

Abstract
The soil bacterial isolate Variovorax sp. strain SRS16 mineralizes the phenylurea herbicide linuron. The proposed pathway initiates with hydrolysis of linuron to 3,4-dichloroaniline (DCA) and N,O-dimethylhydroxylamine, followed by conversion of DCA to Krebs cycle intermediates. Differential proteomic analysis showed a linuron-dependent upregulation of several enzymes that fit into this pathway, including an amidase (LibA), a multicomponent chloroaniline dioxygenase, and enzymes associated with a modified chlorocatechol ortho-cleavage pathway. Purified LibA is a monomeric linuron hydrolase of ∼55 kDa with a Km and a Vmax for linuron of 5.8 μM and 0.16 nmol min−1, respectively. This novel member of the amidase signature family is unrelated to phenylurea-hydrolyzing enzymes from Gram-positive bacteria and lacks activity toward other tested phenylurea herbicides. Orthologues of libA are present in all other tested linuron-degrading Variovorax strains with the exception of Variovorax strains WDL1 and PBS-H4, suggesting divergent evolution of the linuron catabolic pathway in different Variovorax strains. The organization of the linuron degradation genes identified in the draft SRS16 genome sequence indicates that gene patchwork assembly is at the origin of the pathway. Transcription analysis suggests that a catabolic intermediate, rather than linuron itself, acts as effector in activation of the pathway. Our study provides the first report on the genetic organization of a bacterial pathway for complete mineralization of a phenylurea herbicide and the first report on a linuron hydrolase in Gram-negative bacteria.
INTRODUCTION
The phenylurea herbicide linuron is a nonselective pre-emergent herbicide that acts as a photosystem II inhibitor. The herbicide is globally used to control a wide variety of annual and perennial broadleaf and grassy weeds in agricultural land. Microbial degradation is considered an important mechanism in the dissipation of linuron and other phenylurea herbicides in the environment. Several bacterial strains (39, 46), as well as consortia (5, 10), able to degrade and even use the compound as a sole source of carbon and nitrogen have been reported. Although derived from different geographical locations, most of the linuron-catabolizing isolates, either individual strains or key members of linuron-degrading consortia, belong to the genus Variovorax. This suggests that this genus plays an important role in linuron degradation in soil. The proposed pathway of linuron catabolism starts with amide hydrolysis to 3,4-dichloroaniline (DCA) and N,O-dimethylhydroxylamine (N,O-DMHA) (Fig. 1). DCA is harmful and recalcitrant, while N,O-DMHA is not and degraded easily. Several linuron-degrading Variovorax strains, in addition to mediating linuron hydrolysis, are able to use DCA as the sole carbon source and mineralize it. To date, little is known about the genes and enzymes responsible for linuron and DCA degradation. Engelhardt et al. (13) described an arylacyl amidase responsible for conversion of linuron to DCA in Bacillus sphaericus ATCC 12123. In addition, phenylurea hydrolase-encoding genes puhA and puhB were identified in the linuron-degrading actinomycetes Arthrobacter globiformis D47 (52) and Mycobacterium brisbanense JK1 (23), respectively. PuhA and PuhB form a novel branch within the metal-dependent amidohydrolase superfamily (23). Regarding the degradation of DCA, Dejonghe (9) and Breugelmans et al. (6) found indications for the involvement of a multicomponent aniline dioxygenase enzyme in DCA degradation in Variovorax sp. strain WDL1. However, the genes responsible for DCA degradation in linuron-mineralizing bacteria have not yet been identified.
Fig. 1.

Catabolic pathway of linuron degradation in Variovorax sp. SRS16. The catabolic steps specified by libA, dcaQTA1A2B, and ccdCFDE are indicated.
We report here on the identification of the linuron and DCA degradation genes in the linuron-mineralizing strain Variovorax sp. strain SRS16 (46). The enzyme responsible for hydrolysis of linuron was purified and characterized. The expression of the catabolic genes under different conditions and their distribution among other linuron- and/or DCA-degrading strains was analyzed.
MATERIALS AND METHODS
Bacterial strains, cultivation conditions, and chemicals.
Variovorax sp. strains SRS16 (46), WDL1 (10), PBS-H4, PBL-E5, and PBL-H6 and Hydrogenophaga sp. strain PBL-H3 (5) were routinely grown on R2A agar plates supplemented with 20 mg of linuron liter−1 at 26°C. Variovorax sp. strains PBD-E37, PBD-H1, and PBD-E5, Cupriavidus sp. strain PBS-E1, and Afipia sp. strain PBD-E87 (5), Comamonas testosteroni WDL7, and Delftia acidovorans WDL34 (10) were grown in R2A supplemented with DCA (20 mg liter−1). Hyphomicrobium sulfonivorans strains WDL6 (10), PBN-E9, and PBN-H4 (5) were grown in MMO minimal medium supplemented with 1% methanol. R2A and MMO media were prepared as described previously (5, 10). Linuron [3-(3,4-dichlorophenyl)-1-methoxy-1-methyl urea] (99.5%), diuron [3-(3,4-dichlorophenyl)-1,1-dimethyl urea] (99.5%), isoproturon [3-(4-isopropylphenyl)-1,1-dimethyl urea] (99%), metobromuron [3-(4-bromophenyl)-1-methoxy-1-methyl urea] (99.9%), DCA (98%), and aniline were purchased from Sigma-Aldrich, Belgium.
HPLC analysis.
Reverse-phase high-pressure liquid chromatography (HPLC; LaChrom; Merck Hitachi) was used to detect and quantify phenylurea herbicides and their metabolites in cultures containing initial concentrations of 20 to 50 mg liter−1, as previously described (5).
Differential proteomic analysis using isotope-coded protein labeling (ICPL).
SRS16 was cultured in MMO supplemented with succinate (0.2%) on a rotary shaker in the dark at 26°C until an optical density at 600 nm (OD600) of 0.5 was reached. The culture was split in two and MMO, supplemented with either succinate (0.2%) or succinate (0.2%) and linuron (60 mg liter−1), was added to reach a final medium composition of 0.1% succinate or 0.1% succinate and 30 mg of linuron liter−1. The cultures were incubated at 26°C and every 90 min, samples were taken for HPLC-based linuron quantification and OD600 measurement. Degradation of linuron was observed immediately after addition of linuron, indicating that no catabolic repression of linuron degradation occurs when both succinate and linuron are available as carbon sources. After 6 h, when 50% of linuron was degraded and an OD600 of 0.5 was reached, the cultures were centrifuged (3,400 × g, 15 min, 4°C), and the pellets were stored at −80°C for protein extraction. Protein extraction, post-digest ICPL and MuDPIT (Multi-Dimensional Protein Identification Tool) were performed as described previously (27). For each experimental condition, duplicate cultures were analyzed.
Purification and characterization of linuron hydrolase.
SRS16 was cultured in five replicates of 1 liter of MMO supplemented with succinate (0.2%) on a magnetic stirrer in the dark at 26°C until an OD600 of 0.7 was reached. The culture was amended with MMO containing succinate (0.2%) and linuron (60 mg liter−1) to reach a final linuron concentration of 15 mg liter−1. Every 90 min, samples were taken for HPLC-based linuron quantification and OD600 measurement. After 5 h, when 40% of the linuron was degraded and an OD600 of 0.6 was reached, the cultures were centrifuged (3,400 × g, 15 min, 4°C), and the pellets were washed with phosphate-buffered saline (150 mM NaCl, 7 mM K2HPO4, 2.35 mM KH2PO4), suspended in 10 ml of morpholinepropanesulfonic acid (MOPS) buffer (25 mM MOPS, 1 mM dithiothreitol, 5% glycerol; pH 7.6) supplemented with Complete, Mini, EDTA-free protease inhibitor cocktail (Roche Diagnostics GmbH, Germany), and stored at −80°C. The cells in the concentrated cell suspension were lysed by using a French press (Thermo). The crude cell extract was centrifuged (3,400 × g, 15 min, 4°C), and the cell-free supernatant was stored at −80°C. After a 35 to 50% ammonium sulfate precipitation and dialysis against MOPS buffer, the protein extract was loaded on an anion-exchange column (DEAE-Sepharose), equilibrated with MOPS buffer. The proteins were eluted over a linear gradient of 200 ml from 0 to 0.5 M KCl (flow rate, 1 ml min−1). Active fractions were pooled and concentrated by ultrafiltration (molecular mass cutoff, 3 kDa [Millipore]; 4,500 × g, 4°C) before being subjected to size exclusion chromatography (column type G3000SW; Tosoh Corp., Japan). Elution was performed with 150 mM NaCl in MOPS buffer (flow rate, 1 ml min−1), and the oligomeric state of the enzyme was estimated by using the following protein size standards: RNase A (13.7 kDa), ovalbumin (45 kDa), bovine serum albumin (66 kDa), alcohol dehydrogenase (141 kDa), gamma globulin (160 kDa), and thyroglobulin (670 kDa). Protein concentrations were estimated using a Bradford protein assay (Bio-Rad). Protein purity was assessed by means of SDS-PAGE and Coomassie brilliant blue staining. For low protein concentrations, silver staining was used (38). To identify proteins excised from polyacrylamide gels, liquid chromatography-electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS) was used, as previously described by Breugelmans et al. (6). The peptide sequences were analyzed by using BlastP/TBlastN to identify proteins with significant sequence similarity.
To assess linuron hydrolase activity, 10 μl of purified enzyme solution in MOPS buffer was supplemented with 140 μl of linuron solution (60 mg liter−1 [or smaller volumes with the same enzyme/linuron volume ratio]) and incubated at room temperature (15°C) for 60 min. The disappearance of linuron and appearance of DCA was monitored to quantify linuron hydrolase activity. The production of DCA was either detected by a colorimetric reaction or quantified by HPLC. The colorimetric detection of DCA consisted of a diazotization-coupling reaction according to the method of Pease (34) with the absorption measured at 500 nm. In the case of HPLC-based measurement of linuron and DCA, 50 μl of 4 M HCl was added to stop the reaction after 1 h of incubation. Corrections were made for background hydrolysis or absorption. The kinetic parameters Km and Vmax of the purified enzyme solution were determined for linuron by determining the hydrolysis activity by HPLC in the presence of a range of linuron concentrations (200, 120, 80, 40, 32, 24, 16, 8, and 4 μM). The substrate specificity of the linuron hydrolase and the crude protein extract of a linuron-amended Variovorax sp. strain SRS16 culture was examined by HPLC for diuron (50 mg liter−1), isoproturon (50 mg liter−1), and metobromuron (50 mg liter−1) after 24 h of incubation. The activity of the linuron hydrolase at different temperatures (4, 22, 30, 37, and 60°C) was analyzed by HPLC using a linuron concentration of 50 mg liter−1. All tests were performed in duplicate.
De novo sequencing and sequence analysis.
De novo sequencing of the genome of SRS16 was performed by BaseClear (Netherlands) on an Illumina GAIIx platform. CLC Bio Genomic Workbench 3.7 was used to assemble the 50-bp paired-end reads into 354 contigs with an average length of 21.5 kb. The microbial genome annotation system Magnifying Genomes (MaGe) (53) was used to annotate the contigs, while BLAST analyses were used to identify translated open reading frames (ORFs) with a sequence similarity to known proteins in databases (July 2011) (1). Peptide sequences of the amino group transfer protein component of the multicomponent aniline dioxygenase, the maleylacetate reductase, and the suspected linuron hydrolase were used to screen the contigs for the corresponding genes by using a local TBlastN search. For each gene, one contig was identified. The contig containing the multicomponent aniline dioxygenase gene and the contig containing the maleylacetate reductase gene could be assembled based on the sequences of eight fosmid clones selected from an SRS16 genome fosmid library constructed using the CopyControl HTP fosmid library production kit with pCC2FOS and phage T1-resistant EPI300-T1 Escherichia coli plating strain (Epicentre Biotechnologies) according to the recommendations of the manufacturer. These fosmid clones were identified by means of PCR screening of colonies carrying the SRS16 fosmid library using degenerate primer pair AGT-F/AGT-R (see Table S1 in the supplemental material) targeting the gene encoding the amino group transfer protein of the multicomponent aniline dioxygenase. Prior to PCR, a copy-control induction solution was used to induce a high copy number of the fosmids in the clones. Fosmid DNA was isolated with a FosmidMAX DNA purification kit (Epicentre Biotechnologies). The eight fosmid clones were sequenced as a mixture by BaseClear, as reported above for the de novo SRS16 genome sequencing. Mascot 2.2 was used to identify Variovorax sp. SRS16 genes encoding proteins with potential involvement in linuron catabolism.
Southern blotting.
The primer pair LinAmidRF/LinAmidRR was used to synthesize an libA-specific gene probe from genomic SRS16 DNA using a digoxigenin (DIG) labeling mix (Roche Applied Science, Germany). Genomic DNA (10 μg) of Variovorax sp. strains SRS16, PBL-H6, PBL-E5, WDL1, and DSM66 was obtained from pure cultures (OD600 = 0.7) using a Puregene Core Kit A (Qiagen, Belgium) and digested by PstI (New England BioLabs, United Kingdom). Restriction fragments were separated by agarose gel electrophoresis (1.5%, 120 V, 1.5 h). Blotting, hybridization, and chemiluminescent detection were performed as recommended by Roche Applied Science. Details of the procedure are provided as supplemental material.
Transcription analysis.
Variovorax sp. SRS16 was grown in R2A until an OD600 of 0.3 was reached. R2A or R2A supplemented with either linuron, DCA, or aniline was added to the culture at a final concentration of 17 mg liter−1 (0.2, 0.3, and 0.54 mM, respectively). At different time points after addition of the compounds (10, 20, 30, 40, 50, 60, and 120 min), samples were taken for RNA extraction. Degradation of linuron, DCA, and aniline was assessed and confirmed by HPLC analysis. RNA extraction was performed with the RNeasy minikit (Qiagen, Belgium), and contaminating DNA was removed with Turbo DNA-free (Ambion). The RNA concentration in the extracts was determined (Nanodrop), and the RNA extracts were diluted to equal concentrations. RNA was converted to cDNA by reverse transcription using a reverse transcription system (Promega). The cDNA was assessed for the presence of the respective gene sequences by PCR as described below. All experiments were performed in duplicate.
PCR analysis.
Codehop (37) was used to design degenerate primers based on protein sequences. Primer BLAST was used to design primers based on known gene sequences. BLAST and MaGe (53) were used to confirm the selected primer specificity (July 2011). Primers (see Table S1 in the supplemental material) were obtained from Thermo Fisher Scientific (Belgium). PCR was performed in an Eppendorf MasterCycler (Eppendorf, Germany). The PCR mixture compositions and reaction conditions are provided in Table S1 in the supplemental material. Visualization of PCR amplicons was performed by means of agarose gel electrophoresis (1.5% agarose gel, 0.02% ethidium bromide, 90 V for 75 min).
Expression of libA in E. coli.
The libA gene was optimized for E. coli gene expression and cloned into expression vector pJexpress 416 (pJexpress416_libA) by DNA2.0, Inc. A poly-His tag was added at the C-terminal end. As a negative control, the RFP gene was cloned into pJexpress 416 (pJexpress416_RFP_ctrl). Electroporation was used to transform E. coli BL21(DE3) with pJexpress416_libA and pJexpress416_RFP_ctrl. E. coli BL21(DE3) without expression vector, E. coli BL21(DE3) containing pJexpress416_libA, and E. coli BL21(DE3) containing pJexpress416_RFP_ctrl were grown in 50 ml of LB with kanamycin (50 mg liter−1) at 37°C until an OD600 of 0.8 was reached. At this point, the cultures were split into two portions of 25 ml. In one of the portions, expression was induced by adding 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After another 20 h of incubation at 20°C, 1 ml of each bacterial culture was taken for SDS-PAGE analysis, and linuron was added at a concentration of 10 mM. Linuron degradation activity was monitored by HPLC. Then, 1 ml of bacterial culture was prepared for SDS-PAGE by centrifugation (10,000 rpm, 1 min), after which the bacterial pellet was dissolved in 100 μl of SDS-PAGE loading buffer and boiled at 100°C for 3 min. After centrifugation (10,000 rpm, 1 min), 10 μl of the supernatants was used for SDS-PAGE. All treatments were performed in triplicate.
Phylogenetic analysis.
Phylogenetic analysis was performed with Geneious Pro (version 5.4.4) (11) using PHYML (JTT matrix) (19).
GenBank accession numbers.
The nucleotide sequences of contig 1 and contig 2 of the genome of SRS16, of libA in Variovorax sp. strains PBL-H6 and PBL-E5, and of libA in Hydrogenophaga sp. strain PBL-H3 were deposited in GenBank under accession numbers JN104632, JN104633, JN104629, JN104631, and JN104630, respectively.
RESULTS AND DISCUSSION
Differential proteomic analysis using ICPL.
Isotope-coded protein labeling (ICPL) was used to detect differentially expressed proteins in Variovorax sp. SRS16 when grown in the presence of linuron compared to growth without linuron. The linuron-supplemented cultures showed active linuron degradation at the time point of analysis. A higher number of differentially downregulated proteins compared to upregulated proteins was observed in the linuron-amended cultures. Of the downregulated proteins, 45% were membrane proteins. Moreover, upregulation of stress-related proteins such as chaperones GroEL and DnaK was observed in linuron-amended SRS16 cultures (data not shown) as reported in Variovorax sp. strain WDL1 (6). Up- and downregulation of membrane proteins have been previously associated with a stress response (60, 62). As discussed by Breugelmans et al. (6), linuron exposure appears to induce a stress situation in Variovorax.
Interestingly, several upregulated proteins in the linuron-amended culture are associated with catalytic proteins of the proposed linuron degradation pathway. The most strongly upregulated protein in the linuron-amended culture (Table 1) showed significant homology with (putative) amidases, which catalyze the hydrolysis of amide or ester functional groups at carbon and phosphorus centers. In addition, as observed by Breugelmans et al. (6) in Variovorax sp. WDL1, upregulation of several proteins homologous to components of multicomponent aniline dioxygenases involved in (chloro)aniline degradation (amino group transfer protein, glutamine amidotransferase and the large subunit of the dioxygenase) was observed in cultures amended with linuron (Table 1). Expression of the small subunit of a multicomponent aniline dioxygenase was also observed, although it was not significantly different from the control culture. These results imply the involvement of a similar multicomponent dioxygenase in the degradation of DCA in Variovorax sp. SRS16. Furthermore, three upregulated proteins showed significant amino acid similarity to enzymes involved in the modified ortho-cleavage pathway for chlorocatechol degradation, i.e., chlorocatechol dioxygenase responsible for ortho-ring fission of dichlorocatechols to dichloromuconate, carboxymethylenebutenolidase (a dienelactone hydrolase performing dichloromuconate cycloisomerization to chloromaleylacetate) and maleylacetate reductases (responsible for reduction of chloromaleylacetate to oxoadipate via maleylacetate), in chloroaromatic-degrading bacteria (29, 48, 56). These data suggest the involvement of a typical modified ortho-cleavage pathway for chlorocatechol degradation in the further mineralization of DCA by strain SRS16. Other than the discussed proteins, no up- or downregulation of proteins linked to linuron catabolism could be observed.
Table 1.
Overview of the ORFs and their annotations as identified in contigs 1 and 2 (see Fig. 3) and their differential proteomic expressiona
| ORF | Name | Positionb | Possible function | Closest homologous protein (GenBank accession no./% identity) | Organism | Biological replicate 1 |
Biological replicate 2 |
||
|---|---|---|---|---|---|---|---|---|---|
| Score | L/C [SD(geo)] | Score | L/C [SD(geo)] | ||||||
| Contig 1 | |||||||||
| 1 | 3–515 | Cyclase | HMPREF0005_01234 (EFV81793.1/61) | Achromobacter xylosoxidans C54 | |||||
| 2 | 515–1024 | Acyl dehydratase | Rmet_4072 (YP_586209/75) | Cupriavidus metallidurans CH34 | |||||
| 3 | 1021–1944 | Dihydropicolinate synthase | mll1010 (NP_102694.1/52) | Mesorhizobium loti MAFF303099 | |||||
| 4 | 2154–3113 | Extracytoplasmic solute receptor | Reut_A1674 (YP_295884.1/60) | Ralstonia eutropha JMP134 | |||||
| 5 | ccdE | 3151–3873 | Dienelactone hydrolase | Carboxymethylenebutenolidase (YP_001632346.1/68) | Bordetella petrii DSM 12804 | 134 | 2.26 | 216 | 6.37 |
| 6 | ccdD | 3870–5015 | Chloromuconate cycloisomerase | TfdD (AATPP367/53) | Sphingomonas sp. strain tfd44 | ||||
| 7 | ccdF | 5041–6108 | Maleylacetate reductase | TfdF (NP_990894.1/73) | Achromobacter denitrificans | ND | ND | 52 | 3.08 |
| 8 | 6120–6455 | Hypothetical protein | Bpet3729 (YP_001632340.1/85) | Bordetella petrii DSM 12804 | |||||
| 9 | 6601–6837* | FAD-dependent pyridine nucleotide-disulfide oxidoreductase | FAD-dependent pyridine nucleotide-disulfide oxidoreductase (YP_298914.1/56) | Ralstonia eutropha JMP134 | |||||
| 10 | 6825–7124* | Aromatic ring hydroxylating dioxygenase | AndAc (YP_556337.1/79) | Burkholderia xenovorans LB400 | |||||
| 11 | ccdC | 7229–7975 | Chlorocatechol dioxygenase | Chlorocatechol dioxygenase DccAII (CAF32822.1/50) | Sphingobium herbicidovorans | 182 | 3.98 [1.96] | 203 | 3.98 [1.25] |
| 12 | ccdR | 8108–9076 | LysR family transcriptional regulator | LysR family transcriptional regulator Mpe_A3313 (YP_001022501.1/46) | Methylibium petroleiphilum PM1 | ||||
| 13 | 9585–10547 | Extracytoplasmic solute receptor | Extracytoplasmic solute receptor h16_A1254 (YP_725762.1/45) | Ralstonia eutropha H16 | ND | ND | 161 | 2.49 [1.60] | |
| 14 | IS3 orfA | 10659–10925 | Transposase | Putative transposase (NP_395222.1/68) | Yersinia pestis CO92 | ||||
| 15 | IS3 orfB | 10958–11776 | Transposase | Integrase catalytic region AnaeK_1549 (YP_002133907.1/55) | Anaeromyxobacter sp. strain K | ||||
| 16 | 11905–12399 | IclR-type transcriptional protein | Transcriptional regulator Adeg_1855 (YP_003239789.1/31) | Ammonifex degensii KC4 | |||||
| 17 | istB | 12443–13237 | IstB-like ATP-binding protein transposase | IstB protein 22 (YP_195848.1/100) | Achromobacter xylosoxidans A8 | ||||
| 18 | istA | 13227–14783 | IstA transposase | Transposase IstA protein 59 (YP_195883.1/99) | Achromobacter xylosoxidans A8 | ||||
| 19 | dcaR | 14935–15852 | LysR family transcriptional regulator | LysR-type regulator DanR (ABI20712.1/98) | Delftia sp. strain AN3 | 88 | 3.57 | ND | ND |
| 20 | dcaB | 15885–16892 | Aniline dioxygenase reductase | Aniline dioxygenase reductase DanB (ABI20711.1/99) | Delftia sp. strain AN3 | ||||
| 21 | dcaA2 | 16909–17553 | Aniline dioxygenase small subunit | Small subunit of dioxygenase DanA2 (ABI20710.1/100) | Delftia sp. strain AN3 | ND | ND | 128 | 2.5 |
| 22 | dcaA1 | 17550–18896 | Aniline dioxygenase large subunit | Large subunit of dioxygenase TdnA1 (AA038208.1/99) | Delftia acidovorans | 195 | 4.20 [2.23] | 201 | 2.83 [1.44] |
| 23 | dcaT | 18941–19705 | Glutamine amido transferase | Glutamine amido transferase DanT (ABI20709.1/99) | Delftia sp. strain AN3 | ND | ND | 46 | 1.73 |
| 24 | dcaQ | 19725–21230 | Glutamine synthetase | Putative glutamine synthetase DanQ (ABI20708.1/99) | Delftia sp. strain AN3 | 42 | 2.46 | ND | ND |
| 25 | 21515–21658* | Transposase | IS4 family transposase (BAH90225.1/100) | Uncultured bacterium | |||||
| 26 | IS66 orfA | 21742–22083 | Transposase | Transposase IS3/IS911 (YP_003643234/32) | Thiomonas intermedia K12 | ||||
| 27 | IS66 orfB | 22080–22421 | Transposase | Transposase (BAH90226.1/92) | Uncultured bacterium | ||||
| 28 | IS66 orfC | 22501–24084 | Transposase | Transposase ISThsp3 IS66 family (CAZ88005.1/60) | Thiomonas sp. strain sp3As | ||||
| 29 | tnpA | 24388–27300 | Transposase | Transposase TnpA (YP_001967688.1/100) | Comamonas sp. strain CNB-1 | ||||
| 30 | 27378–27842 | Hypothetical protein | MYG1 (XP_002538853.1/56) | Ricinus communis | |||||
| 31 | 28171–28611 | LysR-type transcriptional regulator | LysR-type transcriptional regulator Dtpsy_1784 (YP_002553241/31) | Acidovorax ebreus TPSY | |||||
| 32 | 28717–29250 | No significant match | |||||||
| 33 | 29568–30488* | Transcriptional regulator | Putative transcriptional regulator THI_0620 (CAZ87353.1/57) | Thiomonas sp. strain sp3As | |||||
| Contig 2 | |||||||||
| 41 | 623–1444 | LuxR family transcriptional regulator | Putative transcriptional regulator (YP_789362.1/26) | Pseudomonas aeruginosa UCBPP-PA14 | |||||
| 42 | libA | 1531–2958 | Linuron hydrolase | Putative amidase (YP_002234156.1/52) | Burkholderia cenocepacia J2315 | 2,706 | 13.00 | 3,033 | 8.73 [2.55] |
The last four columns show the genes which were upregulated in Variovorax sp. SRS16 cultures when amended with linuron compared to non-linuron-amended cultures as determined by post-digest ICPL analysis of two biological replicates. For each biological replicate the score is a confidence index of the identification of the peptide sequences with a predicted protein of the Variovorax sp. SRS16 genome. L/C represents the average ratio of the quantity of the protein in the crude protein extract of the linuron-amended culture to the quantity of the protein in the non-linuron-amended culture, calculated as the average of the ratios for each peptide detected for this protein. When L/C is significantly different from 1, the value is marked in boldface. SD(geo), geometrical standard deviation; ND, no data.
*, truncated ORF.
Purification and characterization of a linuron hydrolase.
An enzyme with linuron hydrolase activity was purified from a cell extract of linuron-degrading SRS16 cells. The final purified protein solution with linuron hydrolase activity contained two proteins (∼100 and ∼55 kDa) (Fig. 2). Tandem mass spectrometry showed that the protein of ∼55 kDa corresponded to the upregulated amidase in the linuron-amended SRS16 cultures. The protein of ∼100 kDa was identified as the ribosomal protein elongation factor G (62% sequence coverage). The pI values of both proteins are identical, which explains the difficulty in separating them. Translation elongation factor G, however, is not expected to have amido hydrolase activity. Moreover, no upregulation of this housekeeping protein was observed in the differential proteomic approach. This indicates that the amidase is responsible for linuron hydrolysis in SRS16, and we designated this as LibA (Linuron biodegradation protein A). LibA is a monomeric enzyme with a Km for linuron of 5.8 μM, a Vmax of 0.16 nmol min−1, and a maximum hydrolase activity between 22 and 30°C (see Fig. S1 in the supplemental material). The hexameric phenylurea hydrolase proteins PuhA and PuhB, previously identified in Gram-positive bacteria, show very similar Km values (6.8 and 7.6 μM, respectively) and a maximal linuron hydrolase activity between 30 and 35°C (23). LibA of Variovorax sp. SRS16 showed activity toward linuron but no detectable activity toward other tested phenylurea herbicides such as the N-methoxy-N-methyl phenylurea herbicide metobromuron and the N,N-dimethyl phenylurea herbicides isoproturon and diuron. Sørensen et al. (46) reported diuron mineralization by Variovorax sp. SRS16, but only in case the organism was cultured in media containing growth substrates and nitrogen sources additional to diuron. Based on competition experiments with linuron, the authors of that study concluded that diuron was not hydrolyzed in SRS16 by the linuron hydrolase but instead by a broad-spectrum hydrolase present during active growth The observed inactivity of LibA toward diuron in our study seems to confirm this hypothesis. The narrow phenylurea herbicide specificity of the monomeric LibA enzyme is in contrast with the phenylurea herbicide spectrum of PuhA and PuhB, which show activity toward a wide range of phenylurea herbicides, including N-methoxy-N-methyl phenylureas (underlined) and N,N-dimethyl phenylureas: linuron, chlorbromuron > metobromuron, monolinuron, diuron, chlortoluron > fluomethuron, metoxuron, isoproturon, fenuron (arranged in decreasing order of PuhA/PuhB catalytic efficiency) (23). Also, total protein extracts from the linuron-degrading Bacillus sphaericus ATCC 12123 showed, in addition to linuron hydrolysis, activity toward other phenylurea herbicides (12).
Fig. 2.
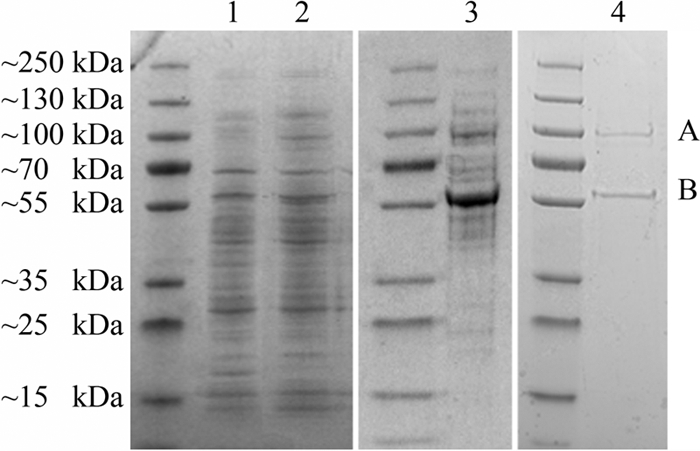
SDS-PAGE analysis of protein extracts showing linuron hydrolase activity, from linuron-amended Variovorax sp. SRS16 cultures at different purification steps. Lane 1, crude cell-free protein extract; lane 2, after 35 to 50% ammonium sulfate precipitation; lane 3, after anion-exchange chromatography; lane 4, after size exclusion chromatography. Band A was identified as transcription elongation factor G, while band B was identified as the translation product of ORF 42 (libA).
Genetic backbone of linuron mineralization by strain SRS16.
The peptide sequences showing similarity to the chloroaniline and chlorocatechol degradation enzymes in the differential proteomic analysis and those obtained from purified LibA could be assigned to ORFs present on two contigs in the draft SRS16 genome sequence, i.e., contig 1 of ±54 kb and contig 2 of ±3.6 kb (Fig. 3 and Table 1).
Fig. 3.

Genetic organization of contigs 1 and 2 containing the identified genes involved in linuron and DCA catabolism in Variovorax sp. SRS16. Contig 1 contains a 54-kb DNA fragment including dcaQTA1A2BR and ccdRCFDE. Upstream of ORF 33, contig 1 includes another 24-kb-long sequenced nucleotide segment. The ORFs of this part of the contig are represented in the corresponding GenBank file JN104632. Contig 2 consists of a 3.5-kb genome fragment containing the linuron hydrolase gene libA (ORF 42). Further information about the different ORFs is shown in Table 1.
Within contig 1, ORFs 19 to 24 compose a cluster of six genes that show more than 98% nucleotide and amino acid similarity with the respective gene product(s) in the danQTA1A2BR gene cluster, a multicomponent aniline dioxygenase in the aniline-degrading strain Delftia sp. AN3. Five of these ORFs (ORFs 19 and 21 to 24) were linuron-dependent upregulated in the differential proteomic analysis. Moreover, the organization of ORFs 19 to 24 is identical to those in strain AN3 and other (chloro)aniline-degrading bacteria. This gene cluster is therefore proposed to be responsible for the dioxygenation of DCA to a chlorocatechol compound in SRS16 and was named dcaQTA1A2BR (Fig. 1). The protein product of dcaR shows significant similarity with LysR-type transcriptional regulators (LTTR), including the protein product of regulatory genes of aniline dioxygenation gene clusters such as tadR in Delftia tsuruhatensis AD9 (16). Therefore, dcaR is expected to perform a similar regulatory role in strain SRS16. dcaQTA1A2BR is surrounded by insertion sequence (IS) elements (i.e., ORFs 14, 15, 17, 18 and 25 to 29) of which one (ORF 14) appears to be truncated. The other eight ORFs belong to four different families of IS elements, i.e., IS66, IS3, IS21 (i.e., IS1600) and Tn3 (i.e., IS1071). This indicates that the dcaQTA1A2BR gene cluster is located within a transposon-like structure and that it was acquired by horizontal gene transfer (HGT) (49). The G+C content of the dcaQTA1A2R gene cluster (65.88%) is similar to the overall G+C content of the SRS16 genome (66.51%), suggesting that in case HGT was involved, it was not acquired recently or the origin of this gene cluster lies in a related bacterium.
Downstream of the dcaQTA1A2BR cluster, a second gene cluster (ORFs 5 to 12) was found which encoded the peptide sequences related to chlorocatechol-degrading proteins. The translation products of ORFs 11, 7, 6, and 5 show significant similarity with a chlorocatechol-1,2-dioxygenase, maleylacetate reductase, chloromuconate cycloisomerase, and dienelactone hydrolase, respectively. Moreover, ORFs 5, 7, and 11 showed linuron-dependent upregulation in the differential proteomic analysis (Table 1). As outlined above, these enzymes compose a classic modified ortho-cleavage pathway for the degradation of chlorocatechols to β-ketoadipate (54) (Fig. 1). Upstream of ORFs 11, 7, 6, and 5, the translation product of ORF 12 showed significant similarity with an LTTR and shows a typical opposite orientation to the operon it regulates (51). LTTRs have been reported to be regulators of chlorocatechol degradation previously (51) (Fig. 4). Therefore, the gene cluster composed of ORFs 11, 7, 6, 5, and 12 is proposed to be responsible for the degradation of DCA to β-ketoadipate and was named ccdRCFDE. This cluster as such complements the dcaQTA1A2BR gene cluster to enable complete degradation of DCA to Krebs cycle intermediates. Interestingly, in the aniline catabolic Delftia sp. strain AN3, the association of an aniline multicomponent dioxygenase with a catechol meta-cleavage pathway (61), typical for degradation of nonchlorinated aromatics (31), was reported. The organization of the ccdRCFDE cluster shows some significant differences with those of gene clusters encoding modified chlorocatechol ortho-cleavage pathways in other organisms (Fig. 4). Most strikingly, in between ccdC and ccdF, three ORFs (ORFs 8, 9, and 10) are located that cannot be directly linked to chlorocatechol degradation and that are not present in most other isofunctional gene clusters (13, 49, 54). The exception is ORF 8, whose translation product shows similarity with a hypothetical protein encoded by a gene present in a chlorocatechol degradation pathway gene cluster in Bordettela petrii (14, 19, 50, 55). ORFs 9 and 10 appear to be truncated genes. Their putative translation products show similarity with a pyridine nucleotide disulfide oxidoreductase in Variovorax paradoxus S110 (20) and an aromatic ring dioxygenase in Burkholderia xenovorans LB400 (7), respectively (Table 1). The ccd gene cluster is flanked by two ORFs (ORF 13 and ORF 4) of which both translation products show similarity with extra-cytoplasmic solute receptors, belonging to the Bug family. Interestingly, ORFs encoding similar hypothetical proteins of unknown function are present in other modified chlorocatechol ortho-cleavage pathway gene clusters (14, 18, 55). A wide range of orthologous bug genes have been identified in the genomes of various Betaproteobacteria, but the function is often unknown (3). Some bug homologues are involved in periplasmic uptake of substrates (3) and a regulatory role has been suggested (2, 35).
Fig. 4.

Organization of the modified chlorocatechol ortho-cleavage pathway gene cluster in chloroaromatic-degrading bacterial strains (14, 18, 36, 48, 50, 55, 56). The arrows indicate the localization, size, and direction of transcription of the genes. Similar shaded patterns indicate isofunctional genes. The following enzymes are shown (the coding genes are in parentheses): 2,4-D transport protein (tfdK), chlorophenol hydroxylases (tfdB), chlorocatechol 1,2-dioxygenases (tfdC, ccdC, clcA, tcbC, tetC, and catA1), chloromuconate cycloisomerases (tfdD, ccdD, clcB, tcbD, tetD, catB4, and catB5), dienelactone hydrolases (tfdE, ccdE, tcbE, tetE, Bpet3735, and Bpet3751), maleylacetate reductases (tfdF, ccdF, clcD, tcbF, tetF, and Bpet3730), transposases (tnpA, Bpet3733, and Bpet3736), and indole acetamide hydrolase (iaaH). The ORFs Bpet3729, Bpet3731, and Bpet3732 code for hypothetical proteins. bug77 codes for a Bug family protein.
The peptide sequences of LibA could be assigned to ORF 42 in contig 2. ORF 42, which is absent in the genomes of V. paradoxus S110 (20) and V. paradoxus EPS (NC_014931), was named libA. Besides libA, contig 2 contains one ORF (ORF 41) directly upstream of libA. Its translation product contains a carboxy-terminal DNA-binding motif typical for regulators of the LuxR superfamily. The absence of a characteristic domain for interaction with a sensor kinase, a quorum-sensing molecule or multiple ligands, suggests that ORF 41 encodes a protein that belongs to the autonomical effector domain regulators, which activate gene expression in the presence of an effector molecule. Possibly, ORF 41 is involved in the regulation of libA.
Although LibA, PuhA, and PuhB all perform linuron hydrolysis, LibA lacks homology with PuhA/PuhB, which both belong to the metal-dependent amidohydrolase superfamily (23), while LibA shows highest homology with members of the amidase signature (AS) family (Fig. 5) that is part of the GGCT-like superfamily (Fig. 5). Although no sequence data of the linuron hydrolase identified in Bacillus sphaericus is available, it was previously proposed to belong to a different family than PuhA/PuhB based on differences in substrate specificity and physicochemical characteristics (23). LibA shows ∼50% amino acid identity with the ω-octalactam hydrolase of Rhodococcus sp. Oct1 (Aaa R-Oct1 in Fig. 5) and orthologues from Nocardia strains (14), as well as with hypothetical proteins from another actinomycete (Pseudonocardia dioxanivorans) and from alpha- and betaproteobacteria (Ruegeria and Burkholderia spp., respectively) The hydrolase of Rhodococcus sp. Oct1, which is the most closely related biochemically characterized homolog of LibA, also shows high activity toward nitroacetanilides (15), which have a linuron-like chemical structure. Many other characterized members of the AS family show substrate specificity toward linuron-resembling molecules (Fig. 5), i.e., mostly aromatic substrates with a side chain containing an amide bond (Fig. 5). Based on the lack of amino acid sequence similarity and the functional and structural differences between LibA and the other phenylurea hydrolase protein families (PuhA/PuhB and linuron hydrolase of Bacillus sphaericus), we can conclude that in Variovorax sp. SRS16 a third phenylurea hydrolyzing protein family has evolved independently.
Fig. 5.

Phylogenetic analysis of LibA and selected members of the amidase signature family. A multiple amino acid alignment of the respective Pfam domains (PF01425) was used to construct a maximum-likelihood tree. The AS domain of LibA (residues 27 to 453) was aligned with the corresponding domain of the following enzymes, with their (preferred) substrates indicated: Aaa R-Oct1 (Rhodococcus sp. strain Oct1, o-nitroacetanilide [15]), Aaa P sp (Pseudomonas sp., p-nitroacetanilide [24]), AmdA R sp (Rhodococcus sp., 2-phenylpropionamide [28]), Ami Ca (Comamonas acidovorans, ketoprofen [58]), Ami Re (Rhodococcus erythropolis PR4, Gly-p-nitroacetanilide [26]), Ami R-N-771 (Rhodococcus sp. strain N-771, benzamide [32]), Ami Vp (Variovorax paradoxus, t-Leu-amide [25]), AtzF P-ADP (Pseudomonas sp. ADP, allophanate [43]), DamA Da (Delftia acidovorans, d-phenylalanine amide [22]), IaaH At (Agrobacterium tumefaciens, indole acetamide [57]), IaaH Pa (Pantoea agglomerans, indole acetamide [8]), IaaH Ps (Pseudomonas savastanoi, indole acetamide [57]), Mae2 Bj (Bradyrhizobium japonicum, malonamide [45]), MdlY Pp (Pseudomonas putida, mandelamide/2-phenylacetamide [17]), NylA (Arthrobacter sp. strain KI72, 6-aminohexanoate cyclic dimer/β-laurolactam [59]), Pam Sm (Stenotrophomonas maltophila, [di]peptides [Phe] [30]), PzaA Ms (Mycobacterium smegmatis, pyrazinamide [4]), and TrzF Ec (Enterobacter cloacae, allophanate [42]). Three hypothetical proteins most similar to LibA were included: HP Bc (Burkholderia cenocepacia J2315 [YP_002234156]), HP Pd (Pseudonocardia dioxanivorans [YP_004335959]), HP Rp (Ruegeria pomeroyi DSS-3 [YP_167742]). The scale bar represents 0.5 substitutions per site. Bootstrap values are represented at the branches.
Together with the dcaQTA1A2BR gene cluster encoding conversion of DCA to chlorocatechol and the ccdRCFDE gene cluster encoding a modified chlorocatechol ortho-cleavage pathway for further metabolism of chlorocatechol, libA provides all of the necessary genetic information for mineralization of linuron in Variovorax sp. SRS16. As such, strain SRS16 is another example of a patchwork assembly of catabolic gene modules (libA, dcaQTA1A2B, and ccdCDEF) in order to use a xenobiotic compound as the sole source of carbon and energy. A similar modular genetic arrangement involving a multicomponent aniline dioxygenase was recently described in the diphenylamine-degrading Burkholderia sp. strain JS667 (44).
Expression of libA in E. coli.
The linuron hydrolysis activity of LibA was confirmed using a recombinant E. coli BL21(DE3) strain containing libA under the control of an IPTG-inducible promoter. In contrast to E. coli BL21(DE3) control strains, IPTG-induced cells of E. coli containing pJexpress416_libA showed linuron degradation (80% after 7 h; 100% in <20 h) and the formation of DCA. Without IPTG induction, a slow linuron degradation was observed (50 and 60% after 24 and 48 h, respectively [data not shown]). SDS-PAGE profiles confirmed the increased expression of a protein of ∼55 kDa in the IPTG-supplemented cultures compared to noninduced cultures (data not shown).
Transcriptomic analysis of linuron degradation genes in Variovorax sp. SRS16.
Transcription analysis was performed to gain a first insight in the regulation of libA, dcaQTA1A2B, and ccdCFDE and their mode of induction. RNA extracts were collected from a Variovorax sp. SRS16 culture which was unamended or supplemented with either linuron, DCA or aniline. Background expression of libA and dca was observed (Fig. 6). Linuron-amended cultures showed a significant increased expression of libA and genes of the dca cluster at all sampling times, as observed in the differential proteomic analysis. Moreover, crude protein extracts from linuron-amended SRS16 cultures degraded 70.45% ± 2.24% of linuron after 18 h of incubation while protein extracts recovered from non-linuron-amended cultures degraded only 10.19% ± 3.26%. Also, DCA and especially aniline caused increased expression of libA and the dca cluster. Previously transcriptional activation of the aniline multicomponent dioxygenase gene cluster tadQTA1A2BR by aniline and several chloroanilines was reported (16). However, the more pronounced increase in expression in the cultures amended with aniline (compared to the cultures supplemented with linuron and DCA) can, in part, be due to the difference in molar concentrations of the added compounds (184 mM aniline, 68 mM linuron, and 102 mM DCA). Different expression intensities as a response to linuron, DCA and aniline can also be due to differences in uptake. Although libA and the dca genes are clearly upregulated in the presence of linuron, the upregulation of libA and the dca cluster in the presence of DCA suggest that DCA or its metabolites, rather than linuron, act as effector(s) of both libA and the dca gene cluster in Variovorax sp. SRS16. Positive regulation of the degradation pathway of (chloro)aromatic compounds by its pathway intermediates has been described previously (33, 41). As such, previously reported linuron induction of linuron mineralization by SRS16 in cultures containing mg-liter−1 linuron concentrations but not in cultures containing μg-liter−1 concentrations (47) can be explained by the observed background expression of libA and its downstream genes, which would allow linuron mineralization at low concentrations even without pre-exposure to linuron. At high concentrations, DCA or other metabolites are produced, resulting in increased expression of libA and dca. A basal enzyme activity at low concentrations versus an enhanced activity at elevated concentrations has been described previously in other xenobiotic-degrading bacteria (21, 40).
Fig. 6.

Transcription analysis of linuron degradation genes in Variovorax sp. SRS16. RNA sampled after 30 min of growth on R2A without (lane 1) or with linuron (lane 2), DCA (lane 3) or aniline (lane 4). A “–” indicates a blank, while “+” indicates a positive control (genomic DNA of SRS16).
No transcripts were detected for the genes of the ccd cluster (Fig. 6) and ORFs 8, 9, and 10 (data not shown) within the sampling time frame, except for ccdR, which showed background expression in each condition. The nondetectable expression of most of the ccd cluster genes is in contrast with the results from the differential proteomic analysis. However, samples for the differential proteomic approach were taken 6 h after addition of linuron, while for the transcription analysis RNA samples were only collected until 2 h after the addition of linuron, DCA, or aniline. In case a metabolite of DCA is the effector for activating transcription of the ccd gene cluster, an increased expression of these genes might only become apparent after longer incubation times, when more of the effector has accumulated.
Presence of libA, dcaQ, and ccdC in other linuron- and/or DCA-degrading bacteria.
Different linuron- and/or DCA-degrading bacterial isolates were screened by PCR for the presence of the linuron and DCA catabolic genes libA, dcaQ (as representative for dcaQTA1A2B), and ccdC (as representative for ccdCDEF) (Table 2). A false-negative result for the presence of a gene may result from differences in nucleotide sequence at the primer annealing sites. Only two other linuron-mineralizing Variovorax strains showed the presence of all three genes (Variovorax sp. strains PBL-H6 and PBL-E5). In contrast, all linuron-degrading strains that can also degrade DCA contained the libA gene, including Hydrogenophaga sp. strain PBL-H3, suggesting that libA has distributed over other (betaproteo)bacteria. The only linuron-mineralizing strain which did not contain libA was Variovorax sp. WDL1, indicating that WDL1 contains a linuron hydrolase gene substantially different from SRS16. This is in agreement with the previously reported absence of an upregulated protein resembling LibA in a linuron-amended Variovorax sp. WDL1 culture (6). The absence of LibA was confirmed by Southern blotting and a local BLAST search in its draft genome sequence (unpublished data). In contrast to SRS16, WDL1 can only efficiently degrade linuron in synergy with other DCA-degrading bacteria (10). The libA amplicons recovered from strains PBL-H6 (100% sequenced), PBL-E5 (97% sequenced), and PBL-H3 (100% sequenced) showed 100% nucleotide identity with libA of SRS16.
Table 2.
Distribution of selected Variovorax sp. SRS16 degradation genes in other relevant bacterial strainsa
| Organism | Degradation capacity |
PCR screening |
||||
|---|---|---|---|---|---|---|
| Linuron | DCA | N,O-DMHA | dcaQ | ccdC | libA | |
| Variovorax sp. strain SRS16 | + | + | – | + | + | + |
| Variovorax sp. strain WDL1 | + | + | – | + | – | – |
| Variovorax sp. strain PBL-H6 | + | + | – | + | + | + |
| Hydrogenophaga sp. strain PBL-H3 | + | + | – | + | – | + |
| Variovorax sp. strain PBL-E5 | + | + | – | + | + | + |
| Variovorax sp. strain PBS-H4 | + | – | – | + | – | – |
| Comamonas testosteroni WDL7 | – | + | – | + | – | – |
| Delftia acidovorans WDL34 | – | + | – | + | – | – |
| Variovorax sp. strain PBD-E5 | – | + | – | + | – | – |
| Variovorax sp. strain PBD-E37 | – | + | – | + | + | – |
| Cupriavidus sp. strain PBS-E1 | – | + | – | – | – | – |
| Afipia sp. strain PBD-E87 | – | + | – | – | – | – |
| Variovorax sp. strain PBD-H1 | – | + | – | + | – | – |
| Hyphomicrobium sp. strain PBN-H4 | – | – | – | – | – | – |
| Variovorax paradoxus DSM66 | – | – | – | – | – | – |
| Hyphomicrobium sp. strain PBN-E9 | – | – | + | – | – | – |
| Hyphomicrobium sulfonivorans WDL6 | – | – | – | – | – | – |
The results are based on PCR analysis. The identity of libA amplicons was confirmed by sequencing.
dcaQ was detected in all linuron-degrading strains and in all DCA-degrading Variovorax strains. Local BLAST search confirmed the presence of a homologous dcaQTA1A2BR gene cluster in the draft genome sequence of WDL1. Some non-Variovorax strains which do not degrade linuron but degrade DCA did not show the presence of dcaQ, whereas dcaQ was detected in Variovorax sp. PBS-H4, which does not degrade DCA. This strain possibly lost the ability to degrade DCA but still contains part of the catabolic pathway.
ccdC was only detected in some of the linuron or DCA-degrading strains. Although strain WDL1 yielded no amplicon with the ccdC primers, a local BLAST search showed the presence of a ccdRCFDE-like gene cluster in strain WDL1. The gene cluster shows a configuration similar to this in SRS16, but the region between ccdF and ccdC differs substantially. In WDL1, this region contains ORF8, but ORF9 and ORF10 are replaced by other unknown genes. Differences in nucleotide sequence at the elongation ends of primers targeting ccdC explain the negative result for PCR detection of ccdC in WDL1.
Our data suggest that in the tested strains, the linuron and DCA degradation genes can differ substantially from those in SRS16, including strains originating from the same environmental sample.
Conclusions.
The linuron catabolic pathway in Variovorax sp. SRS16 is encoded by a patchwork of catabolic gene modules which together allow complete mineralization of linuron. The first gene involved in the pathway encodes a novel phenylurea hydrolase, designated LibA, which converts linuron to DCA, with a high specificity toward linuron. It is the first phenylurea hydrolase reported in Gram-negative bacteria and the first coupled in the same organism with other gene clusters specifying further degradation of DCA. The data suggest that, in accordance with the catabolism of other xenobiotic compounds, the assembly of well-specified existing gene modules in the same organism resulted in the emergence of a novel catabolic pathway. The absence of libA in another linuron-degrading Variovorax strain, i.e., strain WDL1, suggests furthermore that acquisition of linuron degradation by different Variovorax strains involves substantially different linuron hydrolase genes. Current research is focusing on the genes involved in linuron degradation in Variovorax sp. WDL1.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by IWT-Vlaanderen Strategic Basic Research Project 73352, IWT-Vlaanderen Agricultural Research Project LBO 040272, and OT Project OT/10/030. S.R.S. and J.A. were supported by research project MIRESOWA funded by Danish Council for Strategic Research grant 2104-08-0012.
We thank Wen Li for her help and advice in construction of the fosmid library and Caroline Rossier for assistance with the protein purification.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 14 October 2011.
REFERENCES
- 1. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Antoine R., et al. 2005. The periplasmic binding protein of a tripartite tricarboxylate transporter is involved in signal transduction. J. Mol. Biol. 351:799–809 [DOI] [PubMed] [Google Scholar]
- 3. Antoine R., et al. 2003. Overrepresentation of a gene family encoding extracytoplasmic solute receptors in Bordetella. J. Bacteriol. 185:1470–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boshoff H. I. M., Mizrahi V. 1998. Purification, gene cloning, targeted knockout, overexpression, and biochemical characterization of the major pyrazinamidase from Mycobacterium smegmatis. J. Bacteriol. 180:5809–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Breugelmans P., D'Huys P. J., De Mot R., Springael D. 2007. Characterization of novel linuron-mineralizing bacterial consortia enriched from long-term linuron-treated agricultural soils. FEMS Microbiol. Ecol. 62:374–385 [DOI] [PubMed] [Google Scholar]
- 6. Breugelmans P., et al. 2010. Proteomic study of linuron and 3,4-dichloroaniline degradation by Variovorax sp. WDL1: evidence for the involvement of an aniline dioxygenase-related multicomponent protein. Res. Microbiol. 161:208–218 [DOI] [PubMed] [Google Scholar]
- 7. Chain P. S. G., et al. 2006. Burkholderia xenovorans LB400 harbors a multi-replicon, 9.73-Mbp genome shaped for versatility. Proc. Natl. Acad. Sci. U. S. A. 103:15280–15287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chalupowicz L., Barash I., Panijel M., Sessa G., Manulis-Sasson S. 2009. Regulatory interactions between quorum-sensing, auxin, cytokinin, and the Hrp regulon in relation to gall formation and epiphytic fitness of Pantoea agglomerans pv. gypsophilae. Mol. Plant-Microbe Interact. 22:849–856 [DOI] [PubMed] [Google Scholar]
- 9. Dejonghe W. 2003. The importance of genetic and metabolic interactions between bacteria in the degradation of herbicides. Ph.D. thesis University of Gent, Gent, Belgium [Google Scholar]
- 10. Dejonghe W., et al. 2003. Synergistic degradation of linuron by a bacterial consortium and isolation of a single linuron-degrading Variovorax strain. Appl. Environ. Microbiol. 69:1532–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drummond A., et al. 2011. Geneious v5.4.4. Biomatters, Ltd., Auckland, New Zealand [Google Scholar]
- 12. Engelhardt G., Wallnofer P., Plapp R. 1971. Degradation of linuron and some other herbicides and fungicides by a linuron-inducible enzyme obtained from Bacillus sphaericus. Appl. Microbiol. 22:284–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engelhardt G., Wallnofer P., Plapp R. 1973. Purification and properties of an aryl acylamidase of Bacillus sphaericus, catalyzing hydrolysis of various phenylamide herbicides and fungicides. Appl. Microbiol. 26:709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frantz B., Chakrabarty A. M. 1987. Organization and nucleotide-sequence determination of a gene cluster involved in 3-chlorocatechol degradation. Proc. Natl. Acad. Sci. U. S. A. 84:4460–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fukuta Y., Koizumi S., Komeda H., Asano Y. 2010. A new aryl acylamidase from Rhodococcus sp. strain Oct1 acting on omega-lactams: its characterization and gene expression in Escherichia coli. Enzyme Microb. Technol. 46:237–245 [Google Scholar]
- 16. Geng L. Z., et al. 2009. Functional analysis of a putative regulatory gene, tadR, involved in aniline degradation in Delftia tsuruhatensis AD9. Arch. Microbiol. 191:603–614 [DOI] [PubMed] [Google Scholar]
- 17. Gopalakrishna K. N., et al. 2004. Mandelamide hydrolase from Pseudomonas putida: characterization of a new member of the amidase signature family. Biochemistry 43:7725–7735 [DOI] [PubMed] [Google Scholar]
- 18. Gross R., et al. 2008. The missing link: Bordetella petrii is endowed with both the metabolic versatility of environmental bacteria and virulence traits of pathogenic Bordetellae. BMC Genomics 9:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guindon S., Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 20. Han J. I., et al. 2011. Complete genome sequence of the metabolically versatile plant growth-promoting endophyte Variovorax paradoxus S110. J. Bacteriol. 193:1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He Q., Sanford R. A. 2002. Induction characteristics of reductive dehalogenation in the ortho-halophenol-respiring bacterium, Anaeromyxobacter dehalogenans. Biodegradation 13:307–316 [DOI] [PubMed] [Google Scholar]
- 22. Hongpattarakere T., Komeda H., Asano Y. 2005. Purification, characterization, gene cloning and nucleotide sequencing of d-stereospecific amino acid amidase from soil bacterium: Delftia acidovorans. J. Ind. Microbiol. Biotechnol. 32:567–576 [DOI] [PubMed] [Google Scholar]
- 23. Khurana J. L., et al. 2009. Characterization of the phenylurea hydrolases A and B: founding members of a novel amidohydrolase subgroup. Biochem. J. 418:431–441 [DOI] [PubMed] [Google Scholar]
- 24. Ko H. J., et al. 2010. Molecular characterization of a novel bacterial aryl acylamidase belonging to the amidase signature enzyme family. Mol. Cells 29:485–492 [DOI] [PubMed] [Google Scholar]
- 25. Krieg L., Slusarczyk H., Verseck S., Kula M. R. 2005. Identification and characterization of a novel d-amidase gene from Variovorax paradoxus and its expression in Escherichia coli. Appl. Microbiol. Biotechnol. 66:542–550 [DOI] [PubMed] [Google Scholar]
- 26. Lavrov K. V., Zalunin I. A., Kotlova E. K., Yanenko A. S. 2010. A new acylamidase from Rhodococcus erythropolis TA37 can hydrolyze N-substituted amides. Biochemistry (Moscow) 75:1006–1013 [DOI] [PubMed] [Google Scholar]
- 27. Leroy B., et al. 2010. Differential proteomic analysis using isotope-coded protein-labeling strategies: comparison, improvements, and application to simulated microgravity effect on Cupriavidus metallidurans CH34. Proteomics 10:2281–2291 [DOI] [PubMed] [Google Scholar]
- 28. Mayaux J. F., et al. 1991. Purification, cloning, and primary structure of a new enantiomer-selective amidase from a Rhodococcus strain: structural evidence for a conserved genetic coupling with nitrile hydratase. J. Bacteriol. 173:6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muller T. A., Byrde S. A., Werlen C., van der Meer J. R., Kohler H. P. E. 2004. Genetic analysis of phenoxyalkanoic acid degradation in Sphingomonas herbicidovorans MH. Appl. Environ. Microbiol. 70:6066–6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neumann S., Kula M. R. 2002. Gene cloning, overexpression, and biochemical characterization of the peptide amidase from Stenotrophomonas maltophilia. Appl. Microbiol. Biotechnol. 58:772–780 [DOI] [PubMed] [Google Scholar]
- 31. Ogawa N., Miyashita K., Chakrabarty A. M. 2003. Microbial genes and enzymes in the degradation of chlorinated compounds. Chem. Rec. 3:158–171 [DOI] [PubMed] [Google Scholar]
- 32. Ohtaki A., et al. 2010. Structure and characterization of amidase from Rhodococcus sp. N-771: insight into the molecular mechanism of substrate recognition. Biochim. Biophys. Acta Prot. Proteom. 1804:184–192 [DOI] [PubMed] [Google Scholar]
- 33. Parke D. 1993. Positive regulation of phenolic catabolism in Agrobacterium tumefaciens by the pcaQ gene in response to beta-carboxy-cis,cis-muconate. J. Bacteriol. 175:3529–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pease H. L. 1962. Separation and colorimetric determination of monuron and diuron residues. J. Agric. Food Chem. 10:279–281 [Google Scholar]
- 35. Pohlmann A., et al. 2006. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24:1257–1262 [DOI] [PubMed] [Google Scholar]
- 36. Potrawfke T., Armengaud J., Wittich R. M. 2001. Chlorocatechols substituted at positions 4 and 5 are substrates of the broad-spectrum chlorocatechol 1,2-dioxygenase of Pseudomonas chlororaphis RW71. J. Bacteriol. 183:997–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rose T. M., Henikoff J. G., Henikoff S. 2003. CODEHOP (COnsensus-DEgenerate hybrid oligonucleotide primer) PCR primer design. Nucleic Acids Res. 31:3763–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sambrook J., Russell D. W. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 39. Satsuma K. 2010. Mineralization of the herbicide linuron by Variovorax sp. strain RA8 isolated from Japanese river sediment using an ecosystem model (microcosm). Pest Manag. Sci. 66:847–852 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt R., Battaglia V., Scow K., Kane S., Hristova K. R. 2008. Involvement of a novel enzyme, MdpA, in methyl tert-butyl ether degradation in Methylibium petroleiphilum PM1. Appl. Environ. Microbiol. 74:6631–6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shamsuzzaman K. M., Barnsley E. A. 1974. Regulation of naphtalene oxygenase in Pseudomonas. J. Gen. Microbiol. 83:165–170 [DOI] [PubMed] [Google Scholar]
- 42. Shapir N., Cheng G., Sadowsky M. J., Wackett L. P. 2006. Purification and characterization of TrzF: biuret hydrolysis by allophanate hydrolase supports growth. Appl. Environ. Microbiol. 72:2491–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shapir N., Sadowsky M. J., Wackett L. P. 2005. Purification and characterization of allophanate hydrolase (AtzF) from Pseudomonas sp. strain ADP. J. Bacteriol. 187:3731–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shin K. A., Spain J. C. 2009. Pathway and evolutionary implications of diphenylamine biodegradation by Burkholderia sp. strain JS667. Appl. Environ. Microbiol. 75:2694–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shin S., et al. 2003. Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad. J. Biol. Chem. 278:24937–24943 [DOI] [PubMed] [Google Scholar]
- 46. Sorensen S. R., et al. 2005. Elucidating the key member of a linuron-mineralizing bacterial community by PCR and reverse transcription-PCR denaturing gradient gel electrophoresis 16S rRNA gene fingerprinting and cultivation. Appl. Environ. Microbiol. 71:4144–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sorensen S. R., Simonsen A., Aamand J. 2009. Constitutive mineralization of low concentrations of the herbicide linuron by a Variovorax sp. strain. FEMS Microbiol. Lett. 292:291–296 [DOI] [PubMed] [Google Scholar]
- 48. Thiel M., Kaschabek S., Groning J., Mau M., Schlomann M. 2005. Two unusual chlorocatechol catabolic gene clusters in Sphingomonas sp. TFD44. Arch. Microbiol. 183:80–94 [DOI] [PubMed] [Google Scholar]
- 49. Top E. M., Springael D. 2003. The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr. Opin. Biotechnol. 14:262–269 [DOI] [PubMed] [Google Scholar]
- 50. Trefault N., et al. 2004. Genetic organization of the catabolic plasmid pJP4 from Ralstonia eutropha JMP134 (pJP4) reveals mechanisms of adaptation to chloroaromatic pollutants and evolution of specialized chloroaromatic degradation pathways. Environ. Microbiol. 6:655–668 [DOI] [PubMed] [Google Scholar]
- 51. Tropel D., van der Meer J. R. 2004. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol. Mol. Biol. Rev. 68:474–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turnbull G. A., Ousley M., Walker A., Shaw E., Morgan J. A. W. 2001. Degradation of substituted phenylurea herbicides by Arthrobacter globiformis strain D47 and characterization of a plasmid-associated hydrolase gene, puhA. Appl. Environ. Microbiol. 67:2270–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vallenet D., et al. 2006. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 34:53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vandermeer J. R. 1997. Evolution of novel metabolic pathways for the degradation of chloroaromatic compounds. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 71:159–178 [DOI] [PubMed] [Google Scholar]
- 55. Vandermeer J. R., Eggen R. I. L., Zehnder A. J. B., Devos W. M. 1991. Sequence analysis of the Pseudomonas sp. strain P51 tcb gene cluster, which encodes metabolism of chlorinated catechols: evidence for specialization of catechol 1,2-dioxygenases for chlorinated substrates. J. Bacteriol. 173:2425–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vedler E., Koiv V., Heinaru A. 2000. Analysis of the 2,4-dichlorophenoxyacetic acid-degradative plasmid pEST4011 of Achromobacter xylosoxidans subsp denitrificans strain EST4002. Gene 255:281–288 [DOI] [PubMed] [Google Scholar]
- 57. Yamada T., Palm C. J., Brooks B., Kosuge T. 1985. Nucleotide sequences of the Pseudomonas savastanoi indoleacetic acid genes show homology with Agrobacterium tumefaciens T-DNA. Proc. Natl. Acad. Sci. U. S. A. 82:6522–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamamoto K., et al. 1996. Production of R-(−)-ketoprofen from an amide compound by Comamonas acidovorans KPO-2771-4. Appl. Environ. Microbiol. 62:152–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yasuhira K., et al. 2009. X-ray crystallographic analysis of the 6-aminohexanoate cyclic dimer hydrolase: catalytic mechanism and evolution of an enzyme responsible for nylon-6 by-product degradation. J. Biol. Chem. 285:1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yun S. H., et al. 2008. Proteomic analysis of outer membrane proteins from Acinetobacter baumannii DU202 in tetracycline stress condition. J. Microbiol. 46:720–727 [DOI] [PubMed] [Google Scholar]
- 61. Zhang T., Zhang J. L., Liu S. J., Liu Z. P. 2008. A novel and complete gene cluster involved in the degradation of aniline by Delftia sp. AN3. J. Environ. Sci. China 20:717–724 [DOI] [PubMed] [Google Scholar]
- 62. Zhang X., et al. 2011. The R-R-type MYB-like transcription factor, AtMYBL, is involved in promoting leaf senescence and modulates an abiotic stress response in Arabidopsis. Plant Cell Physiol. 52:138–148 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.