensive screening for inhibitors of tumor-related MMPs. On the other hand, cell surface display of a range of active MMPs would provide a basis for estimating and improving inhibitor selectivity and minimizing cross-reactivity with non-MMP targets, which until now has hampered the application of MMP inhibitors as efficient anticancer drugs. Furthermore, the cell-based bioassay developed here might also be useful in a modified and further adapted form to screen for novel MMP substrates; we intend to address this aspect in the near future.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Forschungskommission of Saarland University and is part of patent application number DPA 10 2008 054494.9.
We thank E. Howard (Cell Biology, University of Oklahoma) for kindly providing cDNAs of human MMP-2 and -9.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 14 October 2011.
REFERENCES
- 1. Babine R. E., Bender S. L. 1997. Molecular recognition of protein-ligand complexes: applications to drug design. Chem. Rev. 97:1359–1472 [DOI] [PubMed] [Google Scholar]
- 2. Breinig F., et al. 2006. Cell surface expression of bacterial esterase A by Saccharomyces cerevisiae and its enhancement by constitutive activation of the cellular unfolded protein response. Appl. Environ. Microbiol. 72:7140–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corbitt C. A., Lin J., Lindsey M. L. 2007. Mechanisms to inhibit matrix metalloproteinase activity: where are we in the development of clinically relevant inhibitors? Recent Pat. Anticancer Drug Discov. 2:135–142 [DOI] [PubMed] [Google Scholar]
- 4. Eiden-Plach A., et al. 2004. Viral preprotoxin signal sequence allows efficient secretion of green fluorescent protein by Candida glabrata, Pichia pastoris, Saccharomyces cerevisiae, and Schizosaccharomyces pombe. Appl. Environ. Microbiol. 70:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fingleton B. 2008. MMPs as therapeutic targets—still a viable option? Semin. Cell Dev. Biol. 19:61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gill S. E., Parks W. C. 2008. Metalloproteinases and their inhibitors: regulators of wound healing. Int. J. Biochem. Cell Biol. 40:1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gupta S. P. 2007. Quantitative structure-activity relationship studies on zinc-containing metalloproteinase inhibitors. Chem. Rev. 107:3042–3087 [DOI] [PubMed] [Google Scholar]
- 8. Heussen C., Dowdle E. B. 1980. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal. Biochem. 102:196–202 [DOI] [PubMed] [Google Scholar]
- 9. Imai K., Okada Y. 2008. Purification of matrix metalloproteinases by column chromatography. Nat. Protoc. 3:1111–1124 [DOI] [PubMed] [Google Scholar]
- 10. Johnson W. H., Roberts N. A., Borkakoti N. 1987. Collagenase inhibitors: their design and potential therapeutic use. J. Enzyme Inhib. 2:1–22 [DOI] [PubMed] [Google Scholar]
- 11. Kazmaier U., Schneider C. 1998. Application of the asymmetric chelate-enolate claisen rearrangement to the synthesis of 5-epi-isofagomine. Tetrahedron Lett. 39:817–818 [Google Scholar]
- 12. Kazmaier U., Schneider C. 1998. Application of the asymmetric chelate enolate claisen rearrangement to the synthesis of unsaturated polyhydroxylated amino acids. Synthesis 1998(9):1321–1326 doi:10.1055/s-1998-6104 [Google Scholar]
- 13. Kazmaier U., Schneider C. 1996. Stereoselective synthesis of unsaturated polyhydroxylated amino acids via ester enolate claisen rearrangement. Synlett 1996(10):975–977 doi:10.1055/s-1996-5637 [Google Scholar]
- 14. Kessenbrock K., Plaks V., Werb Z. 2010. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141:52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krebs A., Kazmaier U. 1996. The asymmetric ester enolate claisen rearrangement as a suitable method for the synthesis of sterically highly demanding amino acids. Tetrahedron Lett. 37:7945–7946 [Google Scholar]
- 16. Lee M., Fridman R., Mobashery S. 2004. Extracellular proteases as targets for treatment of cancer metastases. Chem. Soc. Rev. 33:401–409 [DOI] [PubMed] [Google Scholar]
- 17. Miller A., et al. 1997. Inhibition of matrix metalloproteinases: an examination of the S1′ pocket. Bioorg. Med. Chem. Lett. 7:193–197 [Google Scholar]
- 18. Mott J. D., Werb Z. 2004. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 16:558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Okada Y., et al. 1990. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur. J. Biochem. 194:721–730 [DOI] [PubMed] [Google Scholar]
- 20. Porter J. R., et al. 1994. Potent and selective inhibitors of gelatinase-A 1. Hydroxamic acid derivatives. Bioorg. Med. Chem. Lett. 4:2741–2746 [Google Scholar]
- 21. Somerville R. P., Oblander S. A., Apte S. S. 2003. Matrix metalloproteinases: old dogs with new tricks. Genome Biol. 4:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sternlicht M. D., Werb Z. 2001. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Supuran C. T., Scozzafaca A. 2002. Matrix metalloproteinases, p. 35–61 In Smith H. J., Simons C. (ed.), Proteinase and peptidase inhibition: recent potential targets for drug development. Taylor and Francis, New York, NY [Google Scholar]
- 24. Tu G., Xu W., Huang H., Li S. 2008. Progress in the development of matrix metalloproteinase inhibitors. Curr. Med. Chem. 15:1388–1395 [DOI] [PubMed] [Google Scholar]
- 25. Van der Vaart J. M., te Biesebeke R., Chapman J. W., Toschka H. Y., Klis F. M., Verrips C. T. 1997. Comparison of cell wall proteins of Saccharomyces cerevisiae as anchors for cell surface expression of heterologous proteins. Appl. Environ. Microbiol. 63:615–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Visse R., Nagase H. 2003. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92:827–839 [DOI] [PubMed] [Google Scholar]
- 27. Whittaker M., Floyd C. D., Brown P., Gearing A. J. 1999. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 99:2735–2776 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.