

Abstract
Paramyxovirus entry into cells requires the fusion protein (F) and a receptor binding protein (hemagglutinin-neuraminidase [HN], H, or G). The multifunctional HN protein of some paramyxoviruses, besides functioning as the receptor (sialic acid) binding protein (hemagglutinin activity) and the receptor-destroying protein (neuraminidase activity), enhances F activity, presumably by lowering the activation energy required for F to mediate fusion of viral and cellular membranes. Before or upon receptor binding by the HN globular head, F is believed to interact with the HN stalk. Unfortunately, until recently none of the receptor binding protein crystal structures have shown electron density for the stalk domain. Parainfluenza virus 5 (PIV5) HN exists as a noncovalent dimer-of-dimers on the surface of cells, linked by a single disulfide bond in the stalk. Here we present the crystal structure of the PIV5-HN stalk domain at a resolution of 2.65 Å, revealing a four-helix bundle (4HB) with an upper (N-terminal) straight region and a lower (C-terminal) supercoiled part. The hydrophobic core residues are a mix of an 11-mer repeat and a 3- to 4-heptad repeat. To functionally characterize the role of the HN stalk in F interactions and fusion, we designed mutants along the PIV5-HN stalk that are N-glycosylated to physically disrupt F-HN interactions. By extensive study of receptor binding, neuraminidase activity, oligomerization, and fusion-promoting functions of the mutant proteins, we found a correlation between the position of the N-glycosylation mutants on the stalk structure and their neuraminidase activities as well as their abilities to promote fusion.
INTRODUCTION
The Paramyxoviridae are enveloped, negative-strand RNA viruses that infect both humans and animals (24). The family encompasses many clinically and economically important pathogens, including mumps virus, measles virus, parainfluenza viruses 1 to 5 (PIV1 to PIV5), respiratory syncytial virus, Sendai virus, Newcastle disease virus (NDV), Nipah virus, and Hendra virus. To infect cells, the viruses bind to specific receptors, and entry is mediated by fusion of the viral and cellular membranes, releasing the viral genome, in the form of a ribonucleoprotein complex, into the cytoplasm. For nearly all paramyxoviruses, membrane fusion is triggered at the plasma membrane in a receptor-dependent, pH-independent manner. Unlike some enveloped viruses that use a single protein both for binding to cellular receptors and for causing efficient fusion, most paramyxoviruses depend on the concerted actions of two glycoproteins, the attachment protein variously called hemagglutinin-neuraminidase (HN), H, or G and the fusion (F) protein (19, 20, 22, 29, 49). For the paramyxoviruses that use sialic acid as a receptor ligand, the receptor binding protein is known as HN. In addition to fusion promotion, HN also has hemagglutinating and neuraminidase (NA) activities. It is generally thought that binding of HN, H, or G to its ligand on target cells lowers the activation barrier to convert F from a metastable prefusion form to a highly stable postfusion form. This refolding event involves an extensive structural rearrangement and in the process does the work of bringing the viral and target cell membrane together to initiate membrane merger (23). For HN, H, or G to activate fusion, the protein is thought to physically interact with F either before or upon ligand binding; however, the interaction may be weak (5, 19, 22, 29).
Parainfluenza virus 5 (PIV5) HN is a type II membrane protein and has a short N-terminal cytoplasmic tail (residues 1 to 17), a single transmembrane domain (residues 18 to 36), and a large ectodomain (residues 37 to 565). The ectodomain is composed of a globular head that contains a sialic acid binding site that is also the neuraminidase active site and is connected by a helical stalk to the transmembrane domain (21, 47). The atomic structures of the HN, H, or G globular head domains have been determined for PIV5, NDV, Nipah virus, Hendra virus, measles virus, and human parainfluenza virus 3 (hPIV3) (6, 8, 11, 18, 25, 48, 52). The PIV5 atomic structure shows HN as a tetramer consisting of a dimer-of-dimers, and within each dimer, the molecules of HN are linked by a disulfide bond in the stalk region at residue 111 (31, 52).
The globular head of PIV5-HN is related in structure to those of the other paramyxovirus attachment proteins and to other sialidases in general and has a neuraminidase-like fold with a six-β-sheet propeller structure creating the centrally placed active site (52). However, unlike influenza virus NA, which has 4-fold rotational symmetry, the PIV5-HN tetramer exists as a dimer-of-dimers. In the crystal structure, monomers within the dimers are so arranged that the active sites are approximately 90° to each other. Electron microscopy (EM) images show a range of conformations for the HN head (50). The PIV5-HN structure showed that there is minimal change in the subunits upon receptor binding (52).
The stalk region of PIV5-HN is important for forming noncovalent interactions that stabilize the dimer-of-dimers (50, 52). Residues in the transmembrane domain and in the cytoplasmic tail are also possibly involved in such noncovalent associations (31, 32, 35). Biophysical studies have indicated that the stalk is tetrameric and predominantly helical in nature (50). The attachment protein of paramyxoviruses has been implicated in direct interaction with the fusion protein (4, 5, 12, 13, 27, 34, 44, 45), and a variety of mutations in the stalk compromise fusion promotion (4, 5, 10, 15, 27, 28, 39, 44). Interestingly, some of the stalk mutants, which are deficient for fusion, block the attachment protein-fusion protein interaction directly (as assessed by coimmunoprecipitation) (27, 34, 44). Other mutations in the HN stalk in addition to inhibiting fusion also affect receptor binding or neuraminidase activity (4, 10, 28, 44). The fact that modulations in the stalk affect functions within the head domain suggests a structural-functional interplay between the stalk and the globular heads of these attachment proteins.
Direct evidence that F and HN interact has been more difficult to obtain. For human PIV3 (hPIV3), it was found that F and HN cocap in HeLa cells (22), and for NDV and Nipah virus, it has been possible to coimmunoprecipitate F and HN (3, 27, 28, 44). However, coimmunoprecipitation of PIV5 F and HN remains elusive. Another approach toward disrupting an F-HN interaction was taken by Melanson and Iorio (27), who introduced N-linked glycosylation sites into the NDV stalk and observed that many of the mutants abrogated fusion activity. Some of the mutations also caused a reduction in NA activity (27).
Recently, we have determined the atomic structure of the head and a portion of the stalk of NDV-HN by X-ray crystallography (51). It was found that NDV-HN stalk residues 83 to 114 form a four-helix bundle (4HB) that, rather than having a 7-residue repeat (i, i + 3, i + 4), has an 11-residue repeat (i, i + 3, i + 4, i + 4). This structure enabled the mapping of the residues implicated in F protein binding and activation.
To determine the PIV5-HN stalk domain structure in the absence of the head domain, we expressed the PIV5-HN stalk (50) and determined its structure by X-ray crystallography. Similarly to the observed portion of the NDV-HN stalk, the PIV5-HN stalk also forms a 4HB with hydrophobic core residues following an 11-residue repeat in its upper part; however, the hydrophobic core residues switch to a heptad repeat in the lower portion of the PIV5-HN stalk structure. Interestingly, the upper part of the stalk is relatively nonsupercoiled, whereas the lower part of the PIV5-HN stalk 4HB adopts a left-handed superhelical twist, and these structural features are consistent with other 4HB structures based on 11-mer or heptad hydrophobic repeats (17, 42, 43). To probe the structure further and to investigate if PIV5-HN fusion promotion infers a physical interaction with F, we analyzed biological properties of a set of point mutants with N-linked carbohydrate chains introduced into the PIV5-HN stalk sequence. We found that the ability of an N-glycosylation mutant to block fusion had a strong correlation with the position of the glycosylation in the 4HB helix.
MATERIALS AND METHODS
Cells and antibodies.
Vero cells and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). BHK-21F cells were grown in DMEM containing 10% FBS and 10% tryptose phosphate broth. HeLa-CD4-LTR-βGal (AIDS Research and Reference Reagent Program) cells were grown in DMEM supplemented with 10% FBS, 200 μg/ml G418, 100 μg/ml hygromycin B, and 20 mM HEPES (pH 7.4). BSR-T7/5 cells were grown in DMEM containing 10% FBS, with 500 μg/ml G418 added every third passage. Insect Hi5 cells were maintained in Express 5 serum-free medium (Gibco) supplemented with 10% GlutaMax (Gibco), while insect SF9 cell lines were maintained in SF900 II medium containing 10% FBS. Antibodies specific for PIV5-HN included monoclonal antibody (MAb) HN-1b and HN-5a ascites fluids and MAb HN-4b hybridoma supernatant (40) and polyclonal antibody (PAb) R471 serum, raised in rabbits against the purified HN ectodomain expressed by a recombinant baculovirus in insect cells.
Cloning and mutagenesis.
pCAGGS-HN and pCAGGS-F expression constructs harboring the PIV5 (W3A) F and HN genes were used as described previously (38). Mutants in pCAGGS-HN were constructed using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. For mutagenesis, a short segment of pCAGGS-HN, flanked by NheI and SacI restriction sites containing the relevant portion of HN, was subcloned into the pGEM3-5zf(+) vector. This template was used to create mutations bearing the N-glycosylation motif N-X-S/T-X, where X is any amino acid other than proline. Subsequently, the NheI/SacI fragment carrying the desired mutation was cloned back into pCAGGS-HN. The mutants N58, N60, N66, N67, N68, N77, N90, N91, and N102 were named according to the N-glycosylated residue (see Fig. 3C). The nucleotide sequence of the entire HN open reading frame for each of the generated mutants was verified using an Applied Biosystems 3100-Avant automated DNA sequencer (Life Technologies Corp., Carlsbad, CA). pT7-luciferase was obtained from Promega Corp. (Madison, WI). The creation of the PIV5-HN (stalk) domain construct was described previously (50).
Fig. 3.

Design of mutants. (A) Schematic diagram of the PIV5 HN protein and the position of sites introduced for N-linked glycosylation (arrowheads) along the stalk region. CT, cytoplasmic tail, TM, transmembrane domain. (B) Positions of Cα atom of each residue on the PIV5-HN stalk structure that has an additional glycosylation added through mutagenesis (red). The mutants are named according to the residue that harbors the extra carbohydrate moiety. (C) Point mutations introduced in the HN stalk to create the N-linked glycosylation motif Asp-X-Ser/Thr-X, where X is any residue but proline. The point mutations are indicated in bold within the motif.
Protein expression and purification.
The PIV5-HN stalk construct was expressed in Hi5 insect cell lines. The cell cultures were infected (multiplicity of infection [MOI] of 2) with recombinant baculovirus stocks containing the PIV5-HN stalk and harvested 72 h postinfection. Proteins were purified from the supernatants by affinity chromatography using nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen). Ni-NTA columns were washed with 10 mM and 50 mM imidazole to eliminate nonspecific binding. Proteins were eluted in fractions using 250 mM imidazole and were >90% pure by SDS-PAGE and Coomassie brilliant blue staining analysis. The S and His tags were cleaved from the protein using recombinant enterokinase (rEK; Novagen). The sample was dialyzed into rEK cleavage buffer (20 mM Tris, pH 7.4, 50 mM NaCl, and 2 mM CaCl2), and the cleavage reaction was carried out using 0.20 U of EK per 10 μg of protein at a protein concentration of 0.4 mg/ml at room temperature for 14 h. Cleavage was >90% complete by SDS-PAGE analysis. The EK, tags, and remaining uncleaved protein were removed by capture using EKapture agarose (Novagen) followed by Ni-NTA agarose (in the presence of 10 mM imidazole), leaving purified stalk protein in solution. Three residues (SPS) that are not part of the wild-type (wt) HN sequence remain at the N terminus following rEK cleavage, and density for these residues is visible in two of the four chains of the structure.
Crystallization.
The cleaved and purified PIV5-HN stalk (residues 56 to 117) was buffer exchanged into 10 mM Tris, pH 7.4, 50 mM NaCl and concentrated to 10.2 mg/ml. Initial crystals were obtained with the Index crystallization screen (Hampton), by the hanging drop vapor diffusion method, using the Mosquito (TTP LabTech) at the High-Throughput Analysis Lab (Northwestern University, Evanston, IL). After optimization, crystals were grown at room temperature by the sitting drop vapor diffusion method over a reservoir solution containing 4.5% (vol/vol) Tascimate (pH 7.0), 0.09 M HEPES (pH 7.0), and 9% (wt/vol) polyethylene glycol (PEG) MME 5000. Drops consisted of protein and precipitant at a 2:1 ratio. The crystals were flash frozen in liquid nitrogen using 4% (vol/vol) Tascimate (pH 7.0), 0.08 M HEPES (pH 7.0), 8% (wt/vol) PEG MME 5000, and 20% glycerol as the cryoprotectant solution.
Data collection, structure determination, and refinement.
A native data set was collected at the Life Sciences Collaborative Access Team (LS-CAT) beam line at the Argonne National Laboratory Advanced Photon Source and processed to 2.65 Å using HKL2000 (33). The 4HB domain of the NDV-HN ectodomain structure (Protein Data Bank identification [PDB ID]: 3T1E) was used as the search model in molecular replacement to determine initial phases in the P3221 space group (51). A single copy of the 4HB was found in the asymmetric unit, although it was initially fitted in an incorrect helical frame. The PHENIX AutoBuild Wizard software successfully built a portion of the model in the correct helical frame (46). Subsequent model building, structure refinement, and validation were performed with Coot (14), PHENIX Refine (2), and MolProbity (7), respectively. Use of TLS parameters (one chain per group), individual B-factors, and Ramachandran and rotamer restraints during late stages of refinement helped to lower the Rfree values; however, the use of noncrystallographic symmetry restraints increased Rfree values. The data collection and final refinement statistics are shown in Table 1.
Table 1.
Crystallographic data and refinement statistics: HN stalk
| Parameter | Valuea |
|---|---|
| Data collection | |
| Source | Advanced Photon Source |
| Wavelength (Å) | 0.97959 |
| Space group | P3221 |
| Unit-cell parameters | |
| a (Å) | 101.276 |
| b (Å) | 101.276 |
| c (Å) | 85.713 |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 120 |
| Resolution range (Å) | 29.236–2.651 (2.855–2.651) |
| Rmerge (%) | 5.7 (38.2) |
| I/σ (I) | 23.23 (5.2) |
| Completeness (%) | 99.0 (100) |
| Redundancy | 5.4 (5.6) |
| Refinement | |
| Resolution (Å) | 29.24–2.65 |
| No. of used reflections (work/free) | 14,173/757 |
| R factor/Rfree(%) | 20.04/23.71 |
| No. of residues/atoms | 199/1,477 |
| Average B-factors (Å2) | 79.7 |
| RMSDs in bond lengths (Å) | 0.008 |
| RMSDs in bond angles (°) | 1.116 |
| Ramachandran plot statistics | |
| Residues in preferred regions (%) | 95.8 |
| Residues in allowed regions (%) | 4.2 |
Data in parentheses indicate values for outer shell.
Expression of HN and F glycoproteins in mammalian cells.
PIV5-F and HN proteins were expressed from the pCAGGS-F and pCAGGS-HN constructs in Vero, BHK-21F, 293T, and HeLa-CD4-LTR-βGal cells by transient transfection using the Lipofectamine Plus transfection reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Transfections were done in Optimem medium, and the mixtures were incubated for 5 h at 37°C, following which DMEM containing 2% FBS was added and the samples were incubated for an additional 18 h at 37°C. In the mixed oligomer experiments, DNA amounts were adjusted during transfection to result in different wt HN/mutant HN expression ratios. The ratios of wt to mutant used in various experiments were 1:0, 3:1, 1:1, 1:3, and 0:1. The total HN DNA (wt and mutant) in each transfection was always 1 μg.
Immunoprecipitation and SDS-PAGE.
Transfected HeLa-CD4-LTR-βGal cells in 6-well dishes were starved in DMEM deficient in cysteine and methionine for 30 min, followed by labeling with 50 to 75 μCi 35S-label in the same medium for 30 min. The cells were then incubated for 90 min with complete DMEM containing 10% FBS. The cells were subsequently lysed in cold radioimmunoprecipitation assay (RIPA) buffer (37) containing protease inhibitors, 50 mM iodoacetamide, and 2 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was then clarified in a Beckman TLX Ultracentrifuge in a Beckman TLA 120.2 rotor at 55,000 rpm for 10 min at 4°C. Clarified lysates were incubated with a suitable antibody overnight at 4°C, following which protein A-Sepharose beads were added and the samples were further incubated at 4°C for 30 min. Antibody-antigen complexes were washed three times with RIPA buffer (37) containing 0.3 M NaCl, twice with RIPA buffer containing 0.15 M NaCl, and once with 50 mM Tris-HCl, pH 7.4, 0.25 mM EDTA, and 0.15 M NaCl. The proteins were eluted from the beads by boiling for 2 min in protein lysis buffer containing 15% dithiothreitol and separated on a 10% acrylamide gel. Radioactivity was detected using a Fuji FLA-5100 image reader with Multi Gauge v3.0 software (Fuji Medical Systems, Stamford, CT).
Flow cytometry.
To quantify cell surface expression of PIV5-HN and its mutants, 293T cells were seeded onto BD PureCoat amine 6-well dishes (Becton Dickinson, Franklin Lakes, NJ). The cells were transfected as described above with plasmids encoding the wt HN or the HN mutant proteins. At 18 h posttransfection, the monolayers were washed with phosphate-buffered saline (PBS) containing 0.02% sodium azide to prevent internalization of HN. Nonspecific antibody binding was blocked by incubating the monolayers in PBS containing 1% bovine serum albumin (BSA) and 0.02% sodium azide. Plates were then moved to 4°C and incubated with a mixture of HN monoclonal antibodies (HN-1b, HN-4b, and HN-5a) at a dilution of 1:200 each or with a 1:200 dilution of an HN-specific polyclonal antibody (R471) in PBS containing 1% BSA. The monolayers were washed extensively with PBS to remove unbound antibody and incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG (1:200 dilution) or fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1:200 dilution) (Invitrogen, Carlsbad, CA). Cells were washed extensively again with PBS and resuspended in PBS containing 0.5% formaldehyde. The mean fluorescence intensity (MFI) of 10,000 cells was recorded for each sample using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Hemadsorption (HAd) assay.
Monolayers of 293T cells were transfected with pCAGGS-HN and HN mutants (1 μg plasmid each) as described above. Eighteen hours posttransfection, the cells were washed gently with ice-cold PBS, containing calcium and magnesium (PBS+). This was followed by incubation for 2 h with 1% chicken erythrocytes (RBCs) in PBS+ at 4°C to allow receptor binding but not fusion. Subsequently, the monolayers were washed well five times with ice-cold PBS+ to remove unbound RBCs. The remaining bound RBCs were lysed in 0.5 ml ice-cold distilled water and rocked for 2 h at 4°C. The lysate was spun down to remove debris, and the absorbance of the supernatant was read at 540 nm on a Beckman Coulter DU 730 Life Sciences UV/Vis spectrophotometer (Beckman Coulter, Brea, CA).
NA activity assay.
HeLa-CD4-LTR-βGal cells were transfected with wt HN and HN mutants (1 μg plasmid each) as described above using the Lipofectamine Plus system (Invitrogen). Cells were detached from the plates using 500-μl/well 530 μM EDTA in PBS. The cells were then pelleted by centrifugation and resuspended in PBS+, following which they were pelleted for 5 min at 800 rpm at 4°C. The cell pellets were resuspended in 100 μl of 125 mM sodium acetate buffer (pH 4.75) containing 6.25 mM CaCl2. Twenty-five microliters of 5 mM 4-methylumbelliferyl-N-acetyl-α-d-neuraminic acid (Sigma-Aldrich, St. Louis, MO) was added as the neuraminidase substrate. The reaction was allowed to proceed for 30 min at 37°C with occasional mixing. Seventy-five microliters of 20 mM sodium carbonate buffer (pH 10.4) was added to stop the reaction (9). Cells were pelleted at 14,000 rpm, and 180 μl of the supernatant was transferred to a 96-well plate. Fluorescence of the cleaved substrate was measured at excitation and emission wavelengths of 356 and 450 nm, respectively, using a Spectramax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Syncytium formation.
BHK-21F cells were transfected as described above using 1 μg each of pCAGGS-F and pCAGGS-HN or the HN glycosylation mutant plasmids. Eighteen hours posttransfection, the cells were washed with PBS and fixed and stained using a Hema3 staining protocol (Fisher Scientific, Pittsburgh, PA) according to the manufacturer's instructions. The monolayers were photographed using an inverted phase-contrast microscope (Diaphot; Nikon, Melville, NY) connected to a digital camera (DCS 760; Kodak, Rochester, NY). For analyzing fusion activity of the mixed oligomers, DNA ratios of wt HN to mutant HN used in transfections were adjusted as described above and syncytium formation was examined.
Luciferase reporter assay.
To quantitate the fusion observed in the syncytium assay, Vero cell monolayers were transfected with 1 μg each of the pCAGGS-F, pCAGGS-HN, or HN stalk glycosylation mutants and pT7-luciferase, a plasmid that expresses firefly luciferase under T7 polymerase control. BSR-T7/5 cells, expressing T7 RNA polymerase, were overlaid on the Vero cell monolayer at 15 h following transfection and incubated further for 6 to 7 h at 37°C. Reporter lysis buffer (2×; Promega) was used to lyse the cells. Subsequently, the cell lysates were frozen at −80°C overnight to facilitate lysis and release of luciferase. On the following day, cell debris was pelleted from the samples by centrifugation and 150 μl of the cleared lysates was then added to a 96-well dish along with 150 μl of the luciferase assay substrate (Promega). The luciferase activity in relative light units (RLU) was then determined using a SpectraMax M5 plate reader (Molecular Devices). For the luciferase assays examining mixed oligomer titrations, DNA ratios of wt HN to mutant HN used in transfections were adjusted as described above.
Sucrose density gradient centrifugation.
HeLa-CD4-LTR-βGal cells transfected with pCAGGS-HN or HN mutants were labeled with 35S-label as described above. Cells were lysed on ice with 750 μl of MNT buffer (20 mM morpholinoethanesulfonic acid, 30 mM Tris, 100 mM NaCl; pH 5.0) containing 1% Triton X-100 and protease inhibitors. The lysates were clarified as described above, and the supernatant was layered on a 7.5 to 22.5% (wt/vol) sucrose gradient made in MNT buffer containing 0.1% Triton X-100. This gradient was overlaid on a 60% sucrose cushion. The 12-ml gradients were created in 14- by 89-mm Beckman Ultraclear tubes (Beckman, Palo Alto, CA), and after overlaying the cleared cell lysate, the gradients were centrifuged in an SW41 rotor at 37,000 rpm for 19 h at 20°C using an Optima L-80 XP Ultracentrifuge (Beckman Coulter, Brea, CA). Twenty-four 0.5-ml fractions were collected for each sample using an Auto Densi-Flow fraction collector (LabConco, Kansas City, MO). RIPA buffer (2×) with protease inhibitors, iodoacetamide (mM), and PMSF was added to alternate fractions, starting from the top. The proteins were immunoprecipitated from these samples as described above using a mixture of HN monoclonal antibodies and analyzed by SDS-PAGE using 10% acrylamide gels. Imaging of the radioactive bands was done as described above.
Protein structure accession number.
Regarding data deposition, the atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3TSI).
RESULTS
Structure of the PIV5-HN stalk domain.
The PIV5-HN stalk protein was obtained by purification of a soluble stalk construct (residues 56 to 117) expressed using Hi5 cells infected with a recombinant baculovirus. The X-ray crystal structure (Fig. 1A) was solved to 2.65 Å by molecular replacement with the recently reported and closely related NDV-HN stalk structure (51) in the P3221 space group (root mean square deviation [RMSD] = 1.119 over 394 amino acid chain backbone atoms). Electron density was observable for residues 60 to 101 for a single four-helix bundle located in the asymmetric unit (Fig. 1A and B). Though density was observed for residues 56 to 59 and 102 to 108 in at least one out of the four chains, these end residues did not appear to form a distinct 4HB. However, the presence of nonnative residues (enterokinase cleavage artifact) and a significant crystal contact at the N terminus may have disrupted the 4HB in that region. An example of the electron density obtained is shown for a region of the 4HB (Fig. 1D). In the region corresponding to the observed portion of the NDV-HN stalk structure, the PIV5-HN stalk domain similarly contains an 11-mer repeat in which the hydrophobic core residues are located at the “a,” “d,” and “h” positions of the repeating motif (Fig. 1A and B and 2B) (51). However, in the PIV5-HN stalk crystal structure, we observe additional N-terminal residues that form a heptad repeat, with core residues at “a” and “d” positions between amino acid residues 58 and 79.
Fig. 1.

Structure of PIV5-HN stalk domain. (A) Crystal structure of the parainfluenza virus 5 HN stalk domain. Residues in “a,” “d,” and “h” positions of the hydrophobic core of the 4-helix bundle (yellow) are numbered. (B) Top view of the PIV5-HN stalk domain showing the side chain packing of “a,” “d,” and “h” residues (yellow). (C) Portion of the PIV5-HN stalk structure showing a distortion of the helix near serine 79 following which the 4HB shows a slight superhelical twist. The proline at position 84 is also highlighted. (D) Representative electron density (2Fo-Fc map) showing the 4HB structure of the PIV5-HN stalk.
Fig. 2.

Comparison of PIV5-HN and NDV-HN stalk structures. (A) PIV5-HN stalk 4HB in comparison with NDV-HN stalk 4HB. Structural alignment with backbone atoms of PIV5-HN and NDV-HN stalk crystal structures shows a root mean square deviation (RMSD) value of 1.119 over 394 atoms. (B) Sequence alignment showing heptad repeat of the PIV5-HN and NDV-HN stalks with the “a” and “d” positions (red) followed by the 11-mer repeat with the “a,” “d,” and “h” positions (black). Residue 79 is an “a/d” residue. The hydrophobic residues in the heptad repeat are highlighted in olive while those in the 11-mer repeat region are highlighted in yellow.
The 11-mer repeat in the upper part (N-terminal) of the PIV5-HN stalk shifts to a heptad in the lower part (C-terminal), with the apparent transition point occurring at residue 79, which can be incorporated into both the heptad repeat and the 11-mer repeat as an “a” or “d” residue, respectively (Fig. 2B). Interestingly, a strong superhelical twist is observable in the region below residue 79, while there is no obvious supercoiling in the region above this residue (Fig. 1A and C). Conventionally, a left-handed superhelical twist is expected in 4HBs with heptad repeats because the number of residues per turn of helix is greater than the hydrophobic periodicity (17). On the other hand, natural (43) and designed (17, 42) 4HBs with 11-mer repeats have shown that the altered hydrophobic periodicity can give rise to relatively straight segments or even a slightly right-handed superhelix. The PIV5-HN stalk structure is consistent with these previous studies, with the hydrophobic periodicity determining the overall superhelical twist to the 4HB in these two distinct regions. Additional structural features could also facilitate this superhelical transition at the “S79 junction.” First, a helix-disrupting proline (P84) is accommodated in the “i” position of the turn of helix immediately above S79 (Fig. 1C). Second, as a polar residue in the hydrophobic core (“d” position), Ser 79 itself could contribute to the observed shift in superhelical twist (Fig. 1C).
Creation of N-glycosylation site mutants.
To probe the structure of the stalk domain in the multifunctional PIV5-HN protein, a set of point mutants was created, introducing sites for N-glycosylations along the length of the stalk on both the outside and inside of the 4HB (Fig. 3). We changed only a single residue in each mutant to create the N-glycosylation motif Asn-X-Ser/Thr-X (X is any amino acid except for proline) to minimize altering the wt HN sequence (Fig. 3C). However, this restraint reduced our choices of residues available for mutagenesis. Mutants were named according to the residue that was N-glycosylated. The large and bulky N-glycosyl carbohydrate moiety is ideally suited to cause steric hindrance of potential F-HN interactions or of oligomerization of HN. N-glycosylations of N60, N66, N67, N77, and N102 mutants are predicted to be on the outside of a 4HB and carbohydrate chains on N58 and N68 mutants are predicted to be partially or completely in the inner hydrophobic core of the 4HB. Carbohydrate chains on N90 and N91 mutants appear to be neither pointing directly into the core nor completely solvent exposed (Fig. 3B).
N-glycosylation and surface expression of HN stalk mutants.
The synthesis of the N-glycosylation mutants was monitored in HeLa-CD4-LTR-βGal cells (Fig. 4A). 35S-radiolabeled proteins were immunoprecipitated from transfected cell lysates and analyzed by SDS-PAGE. All the mutant proteins were expressed, but there was variability in the expression level among the mutants. The decrease in the electrophoretic mobility of the HN mutants compared to the wt protein indicates that the mutant HN proteins are glycosylated. Mutants N90, N91, and N102 showed a slightly smaller shift in mobility than did the other HN mutants, suggesting that their carbohydrate chains may be processed differently. Glycosylation of the HN mutants was further confirmed by analyzing mutant HN proteins that had been metabolically labeled in the presence of the N-linked glycosylation inhibitor tunicamycin. The shift in protein mobility seen in Fig. 4A was not observed (data not shown).
Fig. 4.

Expression of HN N-glycosylation mutants. (A) Immunoprecipitation of wt HN or N-glycosylation mutants expressed in HeLa-CD4-LTR-βGal cells. The cells were labeled with Tran35S-label for 30 min and then incubated in medium (chased) for 90 min. Cells were then lysed in immunoprecipitation buffer, and proteins were immunoprecipitated using a mixture of HN monoclonal antibodies. Polypeptides were analyzed by SDS-PAGE. Numbers at left are molecular masses in kilodaltons. (B) Detection of proteins at the cell surface of 293T cells transfected with wt HN or HN N-glycosylation mutants using flow cytometry. Surface proteins were detected using a mixture of HN monoclonal antibodies or anti-HN polyclonal antibody R471. The mean fluorescence intensity (MFI) is shown as a percentage of wt protein levels. Results are from four independent experiments.
All the mutant HN proteins exhibited robust cell surface expression levels ranging between 80 and 150% of wt HN protein when measured by flow cytometry (Fig. 4B), indicating that extra glycosylation on HN was not deleterious for cell surface expression. HN mutant N58 showed significantly elevated cell surface levels compared to wt HN, but it is not known whether this increased surface expression is due to increased surface transport or decreased internalization of HN. The difference in the amount of HN observed in a pulse-label experiment and the amount of HN that accumulates at the cell surface, as measured by flow cytometry, can be explained by the different ways in which the experiments were done.
Receptor binding ability of the HN mutants.
A hemadsorption assay was performed to determine if N-linked carbohydrate chains in the stalk mutants affect receptor binding properties of HN. 293T cells transfected with wt HN or mutant HNs were incubated with chicken erythrocytes at 4°C, 18 h posttransfection. Nonspecifically bound RBCs were washed off, remaining bound RBCs were lysed, and released hemoglobin was measured at 540 nm. The mutants N90, N91, and N102 located near the top of the stalk had a reduced receptor binding activity (Fig. 5A). However, when the percent values of hemadsorption were normalized with that for surface expression, none of the mutants were significantly deficient in receptor binding.
Fig. 5.

Functional analyses of the HN N-glycosylation mutants. (A) Receptor binding activity of the HN N-glycosylation mutants. HN wt or N-glycosylation mutants were transfected into 293T cells, and hemadsorption was measured by determining the amount of chicken red blood cell binding. Cells were washed in PBS and then lysed in H2O. The hemoglobin absorbance at 540 nm was expressed as a percentage of wt protein hemadsorption. The receptor binding level was also normalized to the surface expression level (numbers beneath histogram bars). Abs, absorption wavelength; S.E., surface expression. Results are from four independent experiments. (B) Neuraminidase (NA) activities of wt and mutant HN proteins were determined in transfected HeLa-CD4-LTR-βGal cells. NA activity of these proteins was calculated from the readout of cleaved MU-NANA fluorogenic product at an emission wavelength of 450 nm. This was expressed as a percentage of wt HN NA activity. Neuraminidase activity, normalized to surface expression level, is also shown as numbers beneath histogram bars. Em, emission wavelength. (C) Luciferase reporter assay of cell-cell fusion. Vero cells transfected with F, HN, and luciferase under the control of a T7 promoter were overlaid with BSR-T7 cells 15 h posttransfection. After 7 h, the cells were lysed and the extent of cell-cell fusion was obtained as relative luciferase units (RLUs). Fusion activity of the mutants was expressed as a percentage of wt HN fusion. Luc, pT7 luciferase transfected alone. (D) Representative micrographs of syncytia, showing cell-cell fusion in BHK-21 cells transfected with F and wt HN or HN N-glycosylation mutants at 18 h posttransfection.
N-linked carbohydrate chains near the top of the stalk affect neuraminidase activity of HN.
The neuraminidase activities of the N-linked glycosylation stalk mutants were tested using an assay involving cleavage of the neuraminidase substrate MU-NANA into a fluorogenic product. The NA activities of the mutants were determined as a percentage of wt and normalized to surface expression levels of the mutants (Fig. 5B). Interestingly, whereas the mutants near the lower and middle parts of the stalk had wt levels of NA activity, the mutants near the top part of the stalk, N77, N90, and especially N91 and N102, were reduced to 45 to 65% of wt neuraminidase activity, despite being relatively unaffected in receptor binding (Fig. 5A).
HN stalk carbohydrate chains block fusion promotion.
To investigate the ability of HN stalk glycosylation mutants to promote fusion when coexpressed with F protein, the HN mutants were cotransfected with F into Vero cells. Fusion was quantified using a luciferase reporter assay and compared to fusion mediated by wt HN (Fig. 5C). None of the N-glycosylation mutants promoted fusion at any detectable level, except mutant N102, which is located near the top of the stalk. The inability of the mutants to promote fusion was also reflected in their failure to form syncytia in BHK-21F cells beyond that of the F-only-transfected control, which shows low levels of fusion (Fig. 5D).
Oligomerization of the HN glycosylation mutants.
Purified PIV5-HN ectodomain (residues 37 to 565) elutes from a gel filtration column as a tetramer, whereas the purified HN globular head elutes as a monomer (52). A disulfide bond at C111 in the stalk causes the molecule to form a covalently linked dimer (31). The HN tetramer consists of a dimer of disulfide-linked dimers, and tetramerization is stabilized by the stalk domain (52).
To test if the HN N-glycosylation mutants affect oligomerization, six representative mutants were chosen for analysis. N58 is a hydrophobic residue in the N-terminal region of the stalk (Fig. 3B). N67 is located in the polar half of a strongly amphipathic region of the stalk helices, about midway along the length of the stalk. N66 and N68 are also located in this middle region; however, the point mutation to generate the Asp-X-Thr/Ser-X motif is located at residue 68 for both mutants (Fig. 3C). N90 and N91 are in the upper portion of the stalk with their side chains near the hydrophobic-polar boundary of the helix (Fig. 3B).
To examine their oligomeric form, wt HN and the HN mutants were subjected to sucrose density gradient ultracentrifugation (31, 32). The sedimentation pattern of HN was compared to the well-characterized pattern of soluble influenza virus neuraminidase monomers, dimers, and tetramers (36). The bulk of wt HN sedimented in fractions 15 and 17 represents the sedimentation of the tetramer (Fig. 6A). Some wt monomeric HN was observed in fractions 7 and 9, and this is possibly due to protein overexpression in transfected cells, as it is not observed in PIV5-infected cells (32). Mutant N58 sedimented as a monomer (fractions 7 to 9) and dimer (fractions 11 to 13), and only a small amount of HN N58 mutant sedimented in the dimer-of-dimer fractions 15 to 17 (Fig. 6B). Mutants N66, N67, N90, and N91 (Fig. 6C, D, and F and data not shown) showed a sedimentation profile very similar to that of wt HN, indicating that these mutants do not disrupt the tetrameric integrity of the molecule by introducing large N-glycosylation carbohydrate moieties in the stalk. However, the N68 mutant protein (Fig. 6E) sedimented as both a dimer and a tetramer, indicating that either a significant fraction of the tetrameric protein fails to form a tetramer or the tetramer is destabilized and converts to covalently linked dimers in this mutant. Slight differences in the sedimentation of the HN tetramer were observed, and these could be due to altered sedimentation because of the addition of the carbohydrate chains. However, we believe that the differences can be attributed to technical aspects of gradient fractionation.
Fig. 6.

Oligomeric form of wt HN and HN N-glycosylation mutants. Sucrose density gradient ultracentrifugation was used to analyze oligomerization patterns of wt HN and the mutants N58, N66, N67, N68, and N90 expressed in transfected HeLa-CD4-LTR-βGal cells. Eighteen hours posttransfection, Tran35S-labeled proteins were extracted using Triton X-100 and the lysates were subjected to ultracentrifugation on a 7.5% to 22.5% (wt/vol) sucrose density gradient in a SW41 rotor at 37,000 rpm for 19 h. Fractions were collected from the top of the gradient, alternate fractions were immunoprecipitated using an HN monoclonal antibody mixture, and polypeptides were analyzed by SDS-PAGE under nonreducing conditions. Fractions are labeled numerically from top to bottom of the gradient. M, monomer; D, dimer; T, tetramer. HN-S-S-HN, disulfide-linked HN dimer. The figure shows representative gel images of wt-HN (A), HN-N58 (B), HN-N66 (C), HN-N67 (D), HN-N68 (E), and HN-N90 (F).
Mixed oligomers of wt HN and mutant HN behave differently depending on mutation position in the stalk.
Mutants N90, N91, and N102, which are in the upper part of the stalk, had lowered neuraminidase activity compared to mutants N58, N60, N66, N67, and N68, which map to the lower part of the HN stalk (Fig. 5B). Although all the other mutants were blocked in fusion, N102 did not have a fusion block. Thus, fusion properties of the HN stalk mutants were examined further in mixed oligomer experiments to investigate if the mutants had a dominant negative fusion phenotype. Different ratios of wt and mutant DNA were mixed together and transfected into BHK-21 cells together with wt F. For these experiments, wt/mutant ratios of 3:1, 1:1, 1:3, and 0:1 were used. At 18 h posttransfection, syncytia were photographed. As shown in Fig. 7A, the lower mutants showed medium levels of fusion activity at transfection ratios of 3:1 wt to mutant (Fig. 7A, top row). However, at a 1:1 ratio, fusion activation was severely affected for these mutants (Fig. 7A, second row). In contrast, mutants N90 and N91, especially the latter, showed levels of fusion close to wt at a 1:1 mix, and N102 has wt fusion activity. The quantitative luciferase assay for fusion mirrored syncytium formation (Fig. 7B). Note that N90 and N91 showed activity very different from that of the HN mutants located lower in the stalk. To verify that the proteins that were expressed in cells in the mixed oligomer experiments reflected the ratios of transfected DNA, the differential migration properties of wt and N-glycosylated mutant N91 were examined. As shown in Fig. 8 in the lane containing 0.75 μg wt plus 0.25 μg N91 (lane 2), the majority of the protein was wt, as opposed to the reverse ratio in lane 4, where N91 predominated. In lane 3, where there was an equal proportion of wt and mutant transfected, the broad HN band reflected this ratio. Thus, HN that was synthesized emulated the input transfected DNA ratio even though the exact composition of oligomers could not be determined from these data.
Fig. 7.

Fusion activity of HN N-glycosylation mutants in mixed oligomers. (A) Representative micrographs of syncytia, demonstrating cell-cell fusion in BHK-21 cells transfected with F and titrated mixtures of wt HN and mutant HN constructs. One microgram of F DNA was cotransfected with a 1-μg mixture of wt and mutant HN DNAs at different ratios. Mutants are arranged from left to right according to their position in the PIV5-HN stalk, with the mutant toward the bottom of the stalk on the left and the mutant toward the top of the stalk on the right. (B) Quantitative luciferase reporter assay of cell-cell fusion. Fusion activity of HN N-glycosylation mutants was characterized by forming different combinations of tetramers, which would include wt and mutant monomers in various ratios. These different DNA wt/mutant HN ratios, F, and luciferase under the control of the T7 promoter were expressed in Vero cells, which were overlaid with BSR-T7 cells 15 h posttransfection. Seven hours postoverlay, the cells were lysed and luciferase activity (expressed in RLU) was determined. Fusion-inducing capabilities of the HN mutants in mixed oligomers are shown, with wt fusion considered 100%.
Fig. 8.
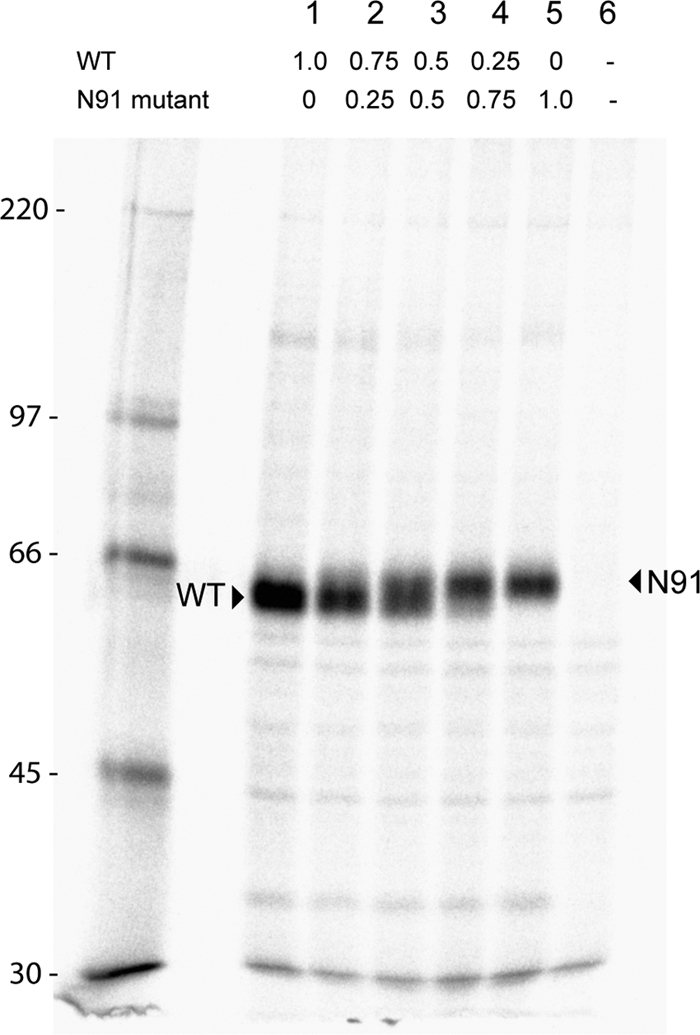
Expression of proteins in mixed oligomer experiments. Immunoprecipitations of mixed oligomer proteins of HN wt and the N91 mutant were carried out from transfected HeLa cell lysates. Transfected cells were pulsed with Tran35S-label for 30 min and then chased with complete medium for 90 min. The cells were lysed, and immune complexes were detected using an HN-specific monoclonal antibody mix. The proteins were resolved on an 8% SDS-PAGE gel. The arrowheads indicate the shift in size between the wt and the N91 mutant, the latter carrying an N-glycosyl carbohydrate chain. Numbers above the gel indicate the μg of DNA of wt and N91 transfected. Numbers at left indicate molecular masses in kilodaltons.
DISCUSSION
Paramyxovirus glycoproteins coordinate their functions to initiate the fusion process. Receptor binding presumably causes a conformational change in the attachment protein transmitting a signal to the fusion protein, allowing its refolding and resulting in membrane fusion. Though direct interaction between the fusion protein and attachment protein has been characterized through coimmunoprecipitation for some paramyxoviruses (4, 12, 26, 27, 28, 44), results for PIV5 have been variable, suggesting a comparatively weaker or more transient interaction. Based on these and other studies, the stalk domain of the receptor binding proteins is believed to be critical for F interaction and F activation. Recently, the atomic structure of the NDV-HN ectodomain, including a portion of the stalk domain, was determined (51). Here we report the crystal structure of the parainfluenza virus 5 HN stalk domain at a resolution of 2.65 Å and show that, like NDV, the stalk forms a 4HB and that this structure is maintained even in the absence of residues 1 to 55 and the globular head. Additionally, to correlate the function of the stalk domain with its crystal structure, we designed a set of mutants that introduce N-glycosylations along the PIV5-HN stalk (Fig. 3B). Large oligosaccharides like N-glycans have been used previously in studies that require shielding or steric disruption of interactions among proteins or their domains (1, 3, 16, 27). We designed these mutants in the stalk in order to disrupt putative interactions between F and HN, thus preventing HN from triggering F. Among the residues chosen for N-glycosylation, 60, 66, 67, 77, and 102 have side chains that are normally surface exposed in the wild-type protein (Fig. 3B and 9), whereas side chains of 58, 68, 90, and 91 partially or wholly extend into the hydrophobic core of the 4HB (Fig. 3B).
Fig. 9.
Mapping of mutants implicated in F interaction and affecting NA activity. (Left) PIV5-HN mutations (pink) and corresponding residues of NDV-HN mutations (lilac) believed to block F interaction (category I mutants) are mapped onto the PIV5-HN stalk structure. Residues that affect only fusion in PIV5-HN and NDV-HN are shown in red. (Right) Mutations in PIV5-HN (olive) and residues corresponding to NDV-HN mutations (orange) implicated in influencing functions of the head in addition to fusion (category II mutants) are mapped on the PIV5-HN stalk structure. Residues that affect fusion and NA activity in both PIV5-HN and NDV-HN are shown in yellow.
The crystal structure of the PIV5-HN stalk reveals a 4HB with an 11-mer hydrophobic repeat in a relatively straight region (residues 80 to 101) that is structurally very similar to the observed portion of the NDV-HN stalk (51) (Fig. 2A). The rest of the stalk below residue Ser79 maintained the 4HB but was found to possess a heptad repeat within a left-handed supercoiled region. In the PIV5-HN stalk, 4HB packing of hydrophobic side chains in the core may provide the thermostability of this helical domain (41, 50) and induce tetramerization of the molecule. This notion is supported by the observation that engineered disulfide bonds in the NDV stalk enhance stability (51) and by the mutation data presented here demonstrating that the mutations N58 and N68 in the hydrophobic core disrupt the HN tetramer and F activation (Fig. 6). Additionally, we reported that NDV-HN forms a weaker tetramer than does PIV5-HN (50, 51, 52). A possible explanation for this relative instability is that NDV-HN has three polar residues (Tyr85, Ser92, and Thr99) and potentially a fourth (Asn77) in the hydrophobic core of the region of the 4HB observed here for PIV5-HN, whereas PIV5-HN has only one such residue (Ser79). Notably, one of the engineered disulfide bonds at Ser92 of NDV-HN stabilizes the dimer and forms a possible tetramer, indicated by a leading edge shoulder peak on gel filtration chromatography (51).
Our mutagenesis data reveal that N-glycosylation mutants N60, N66, and N67 do not affect biochemical functions associated with the globular head and yet are unable to promote fusion (Fig. 5A to D). In this regard, these mutants are similar to a subset of mutants in the HN protein of NDV (27, 28, 44) that have been classified as category I mutants (51). We aligned the PIV5-HN and NDV-HN protein sequences and mapped NDV category I mutants (including 3 mutants for which no density was observed in the NDV-HN ectodomain structure) onto corresponding PIV5 residues in the PIV5-HN stalk structure along with our PIV5 stalk mutants (Fig. 9). PIV5 stalk mutations N60, N66, and N67 are present on the surface in an overlapping region that possibly extends the putative F-activation region seen in the NDV-HN stalk (Fig. 9). Notably, this subset of mutants in PIV5-HN is present in a region below residue Ser79, which is the heptad repeat containing the left-handed supercoiled part of the stalk (Fig. 1A). In another set of NDV-HN mutants (category II) (51), characterized by Melanson and Iorio (27, 28) and Stone-Hulslander and Morrison (44), both F-activation and NA activity were decreased. Our N77, N90, and N91 mutants are similar to these category II mutants. Interestingly, there is also overlap between PIV5-HN and NDV-HN regions harboring category II mutants that also affect NA activity of the globular head (Fig. 9). The crystal structure of the NDV-HN ectodomain shows a contact between the NA head domain and this region of the stalk, suggesting a functional interface between the head and the upper stalk, possibly important for both NA activity and F activation upon receptor binding (51).
Previously, NA activity was found to have a correlation with an altered sucrose density gradient sedimentation profile (27). We did not observe the same correlation for our PIV5-HN mutants, as mutants N90 (Fig. 6F) and N91 (data not shown) sedimented primarily as a dimer-of-dimers but had NA activity at 50% of wt (Fig. 5B). A possible explanation for this pattern is that the carbohydrate chains on N90 and N91 could potentially be surface exposed since the alpha carbon of each residue lies at the hydrophobic/polar boundary of the 4HB, which may allow their side chains to point either toward the hydrophobic core or toward the solvent (Fig. 3B).
All the mutants made for this study except N102 blocked fusion. N102, which is located closest to the globular head, showed close to wt levels of fusion despite having NA activity at 50% of wt (Fig. 5B to D). To examine more closely how carbohydrate shielding disrupts function in the tetrameric HN stalk, we expressed mixed oligomers of wt and mutant HN in different ratios in cells. The differential migration properties of wt and mutant proteins on an SDS-PAGE gel showed that variable ratios of transfected DNA corresponded to the ratios of synthesized polypeptides (Fig. 8). With the knowledge that the amount of HN expressed is correlated to the amount of observed fusion (9), we conducted fusion assays with these mixed oligomer transfections. Both BHK cell fusion (Fig. 7A) and luciferase reporter syncytium assays (Fig. 7B) clearly showed that the mutants lower down the stalk acted in a dominant negative manner in disrupting fusion in comparison to mutants N90, N91, and N102 near the top of the stalk, which had the characteristics of a recessive mutation. N91 required at least 75% of the mutant monomer in the population to disrupt fusion effectively, while a 100% N102 mutant protein population did not disrupt fusion at all. N90 showed a fusion phenotype intermediate between that of the lower mutants and that of N91. In contrast, the lower mutants reduced fusion significantly even when only 25% of the mutant was present in the mixture. These results and the fact that mutant N90 did not disrupt oligomerization while N68 did make the tetramer fall apart, despite their both being core residues, may suggest a degree of flexibility near the top of the stalk. Additionally, D105, near the top of the stalk, is in a core position (“h”); however, the lack of density in this region for three of the four chains may suggest that while these residues can adopt a helical configuration, the 4HB is disrupted by D105 and may not propagate beyond L101 (Fig. 1A). These results suggest flexibility in the region beyond residue L101, providing support for the hypothesis that the receptor binding protein heads may translocate upon receptor binding and/or fusion activation (30, 52).
Overall, our studies of the PIV5-HN stalk show that residues 60 to 101 form a 4HB based with a relatively nonsupercoiled upper region and a heptad repeat with a left-handed supercoil in its lower part. Addition of glycosylation sites on the surface of the 4HB between residues 60 and 77 has little effect on receptor binding or neuraminidase activity but completely blocks fusion activity, data which are consistent with blocking of the putative direct interaction between F and the HN stalk and expand our view of the F-HN interaction site. We show by sequence and structure comparison analysis between NDV-HN and PIV5-HN that mutants that affect NA activity in addition to fusion map to very similar regions on the HN stalk for the two viruses—a region that interacts with the NA domain in the NDV-HN ectodomain crystal structure. Additionally, we show evidence for a degree of flexibility at the top of the stalk, supporting the idea that receptor binding protein heads move during fusion activation.
ACKNOWLEDGMENTS
This research was supported in part by National Institutes of Health research grants AI-23173 (to R.A.L.) and GM-61050 (to T.S.J.). B.D.W. is an Associate and R.A.L. is an Investigator of the Howard Hughes Medical Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CHI1357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (grant 085PI000817).
We thank Alfonso Mondragon and Heather Pinkett for useful advice and discussions.
Footnotes
Published ahead of print on 12 October 2011.
REFERENCES
- 1. Abe Y., et al. 2004. Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J. Virol. 78: 9605–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams P. D., et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aguilar H. C., et al. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 80: 4878–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop K. A., et al. 2007. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol. 81: 5893–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bousse T., Takimoto T., Gorman W. L., Takahashi T., Portner A. 1994. Regions on the hemagglutinin-neuraminidase proteins of human parainfluenza virus type-1 and Sendai virus important for membrane fusion. Virology 204: 506–514 [DOI] [PubMed] [Google Scholar]
- 6. Bowden T. A., et al. 2008. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J. Virol. 82: 11628–11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen V. B., et al. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colf L. A., Juo Z. S., Garcia K. C. 2007. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 14: 1227–1228 [DOI] [PubMed] [Google Scholar]
- 9. Connolly S. A., Leser G. P., Jardetzky T. S., Lamb R. A. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83: 10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corey E. A., Iorio R. M. 2007. Mutations in the stalk of the measles virus hemagglutinin protein decrease fusion but do not interfere with virus-specific interaction with the homologous fusion protein. J. Virol. 81: 9900–9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crennell S., Takimoto T., Portner A., Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7: 1068–1074 [DOI] [PubMed] [Google Scholar]
- 12. Deng R., et al. 1999. Mutations in the Newcastle disease virus hemagglutinin-neuraminidase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion. Virology 253: 43–54 [DOI] [PubMed] [Google Scholar]
- 13. Deng R., Wang Z., Mirza A. M., Iorio R. M. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209: 457–469 [DOI] [PubMed] [Google Scholar]
- 14. Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- 15. Ennis M. K., et al. 2010. Mutations in the stalk region of the measles virus hemagglutinin inhibit syncytium formation but not virus entry. J. Virol. 84: 10913–10917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallagher P., Henneberry J., Wilson I., Sambrook J., Gething M.-J. 1988. Addition of carbohydrate side chains at novel sites on influenza hemagglutinin can modulate the folding, transport, and activity of the molecule. J. Cell Biol. 107: 2059–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harbury P. B., Plecs J. J., Tidor B., Alber T., Kim P. S. 1998. High-resolution protein design with backbone freedom. Science 282: 1462–1467 [DOI] [PubMed] [Google Scholar]
- 18. Hashiguchi T., et al. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U. S. A. 104: 19535–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heminway B. R., Yu Y., Galinski M. S. 1994. Paramyxovirus mediated cell fusion requires co-expression of both the fusion and hemagglutinin-neuraminidase glycoproteins. Virus Res. 31: 1–16 [DOI] [PubMed] [Google Scholar]
- 20. Horvath C. M., Paterson R. G., Shaughnessy M. A., Wood R., Lamb R. A. 1992. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol. 66: 4564–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu M.-C., Scheid A., Choppin P. W. 1979. Reconstitution of membranes with individual paramyxovirus glycoproteins and phospholipid in cholate solution. Virology 95: 476–491 [DOI] [PubMed] [Google Scholar]
- 22. Hu X., Ray R., Compans R. W. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 66: 1528–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lamb R. A., Jardetzky T. S. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17: 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lamb R. A., Parks G. D. 2007. Paramyxoviridae: the viruses and their replication, p. 1449–1496 In Knipe D. M., et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 25. Lawrence M. C., et al. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335: 1343–1357 [DOI] [PubMed] [Google Scholar]
- 26. Lee J. K., et al. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283: 16561–16572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Melanson V. R., Iorio R. M. 2006. Addition of N-glycans in the stalk of the Newcastle disease virus HN protein blocks its interaction with the F protein and prevents fusion. J. Virol. 80: 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melanson V. R., Iorio R. M. 2004. Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78: 13053–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morrison T., McQuain C., McGinnes L. 1991. Complementation between avirulent Newcastle disease virus and a fusion protein gene expressed from a retrovirus vector: requirements for membrane fusion. J. Virol. 65: 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Navaratnarajah C. K., et al. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat. Struct. Mol. Biol. 18: 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ng D. T., Hiebert S. W., Lamb R. A. 1990. Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin-neuraminidase. Mol. Cell. Biol. 10: 1989–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ng D. T., Randall R. E., Lamb R. A. 1989. Intracellular maturation and transport of the SV5 type II glycoprotein hemagglutinin-neuraminidase: specific and transient association with GRP78-BiP in the endoplasmic reticulum and extensive internalization from the cell surface. J. Cell Biol. 109: 3273–3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326 [DOI] [PubMed] [Google Scholar]
- 34. Paal T., et al. 2009. Probing the spatial organization of measles virus fusion complexes. J. Virol. 83: 10480–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parks G. D., Lamb R. A. 1990. Folding and oligomerization properties of a soluble and secreted form of the paramyxovirus hemagglutinin-neuraminidase glycoprotein. Virology 178: 498–508 [DOI] [PubMed] [Google Scholar]
- 36. Paterson R. G., Lamb R. A. 1990. Conversion of a class II integral membrane protein into a soluble and efficiently secreted protein: multiple intracellular and extracellular oligomeric and conformational forms. J. Cell Biol. 110: 999–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paterson R. G., Lamb R. A. 1993. The molecular biology of influenza viruses and paramyxoviruses, p. 35–73 In Davidson A., Elliott R. M. (ed.), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 38. Paterson R. G., Russell C. J., Lamb R. A. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270: 17–30 [DOI] [PubMed] [Google Scholar]
- 39. Porotto M., Murrell M., Greengard O., Moscona A. 2003. Triggering of human parainfluenza virus 3 fusion protein (F) by the hemagglutinin-neuraminidase (HN) protein: an HN mutation diminishes the rate of F activation and fusion. J. Virol. 77: 3647–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Randall R. E., Young D. F., Goswami K. K. A., Russell W. C. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 68: 2769–2780 [DOI] [PubMed] [Google Scholar]
- 41. Robach J. G., Lamb R. A. 2010. Analysis of parainfluenza virus-5 hemagglutinin-neuraminidase protein mutants that are blocked in internalization and degradation. Virology 406: 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sales M., Plecs J. J., Holton J. M., Alber T. 2007. Structure of a designed, right-handed coiled-coil tetramer containing all biological amino acids. Protein Sci. 16: 2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stetefeld J., et al. 2000. Crystal structure of a naturally occurring parallel right-handed coiled coil tetramer. Nat. Struct. Biol. 7: 772–776 [DOI] [PubMed] [Google Scholar]
- 44. Stone-Hulslander J., Morrison T. G. 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol. 73: 3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tanabayashi K., Compans R. W. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70: 6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Terwilliger T. C., et al. 2008. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 64: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson S. D., Laver W. G., Murti K. G., Portner A. 1988. Isolation of a biologically active soluble form of the hemagglutinin-neuraminidase protein of Sendai virus. J. Virol. 62: 4653–4660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu K., et al. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105: 9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yao Q., Hu X., Compans R. W. 1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol. 71: 650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yuan P., Leser G. P., Demeler B., Lamb R. A., Jardetzky T. S. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378: 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yuan P., et al. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108: 14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan P., et al. 2005. Structural studies of the parainfluenza virius 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13: 803–815 [DOI] [PubMed] [Google Scholar]