

Abstract
Promyelocytic leukemia (PML) protein is the organizer of nuclear matrix-associated nuclear bodies (NBs), and its conjugation to the small ubiquitin-like modifier (SUMO) is required for the formation of these structures. Several alternatively spliced PML transcripts from a single PML gene lead to the production of seven PML isoforms (PML isoform I [PMLI] to VII [PMLVII]), which all share a N-terminal region that includes the RBCC (RING, B boxes, and a α-helical coiled-coil) motif but differ in the C-terminal region. This diversity of PML isoforms determines the specific functions of each isoform. There is increasing evidence implicating PML in host antiviral defense and suggesting various strategies involving PML to counteract viral production. We reported that mouse embryonic fibroblasts derived from PML knockout mice are more sensitive than wild-type cells to infection with encephalomyocarditis virus (EMCV). Here, we show that stable expression of PMLIV or PMLIVa inhibited viral replication and protein synthesis, leading to a substantial reduction of EMCV multiplication. This protective effect required PMLIV SUMOylation and was not observed with other nuclear PML isoforms (I, II, III, V, and VI) or with the cytoplasmic PMLVII. We demonstrated that only PMLIV interacted with EMCV 3D polymerase (3Dpol) and sequestered it within PML NBs. The C-terminal region specific to PMLIV was required for both interaction with 3Dpol and the antiviral properties. Also, depletion of PMLIV by RNA interference significantly boosted EMCV production in interferon-treated cells. These findings indicate the mechanism by which PML confers resistance to EMCV. They also reveal a new pathway mediating the antiviral activity of interferon against EMCV.
INTRODUCTION
Interferons (IFNs) play a central role in mediating antiviral innate immunity. IFN signaling is initiated by IFNs binding to specific receptors at the cell surface, followed by the activation of Janus-activated kinase (Jak)/Stat pathway and the induction of interferon-stimulated genes (ISGs). These genes have diverse antiviral activities, including translational control, regulation of RNA stability, and effects on protein transport and turnover (22, 31).
Several pathways, including 2′,5′ oligoadenylate (2′5′A) synthetase/RNase L, double-stranded RNA-activated kinase (protein kinase R [PKR]), Mx proteins, and promyelocytic leukemia (PML) protein, which has also been called tripartite motif 19 (TRIM19), have been implicated in mediating resistance to infection by viruses of various families (7, 13, 14, 17, 24, 32, 34, 37). PML functions as the organizer of PML nuclear bodies (NBs), which are dynamic nuclear structures harboring numerous proteins, some transiently and some permanently (23, 27). These structures are involved in the transient recruitment/sequestration of several transcriptional regulators and proteins and have a role in both the antiviral response and cancer. PML NB formation requires the RBCC (RING, B boxes, and a α-helical coiled-coil) motif and covalent small ubiquitin-like modifier (SUMO) modification at lysines at positions 65, 160, and 490 (5, 20, 21). Seven PML isoforms, PML isoform I (PMLI) to PML isoform VII (PMLVII), result from alternative splicing from a single gene. They share a N-terminal region (exons 1 to 3), which includes the RBCC motif, but differ in their C-terminal regions according to alternative splicing of exons 4 to 9. The C termini of all nuclear PML isoforms contain a nuclear localization signal (NLS). The variability of the C-terminal regions of PML isoforms explains why each recruits its own subset of interaction partners, and consequently why they display different functions (15, 23).
Some PML isoforms impair replication of RNA viruses from different families (13, 16). PMLIII confers resistance to human foamy virus (HFV), vesicular stomatitis virus (VSV), influenza virus, and poliovirus (9, 30, 35), whereas only PMLIV confers resistance to rabies virus infection (3). PML knockout mice are more sensitive than wild-type mice are to lymphocytic choriomeningitis virus (LCMV) and VSV infections (4), confirming the antiviral effect of PML in vivo. Also, cells are more sensitive to encephalomyocarditis virus (EMCV) infection in the absence of PML (12).
Resistance to EMCV has been described in cells stably expressing other ISG products, notably 2′,5′ oligoadenylate synthetase (8, 10), PKR (26), guanylate binding protein 1 (2), and TRIM22 protein (11).
EMCV is the prototype of the cardiovirus subgroup of picornaviruses. Its genome is a single-stranded, positive-sense RNA with a single long open reading frame (ORF) (29). This ORF encodes a precursor polyprotein that is cleaved by its own virus-encoded protease to produce the viral capsid and nonstructural proteins. One of these proteins, the RNA-dependent RNA polymerase (3D polymerase [3Dpol]), is required for the elongation of positive- and negative-stranded viral RNA. 3Dpol forms a replication complex with the virion protein genome-linked protein (Vpg) and precursor proteins 3AB and 3CD to initiate Vpg uridylylation; the resulting product acts as a primer for positive- and negative-strand RNA replication by 3Dpol (38, 39). EMCV replicates in the cytoplasm, but viral proteins 2A, 3B, 3Cpro, and 3Dpol are found, early postinfection, as punctate spots in the nucleus (1).
We studied the roles of the PML isoforms (PMLI to PMLVII) during EMCV infection. We report that cells stably expressing PMLIV or PMLIVa (missing exon 5) conferred resistance to EMCV infection, whereas other nuclear PML isoforms (I, II, III, V, and VI) and cytoplasmic isoform PMLVII did not inhibit the virus. The PMLIV-3KR mutant, which cannot be SUMOylated, did not impair EMCV production, indicating that PMLIV SUMOylation is required for the protective effect against EMCV. The unique C-terminal region of PMLIV, encoded by exon 8b, interacted with 3Dpol and recruited it to PML NBs. RNA interference abolishing PMLIV activity was associated with increased EMCV yield in alpha interferon (IFN-α)-treated cells.
MATERIALS AND METHODS
IFN and antibodies.
Recombinant human IFN-α2 was from Schering. Mouse monoclonal anti-PML (clone PGM3) and rabbit polyclonal anti-PML (clone H-238) antibodies were from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)-conjugated mouse monoclonal anti-β-actin (clone AC-15) antibody was from Sigma. Rabbit anti-EMCV and mouse monoclonal anti-3Dpol antibodies were from Ann Palmenberg (1). Rabbit anti-VSV antibodies were from Danielle Blondel.
Cell cultures and cell treatments.
U373MG, HEK293, L929, and Chinese hamster ovary (CHO) cells were grown at 37°C in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). CHO cells transfected with the empty vector (EV) or stably expressing PMLIII were kept in medium supplemented with 0.5 mg/ml hygromycin (9). CHO and U373MG cells stably expressing PMLIVa were grown in medium supplemented with 0.2 mg/ml zeomycin. U373MG cells transfected with empty vector or stably expressing PMLI, PMLII, PMLIII, PMLIV, PMLV, PMLVI, or PMLVII were kept in medium supplemented with 0.5 mg/ml of G418 as described previously (3).
Virus stocks.
L929 cells (2 × 107) seeded in culture flasks, were infected with EMCV at a multiplicity of infection (MOI) of 1 by adsorption in 1 ml medium without serum. After 1 h, the medium containing virus was removed and replaced with 10 ml of medium containing 2% FCS, and the cells were incubated at 37°C for 24 h. The cultures were then frozen, thawed three times, centrifuged to remove cellular debris, and the supernatants were saved. Viral titers were determined on these cells by measuring the 50% tissue culture infective dose (TCID50).
Stable expression of PML isoforms and PML mutants.
The accession numbers (GenBank) for PML isoforms are as follows: M79462 (PMLI), M79464 (PMLII), S50913 (PMLIII), X63131 (PMLIV), AF230411 (PMLIVa), M79463 (PMLV), AF230405 (PMLVI), and AF230408 (PMLVII). In the PMLIV-3KR mutant, the three SUMO-target lysines (at positions 65, 160, and 490) were replaced with arginines. U373MG cells stably expressing PMLI, PMLII, PMLIII, PMLIV, PMLV, PMLVI, and PMLVII and PMLIV-3KR cells were obtained by transfection with the corresponding constructs (pcDNA3.1 PMLI, pcDNA3.1 PMLII, pcDNA3.1 PMLIII, pcDNA3.1 PMLIV, pcDNA3.1 PMLV, pcDNA3.1 PMLVI, pcDNA3.1 PMLVII, and pcDNA3.1 PMLIV-3KR) and selection on neomycin (final concentration, 0.5 mg/ml). U373MG and CHO cells stably expressing PMLIVa were generated by transfection with pcDNA4.1 PMLIVa (a gift from P. G. Pelicci) and selection on zeomycin (0.2 mg/ml). CHO-PMLIII was generated as previously described (9). The PMLIV-Δ8b and PMLIV-Δ8ab mutants (40) were expressed in U373MG cells. U373MG and CHO cells were also transfected with the empty vectors and used as controls.
Real-time quantification of mRNA.
The amount of viral RNA was determined by quantifying the 3Dpol region in an assay. Quantification of PMLIII, PMLIV, and 3Dpol transcripts was performed using reverse transcription-PCR with the FastStart DNA Master SYBR green I kit and the LightCycler apparatus, according to the manufacturer's instructions (Roche Molecular Biochemicals, Indianapolis, IN), with the following oligonucleotide pairs: 5′-CCCTACCTCACGGAATGGGGCAAAG-3′ and 5′-GGTGAGAGCAAGCCTCGCAAAGACAG-3′ for 3Dpol; 5′-CCCGTCATACGAAGTGAGGT-3′ and 5′-AGACTGAGGGCTGGAAGAGA-3′ for PMLIII; 5′-TGGACGAGAACCTTGCTGAC-3′ and 5′-CCAGGAGAACCCACTTTCAT-3′ for PMLIV; and 5′-AGCTCACTGGCATGGCCTTC-3′ and 5′-ACGCCTGCTTCACCACCTTC-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The results were normalized to those for GAPDH.
DNA and siRNA transfection.
The cells were seeded in six-well plates and transfected with 2 μg of plasmid DNA or small interfering RNA (siRNA) using the Lipofectamine 2000 and Lipofectamine RNAiMax transfection reagents, respectively (Invitrogen), according to the manufacturer's recommendations. The cells were transfected with siRNAPMLc (common to all PML isoforms), corresponding to nucleotides 362 to 381, siRNAPMLIII corresponding to nucleotides 1768 to 1749, siRNAPMLIV corresponding to nucleotides 1857 to 1876, or siRNA scramble. Transfected cells were then prepared for Western blot analysis.
Immunofluorescence staining and confocal microscopy.
Transfected cells were fixed in 4% paraformaldehyde for 15 min at 4°C and permeabilized for 5 min with 0.1% Triton X-100 in phosphate-buffered saline (PBS). They were then prepared for double-immunofluorescence staining and analyzed by confocal microscopy. The cells were stained with rabbit anti-PML and monoclonal anti-3Dpol antibodies. They were washed twice and incubated for 1 h with the appropriate Alexa Fluor-conjugated secondary antibody (Molecular Probe, Inc.). Coverslips were mounted using Vectashield (Vector Laboratory). For confocal analysis, images were sequentially collected with a Zeiss LSM510 confocal laser scanning microscope.
Western blot analysis.
Cells were washed and resuspended in PBS, lysed in hot Laemmli sample buffer, and boiled for 5 min. These samples were run on a 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane. The membranes were blocked with 10% skim milk in Tris-buffered saline (TBS) for 2 h and incubated overnight at 4°C with rabbit polyclonal anti-PML, monoclonal anti-3Dpol, antiviral proteins, or antiactin antibodies. The blots were then washed extensively in PBS containing Tween 20 (PBS-Tween) and incubated for 1 h with the appropriate peroxidase-coupled secondary antibodies (Amersham). On all blots, antibody binding was revealed by chemiluminescence (ECL kit; Amersham). To estimate the apparent molecular mass of the polypeptides, the proteins of interest were compared to prestained molecular weight standards included on each gel (Bio-Rad Laboratories, Richmond, CA).
Immunoprecipitation assays.
Transfected cells (107) were incubated for 30 min at 4°C in 0. 5 ml of buffer containing 20 mM Tris-HCl (pH 7.4), 1 M NaCl, 5 mM MgCl2, 1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride (PMSF). After cell lysis, 1. 25 ml of immunoprecipitation buffer (IB) (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% deoxycholate [DOC], 1% Triton X-100, 0.1% SDS, and 1 mM EDTA) were added. The samples were used for PML and 3Dpol immunoprecipitation in separate experiments as follows. Rabbit anti-PML or anti-3Dpol antibodies were added, and the samples were incubated overnight at 4°C. Protein G beads (Sigma) in IB were then added, and the samples were mixed for 2 h at room temperature. The beads were collected and washed four times with modified IB (IB plus 150 mM NaCl), and the bound proteins were subjected to Western blotting.
RESULTS
Effects of the different PML isoforms on EMCV protein expression.
Mouse embryonic fibroblasts (MEFs) derived from knockout PML mice are more sensitive than MEFs from wild-type mice to EMCV infection (12). To determine whether particular PML isoforms inhibit EMCV production, we stably expressed each of the different PML isoforms (PMLI to PMLVII) in U373MG cells. The structures of these PML isoforms are presented in Fig. 1A. The PML isoforms were detected by Western blotting at the expected sizes, and in each case, the lower band corresponds to the unmodified SUMO isoform with special forms migrating above representing the SUMOylated species (Fig. 1B). In contrast, no bands of higher molecular weights were observed for PMLVII, indicating that this cytoplasmic isoform was not SUMOylated (Fig. 1B). U373MG cells stably expressing each of the PML isoforms (PMLI to PMLVII) were infected with EMCV at an MOI of 0.2 and incubated for 12 h. Extracts from these cells were then tested for viral proteins (VP1, VP2, and VP3) by Western blotting (Fig. 1C). EMCV proteins were detected in similar amounts in extracts from U373MG cells transfected with empty vector (U373MG-EV) and in extracts from cells expressing nuclear isoform PMLI, PMLV, or PMLVI or the cytoplasmic isoform PMLVII. Thus, these PML isoforms did not inhibit EMCV infection. Somewhat less viral protein was detected in extracts from cells expressing PMLII or PMLIII. In contrast, viral proteins were undetectable in extracts from cells stably expressing PMLIV) (Fig. 1C), suggesting that this particular isoform may have a protective effect against EMCV infection.
Fig. 1.

PMLIV is the only PML isoform that inhibits production of EMCV proteins. (A) Schematic representations of the domain structures of PML isoforms. All PML isoforms have the same N terminus, but they have different C termini due to alternative splicing of exons 4 to 9. All isoforms contain the RING finger (R), the B1 and B2 boxes, and the coiled-coil (CC) motif. The NLS is present in all nuclear PML isoforms (PMLI to PMLVI at positions 476 to 490). PML contains three SUMOylation sites at position 65, 160, and 490. PMLI to PMLV contain a SUMO-interacting motif (SIM) (at positions 508 to 511). aa, amino acids. (B) Extracts from U373MG-EV cells and cells stably expressing PMLI, PMLII, PMLIII, PMLIV, PMLV, PMLVI, or PMLVII were analyzed by Western blotting using anti-PML antibody. (C) The cells described above for panel B were infected with EMCV (+) at an MOI of 0.2 for 12 h, and their extracts were analyzed by Western blotting using antiviral proteins or antiactin antibodies.
PMLIV inhibits viral RNA replication and EMCV production.
To confirm the antiviral effect of PMLIV, U373MG cells expressing PMLIV (U373MG-PMLIV), and cells expressing PMLIII as negative controls, were infected at higher MOIs with EMCV. U373MG-EV cells and cells stably expressing PMLIII or PMLIV were infected at MOIs of 0.2, 0.5, or 1 for 12 h. Almost 100% of U373MG-EV cells and cells expressing PMLIII died, whereas cells expressing PMLIV were totally protected against cell lysis (Fig. 2A). Cell extracts were studied by Western blotting; the abundance of viral proteins in extracts from control cells was dependent on the MOIs with EMCV, whereas no viral protein was detected in extracts from cells expressing PMLIV (Fig. 2B). Thus, viral protein synthesis was blocked by PMLIV even after infection at high MOIs (Fig. 2B). The antiviral effect of PMLIV against EMCV was also observed in transiently transfected HEK293 cells (data not shown). Viral protein production appeared to be weakly inhibited in cells expressing PMLIII (Fig. 2B).
Fig. 2.

PMLIV inhibits viral RNA replication and EMCV production. (A) PMLIV protects cells from EMCV-induced cell lysis. U373MG-EV cells and cells stably expressing PMLIII or PMLIV were not infected (−) or infected (+) with EMCV at an MOI of 0.5 for 12 h. The phase picture was acquired using a Nikon Eclypse TS100 microscope and Coolpix MDC lens camera. (B) U373MG-EV cells and cells stably expressing PMLIII or PMLIV were infected at various MOIs for 12 h. Cell extracts were analyzed by Western blotting for viral proteins. (C) PMLIV inhibits EMCV RNA synthesis. U373MG-EV cells and cells stably expressing PMLIV were infected with EMCV at an MOI of 0.5. RNA was extracted 2 h and 12 h later, and 3Dpol RNA was assayed by real-time PCR. Results were normalized to those for GAPDH. The means plus standard deviations (error bars) of three independent assays are shown. (D) U373MG-EV cells and cells stably expressing PMLIII or PMLIV were infected at various MOIs for 12 h and used for determination of the viral titers as described in Materials and Methods. (E) Inhibition of EMCV production in IFN-treated cells and in cells expressing PMLIV. U373MG-EV cells were treated for 24 h with 10, 50, 200, or 1,000 units/ml of IFN-α. These cells and U373MG-PMLIV cells were then infected for 14 h with EMCV at an MOI of 0.5. Viral titers were determined as described in Materials and Methods. (F) PMLIV-expressing cells do not produce more IFN than control cells during EMCV infection. U373MG-EV and U373MG-PMLIV cells were infected with EMCV at an MOI of 0.5 for 12 h. Culture supernatants (from EV cells [S1] and PMLIV-expressing cells [S2]) were treated with acid buffer to inactivate virus, and the pH was then neutralized. U373MG cells were left untreated as a control (C) or treated for 24 h with S1 or S2. One series of cells was analyzed by Western blotting using anti-STAT1, anti-PKR, and antiactin antibodies (top panel). The cells in the second series were infected with VSV at an MOI of 1 for 8 h and were then analyzed by Western blotting using anti-VSV antibodies to detect viral N and M proteins (bottom panel).
To determine how PMLIV inhibits EMCV infection, we used quantitative reverse transcription-PCR of the 3Dpol region to assay the viral RNA in U373MG-EV cells and U373MG-PMLIV cells 2 and 12 h postinfection. Infected U373MG cells stably expressing PMLIV contained substantially less viral RNA than infected control cells (Fig. 2C), suggesting that PMLIV blocked EMCV RNA synthesis.
Inhibition of viral RNA and protein synthesis in cells expressing PMLIV was accompanied by a high decrease in EMCV production as shown by the viral titers (Fig. 2D). Indeed, compared to control infected cells, EMCV multiplication in U373MG-PMLIV cells was 3 to 4 log units lower.
To compare inhibition of EMCV by PMLIV with that by IFN, U373MG cells were treated for 24 h with 10, 50, 200, and 1,000 units/ml of IFN-α. U373MG cells treated with IFN-α or stably expressing PMLIV were then infected with EMCV at an MOI of 0.5 for 14 h. Inhibition of EMCV multiplication in U373MG-PMLIV cells was comparable to that in cells treated with 200 units/ml of IFN-α (Fig. 2E). Note that in U373MG cells treated with IFN-α, the expression of several isoforms was enhanced by Western blotting (9) (see Fig. 7A below), and the level of PMLIV transcript was increased by quantitative reverse transcription-PCR (see Fig. 7B below).
Fig. 7.
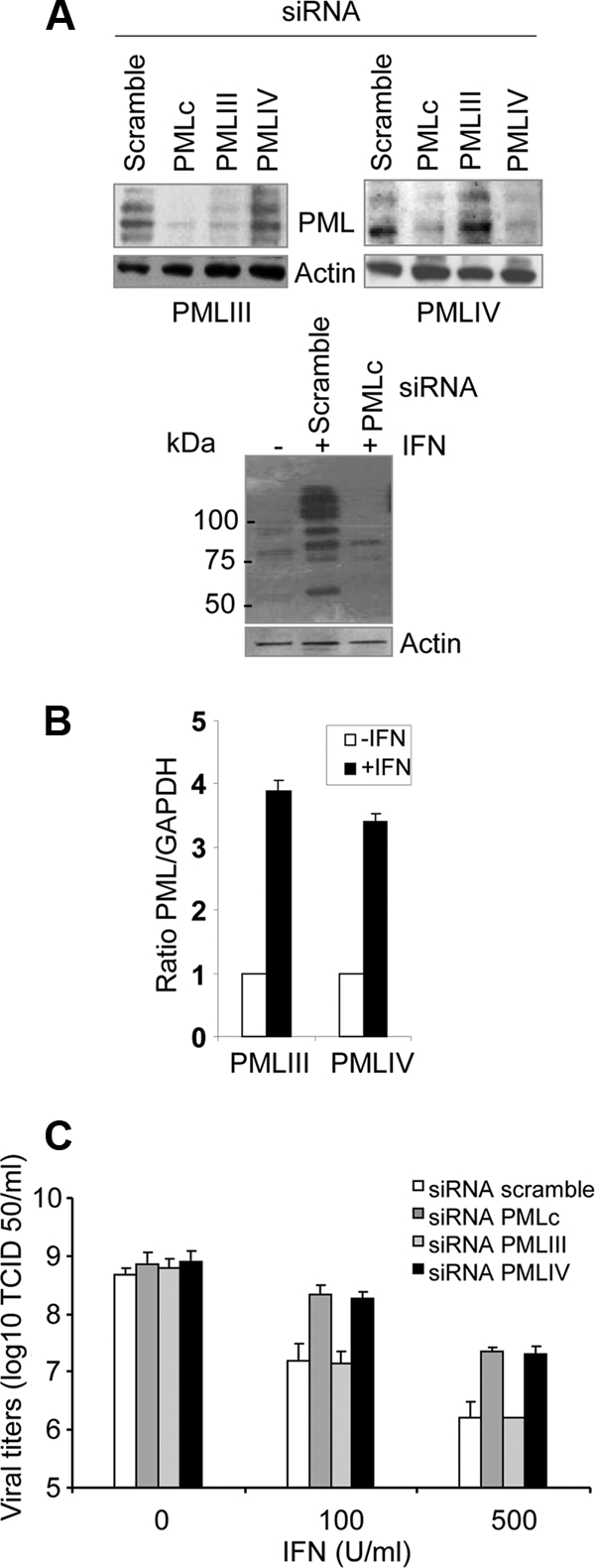
PMLIV depletion boosts EMCV production in IFN-α-treated cells. (A) Efficacy of siRNA PMLc, siRNA PMLIV, and siRNA PMLIII. U373MG cells stably expressing PMLIII or PMLIV were transfected with scramble siRNA, PMLc siRNA (common to all PML isoforms), PMLIV-specific siRNA, or PMLIII-specific siRNA (top blots). U373MG cells, transfected with scramble siRNA or PMLc siRNA, were treated 1 day posttransfection with 500 units/ml of IFN-α for 24 h (bottom blots). Western blot analysis was performed using anti-PML and antiactin antibodies. (B) Increase of PMLIII and PMLIV expression in IFN-treated U373MG cells. U373MG cells were left untreated or treated with 100 units of IFN-α. RNA was extracted in all samples, and PMLIII and PMLIV expression was tested by the real-time PCR method. Results were normalized to GAPDH. The averages plus standard deviations (error bars) for three independent assays are shown. The statistical significance (P values) was calculated using a Student's t test. (C) Knockdown of PMLIV by PMLIV-specific siRNA impairs IFN-α-induced antiviral activity. U373MG cells were transiently transfected for 12 h with siRNA scramble, siRNA PMLc, siRNA PMLIV, or siRNA PMLIII, treated with IFN-α for 24 h, and then infected with EMCV at an MOI of 1 for 14 h. Viral titers were determined as described in Materials and Methods.
It was therefore possible that PMLIV induces IFN secretion during EMCV infection, which may in turn inhibit viral protein expression. Culture supernatants from EMCV-infected U373MG-EV cells (S1) and U373MG-PMLIV cells (S2) were tested for their capacity to induce the expression of PKR and signal transducer and activator of transcription factor 1 (STAT1), two ISG products and to inhibit VSV protein synthesis. STAT1 and PKR concentrations as well as VSV N- and M-protein expression were similar in extracts from cells treated with culture supernatants S1 and S2 (Fig. 2F). Similarly, both supernatants contained 4 IU/ml of IFN (data not shown). STAT1 and PKR expression was enhanced, and VSV proteins were inhibited when these cells were incubated with 50 units/ml of IFN (data not shown). This demonstrates that resistance in PMLIV-expressing cells was not due to increased IFN production during EMCV infection. This finding is in agreement with a previous report showing that little, if any, IFN-α/β is produced following infection with EMCV, due to L-protein inhibition of interferon regulatory factor 3 (IRF-3) activation (18).
To confirm the antiviral effect of PMLIV, we stably expressed in U373MG and CHO cells a PMLIV variant missing exon 5 (PMLIVa). Expression of PMLIVa in U373MG cells by Western blotting (Fig. 3A) and in CHO cells by immunofluorescence analysis is shown (Fig. 3C). These cells were infected with EMCV, and their extracts were tested by Western blotting for viral proteins (Fig. 3A and C) and for determination of viral titers (Fig. 3B and D). Like PMLIV, PMLIVa expression in two different cell lines inhibited viral protein synthesis and EMCV multiplication (Fig. 3). Thus, PMLIV and its variant PMLIVa with the same C-terminal region unique to this isoform conferred resistance to EMCV.
Fig. 3.

PMLIVa stably expressed in U373MG or CHO cells inhibits EMCV multiplication. (A and B) U373MG-EV cells and U373MG cells stably expressing PMLIVa were infected with EMCV at an MOI of 0.5 for 12 h, and the cells were analyzed by Western blotting for viral proteins, PML, and actin (A) or the viral titers were determined (B). (C and D) Immunofluorescence (red) of PML in CHO-PMLIVa and CHO-EV is shown in the top micrographs. CHO cells containing EV or stably expressing PMLIII or PMLIVa, prepared in duplicate, were similarly infected with EMCV. One series of cells was analyzed by Western blotting for viral antigens and actin expression (bottom panel). The second series was used for the determination of viral titers (D). (E) Fate of PMLIII and PMLIV during EMCV infection. U373MG cells stably expressing PMLIII or PMLIV were infected for 12 h at various MOIs, and cell extracts were then analyzed by Western blotting using anti-PML and antiactin antibodies.
As observed in U373MG cells (Fig. 2B), viral protein expression was weakly inhibited in CHO cells expressing PMLIII (Fig. 3C). We suggested previously that EMCV counteracts the antiviral property of PMLIII by inducing proteasome- and SUMO-dependent degradation of this isoform (12). We tested whether PMLIV levels were altered during infection with EMCV at various MOIs. Consistent with the previous report, PMLIII was degraded during EMCV infection, whereas the abundance of PMLIV was not affected (Fig. 3E).
We conclude that PMLIV-expressing cells do not produce more IFN than control cells and thus that the protective effect of PMLIV is not due to induction of IFN. Therefore, PMLIV displays an intrinsic antiviral activity against EMCV.
SUMOylation of PMLIV is required for its antiviral property.
PML is covalently linked to SUMO at lysines 65, 160, and 490 (21). This modification is required for PML NB formation. To investigate the role of SUMOylation in the antiviral property of PMLIV, we infected U373MG cells stably expressing PMLIV-3KR in which the three SUMO target lysines (positions 65, 160, and 490) were replaced with arginines. Immunofluorescence analysis for PML and viral proteins revealed that PMLIV inhibited viral protein production, whereas PMLIV-3KR did not (Fig. 4A). Note that PMLIV-3KR (3), like PMLIII-3KR (25), presented punctate dots in the nucleus. PML and its SUMOylated forms were indeed produced in cells expressing PMLIV, but as expected, only the unmodified form was detected in cells expressing PMLIV-3KR (Fig. 4B). The expression of PMLIV and PMLIV-3KR was not altered during viral infection (Fig. 4B, top blot). Western blot analysis of extracts from infected U373MG-EV, U373MG-PMLIV, and U373MG-PMLIV-3KR cells confirmed that viral protein production was inhibited by PMLIV but was not affected by PMLIV-3KR (Fig. 4B, bottom blot). Next, U373MG-EV cells and cells expressing PMLIV or PMLIV-3KR were infected with EMCV at an MOI of 0.5 for 12 h, and virus yields were then determined. Viral multiplication was inhibited in cells expressing PMLIV but not in cells expressing PMLIV-3KR (Fig. 4C).
Fig. 4.

PMLIV SUMOylation is required for resistance against EMCV. U373MG-EV cells and cells stably expressing PMLIV or PMLIV-3KR were infected with EMCV at an MOI of 0.5 for 12 h. (A) Double immunofluorescence staining for PML (red) and EMCV proteins (green) was performed. (B) Cell extracts were analyzed by Western blotting using anti-PML, anti-viral protein, and antiactin antibodies. (C) Viral titers were determined as described in Materials and Methods.
These results demonstrate that SUMO modification of PMLIV is required to confer resistance against EMCV.
PMLIV interacts with 3Dpol and sequesters it in PML NBs.
EMCV replicates in the cytoplasm, but 3Dpol is found as punctate dots in the nucleus early postinfection (1), suggesting a possible interaction between PML and 3Dpol. Therefore, we used immunofluorescence to test whether 3Dpol colocalized with PML in NBs. U373MG cells were transiently cotransfected with constructs encoding 3Dpol and a PML isoform (PMLI to PMLVII), and double immunofluorescence staining was used to reveal PML (red) and 3Dpol (green). As previously reported (6), all the nuclear isoforms were found in punctate NBs, whereas PMLVII was cytoplasmic (Fig. 5A). Confocal microscopy revealed that PMLIV and 3Dpol colocalized within the NBs, but that no such colocalization was observed with the other PML isoforms (Fig. 5A). Additionally, PMLIV-3KR, which did not alter EMCV replication, did not colocalize with 3Dpol (Fig. 5B).
Fig. 5.

PMLIV interacts with the viral 3Dpol and sequesters it within the NBs. (A) PMLIV recruits 3Dpol to PML NBs. U373MG cells were transiently transfected with constructs encoding 3Dpol and either PMLI, PMLII, PMLIII, PMLIV, PMLV, PMLVI, or PMLVII. The cells were fixed, stained for PML (red) and 3Dpol (green) and with 4′,6′-diamidino-2-phenylindole (DAPI) (blue), and analyzed by confocal microscopy. The Merge panels show the two-color overlay results, demonstrating colocalization of 3Dpol and PMLIV proteins (yellow). (B) U373MG cells were transfected with constructs encoding 3Dpol or 3Dpol and PMLIV-3KR. The cells were fixed, stained for PML (red) or 3Dpol (green) and with DAPI (blue) and analyzed by confocal microscopy. (C) 3Dpol interacts with PMLIV. HEK293 cells were cotransfected with constructs encoding 3Dpol and either PMLIV or PMLIII. One aliquot of cell extract was immunoprecipitated (IP) with polyclonal anti-PML antibody, and the precipitate was analyzed by Western blotting using anti-3Dpol antibody. A second aliquot was immunoprecipitated with anti-3Dpol antibody, and the precipitate was analyzed by Western blotting using anti-PML antibody.
Given that 3Dpol and PML proteins colocalize in the nucleus, we investigated whether PMLIV and 3Dpol interact by coimmunoprecipitation assays. Cells were cotransfected with constructs encoding 3Dpol and either PMLIV or PMLIII. One aliquot of the cell extract was immunoprecipitated with polyclonal anti-PML antibody and analyzed by Western blotting using anti-3Dpol antibody. Another aliquot was immunoprecipitated with anti-3Dpol antibody and analyzed by Western blotting using anti-PML antibody (Fig. 5C). PML antibody precipitated 3Dpol, and reciprocally, the 3Dpol antibody precipitated PML from extracts derived from cells transfected with PMLIV and 3Dpol. No such interactions were observed in extracts from cells transfected with PMLIII and 3Dpol. Therefore, 3Dpol protein specifically interacts with PMLIV in the absence of other EMCV proteins and is recruited into PML NBs.
Both PMLIV-mediated sequestration of 3Dpol and antiviral activity require the C-terminal region specific to PMLIV.
PMLIV differs from the other five nuclear PML isoforms by the presence of exons 8a and 8b in its C-terminal region. To map the region of PMLIV involved in the interaction with 3Dpol, we used PMLIV mutants with exon 8b deleted (PMLIV-Δ8b) or both exons 8a and 8b deleted (PMLIV-Δ8ab) (Fig. 6A) (40) and tested their interactions with 3Dpol. Double immunofluorescence staining revealed that 3Dpol colocalized with PMLIV but not with the PMLIV-Δ8b or PMLIV-Δ8ab mutant (Fig. 6B), indicating that exon 8b is essential for sequestration of 3Dpol into PML NBs. Similarly, coimmunoprecipitation experiments confirmed that, unlike PMLIV, both PMLIV-Δ8b and PMLIV-Δ8ab were unable to interact with 3Dpol (Fig. 6C), confirming that the C-terminal region of PMLIV is needed for the specific interaction with viral polymerase.
Fig. 6.

The C-terminal region of PMLIV is required for interaction with and recruitment to NBs of 3Dpol. (A) The structures of PMLIV and of PMLIV-Δ8b and PMLIV-Δ8ab (carrying C-terminal mutations) are shown. (B) U373MG cells, cotransfected with constructs encoding 3Dpol and PMLIV, PMLIV-Δ8b, or PMLIV-Δ8ab, were fixed, stained for PML (red) or 3Dpol (green) and with DAPI (blue), and were examined by confocal microscopy. The merge (yellow) indicates the colocalization between the two proteins. (C) The C-terminal region of PML is required for the interaction with 3Dpol. HEK293 cells were cotransfected with constructs encoding 3Dpol and PMLIV, PMLIV-Δ8b, or PMLIV-Δ8ab. Cell extracts were immunoprecipitated (IP) with anti-3Dpol antibody and analyzed by Western blotting using anti-PML antibody. Ab, antibody. (D and E) U373MG-EV cells and cells expressing PMLIV, PMLIV-Δ8b, or PMLIV-Δ8ab were infected with EMCV at an MOI of 0.5 for 12 h and were analyzed by Western blotting for viral proteins, PML, and actin (D), or viral titers were determined (E).
We tested whether the inability of PMLIV-Δ8b and PMLIV-Δ8ab mutants to interact with 3Dpol was associated with the loss of antiviral activity. U373MG cells expressing PMLIV, PMLIV-Δ8b, or PMLIV-Δ8ab were infected with EMCV in order to analyze EMCV proteins by Western blotting and to determine viral production. Viral protein expression was inhibited by PMLIV but unaffected by PMLIV-Δ8b and PMLIV-Δ8ab (Fig. 6D). Similarly, viral production was inhibited by PMLIV but not by PMLIV C-terminal mutants (Fig. 6E). Thus, the unique C-terminal region of PMLIV is required for the antiviral activity against EMCV.
PMLIV depletion boosts EMCV production in IFN-α-treated cells.
We analyzed EMCV production in IFN-α-treated cells depleted of PMLIV. First, we verified the efficacy of the siRNA to deplete PML. U373MG cells stably expressing PMLIII or PMLIV were transfected with siRNA scramble, siRNA PMLc (common to all PML isoforms), siRNA PMLIII, and siRNA PMLIV; PMLIII was depleted by each siRNA PMLIII and siRNA PMLc, but not by siRNA PMLIV, and PMLIV was depleted by each siRNA PMLIV and siRNA PMLc, but not by siRNA PMLIII (Fig. 7A, top blots). These tests confirmed the specificity of the siRNAs. Knockdown of PMLIV in U373MG cells stably expressing this isoform is accompanied by a loss of antiviral activity (data not shown). Also, the expression of all PML isoforms was depleted by siRNA PMLc in U373MG cells treated with IFN-α (Fig. 7A, bottom blot). To determine whether PMIII and PMLIV transcripts were increased in response to IFN, we measured the levels of PMLIII and PMLIV by quantitative reverse transcription-PCR in U373MG cells treated for 10 h with 100 units/ml of IFN-α. As shown in Fig. 7B, PMLIII and PMLIV transcripts were increased in IFN-treated U373MG cells. To determine the effect of PMLIV depletion on the antiviral action of IFN, U373MG cells were transfected for 12 h with siRNA scramble, siRNA PMLc, siRNA PMLIV, or siRNA PMLIII and treated with various concentrations (100 and 500 U/ml) of IFN-α for 24 h. The cells were then infected with EMCV at an MOI of 1 for 14 h. As expected, EMCV production was inhibited in an IFN concentration-dependent manner (Fig. 7B). This antiviral effect was not impaired by depletion of PMLIII at any IFN concentration. Interestingly, the capacity of IFN to inhibit EMCV was similarly reduced by depletion of PMLIV or PMLc, demonstrating that PMLIV plays a role in conferring resistance to EMCV in IFN-treated cells.
DISCUSSION
Here, we report that expression of PMLIV or PMLIVa (missing exon 5) in U373MG or CHO cells confers resistance to EMCV by blocking synthesis of viral RNA and proteins, resulting in an EMCV yield 3 to 4 log units lower than in control cells. In contrast, other nuclear PML isoforms (PMLI, PMLII, PMLV, and PMLVI) and the cytoplasmic PMLVII had no significant effects on EMCV multiplication. A small decrease in viral protein expression was seen in cells expressing PMLIII. We have shown previously that EMCV induces a proteasome- and SUMO-dependent degradation of PMLIII (12). This suggests that the antiviral property of this isoform is counteracted during viral infection by proteolysis. In contrast, the level of PMLIV remained constant during EMCV infection. PMLIV inhibition of EMCV was not due to increased IFN synthesis by cells expressing PMLIV during infection, demonstrating that PMLIV has intrinsic antiviral properties effective against EMCV. In addition, PMLIV conferred resistance to poliovirus, another member of picornavirus family (unpublished data).
SUMOylation of PML, essential for PML NB formation and recruitment of various proteins (19, 41), is also required for the antiviral activity of PMLIV. Indeed, PMLIV-3KR, in which the three SUMOylation sites have been mutated, was not able to impair EMCV replication. The diversity of the C-terminal regions of the PML isoforms is important for recruitment of partners that interact with PML (15, 23). Thus, it seemed likely that PMLIV and PMLIVa, which have the same C-terminal region, recruit a viral factor crucial for EMCV multiplication. Indeed, we demonstrated in transfected cells that PMLIV interacted with 3Dpol and sequestered it within PML NBs. In contrast, none of the other nuclear or cytoplasmic isoforms of PML was able to recruit 3Dpol to PML NBs. Such interactions between PMLIV and 3Dpol could not be visualized in infected cells, due to inhibition of viral protein expression by PMLIV. The unique C-terminal domain of PMLIV, which is encoded by exons 8a and 8b of the PML gene, differentiates this isoform from the other six PML isoforms that failed to sequester 3Dpol. Furthermore, PMLIV-Δ8b and PMLIV-Δ8ab mutants (C-terminal sequences deleted in both mutants) neither colocalized nor interacted with 3Dpol and failed to inhibit viral protein synthesis and EMCV production. Therefore, the C-terminal portion of PMLIV, which differs from those of the other PMLs, is essential both for interaction with 3Dpol and antiviral activity. Therefore, our experimental results show that PMLIV and PMLIVa exert the isoform-specific anti-EMCV function through their common C-terminal region.
Additionally, PMLIV has antiviral activity against varicella-zoster virus (VZV), an alphaherpesvirus and member of a DNA virus family that replicates in the host cell nucleus (36). Among the six nuclear PML isoforms, only PMLIV sequesters VZV nucleocapsids, which requires the C-terminal domain of PMLIV (36). Thus, the specific antiviral function of PMLIV against EMCV and VZV was associated with a unique capacity to sequester viral proteins crucial for viral production, which suggests that PMLIV NBs perform an important role in intrinsic host defense against infection of RNA and DNA viruses.
PML−/− MEFs are more sensitive than wild-type MEFs to infection with EMCV (12). However, IFN-α inhibits EMCV production in PML−/− MEFs (data not shown), indicating that PML is not the sole mediator and that other IFN-induced proteins are involved in this process. The 2′,5′A synthetase is induced by IFN and activated by double-stranded RNA (dsRNA) (33). The replication of EMCV, which produces dsRNA during its life cycle, is suppressed in IFN-treated cells as a direct result of RNA decay by the activated 2′,5′A-dependent RNase L (33). PKR is also activated during EMCV infection resulting in phosphorylation of the small subunit of protein synthesis initiation factor eIF2 and reduction of EMCV production (26). However, IFN still inhibits EMCV production in cells derived from PKR, RNase L, and Mx triply deficient mice (42), implicating other suppression mechanisms. In addition to PML/TRIM19, several members of the TRIM superfamily are involved in innate immunity (28). For example, TRIM22 confers resistance to EMCV by targeting the viral 3Cpro for ubiquitin-mediated degradation (11).
Several reports describe inhibitory effects of PML against DNA and RNA viruses (13, 16, 34). Focusing on RNA viruses, PMLIII confers resistance to poliovirus in a p53-dependent manner by inducing PML SUMOylation, leading to PML-dependent p53 activation and to apoptosis in infected cells (30). In cells in which p53 is inactive, PMLIII confers resistance to influenza virus, VSV, and HFV, and PMLIV inhibits rabies virus. The mechanism by which PML inhibits VSV, influenza virus, and rabies virus is unknown, whereas that conferring resistance to HFV has been elucidated (35): PMLIII represses HFV transcription by complexing the HFV transactivator, Tas, preventing its binding to DNA and thereby inhibiting HFV replication (35). Thus, generating several PML isoforms with differences in their C-terminal region from a single gene could provide the host with a mechanism of defense active against viruses of different families. Understanding the specific role of PML isoforms in the diverse functions should be a specific focus of future studies.
In this report, we reveal a new intrinsic defense mechanism in which PMLIV, through its specific C-terminal region, interacts with 3Dpol, and recruits it to NBs. This sequestration within the NBs may inhibit the viral polymerase and consequently prevent viral RNA synthesis and EMCV replication. This is the first evidence that a specific PML isoform, PMLIV, contributed to a cellular antiviral response against an RNA virus that replicates in the cytoplasm by promoting nuclear sequestration of viral polymerase within PML NBs. Importantly, specific depletion of PMLIV reduced the capacity of IFN-α to protect cells from EMCV infection, a finding that implicates PMLIV as an IFN antiviral mediator in the mechanism of resistance against EMCV.
ACKNOWLEDGMENTS
We thank Ann Palmenberg for the gift of anti-EMCV antibodies, anti-3Dpol antibodies, and the 3Dpol-expressing vector, Pier Guiseppe Pelicci for PMLIVa, and Issay Kitabayashi for PMLIV C-terminal mutants. Also, we thank Danielle Blondel, Marie-Claude Geoffroy, and Laurent Dianoux for critically reading the manuscript.
This study was supported by a grant from the Association pour la Recherche sur le Cancer.
We have no conflicts of interest.
Footnotes
Published ahead of print on 12 October 2011.
REFERENCES
- 1. Aminev A. G., Amineva S. P., Palmenberg A. C. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 95: 59–73 [DOI] [PubMed] [Google Scholar]
- 2. Anderson S. L., Carton J. M., Lou J., Xing L., Rubin B. Y. 1999. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 256: 8–14 [DOI] [PubMed] [Google Scholar]
- 3. Blondel D., Kheddache S., Lahaye X., Dianoux L., Chelbi-Alix M. K. 2010. Resistance to rabies virus infection conferred by the PMLIV isoform. J. Virol. 84: 10719–10726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonilla W. V., et al. 2002. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 76: 3810–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borden K. L. 1998. RING fingers and B-boxes: zinc-binding protein-protein interaction domains. Biochem. Cell Biol. 76: 351–358 [DOI] [PubMed] [Google Scholar]
- 6. Brand P., Lenser T., Hemmerich P. 2010. Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys. 3: 3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chakrabarti A., Jha B. K., Silverman R. H. 2011. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 31: 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chebath J., Benech P., Revel M., Vigneron M. 1987. Constitutive expression of (2′-5′) oligo A synthetase confers resistance to picornavirus infection. Nature 330: 587–588 [DOI] [PubMed] [Google Scholar]
- 9. Chelbi-Alix M. K., Quignon F., Pelicano L., Koken M. H., de The H. 1998. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 72: 1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coccia E. M., et al. 1990. A full-length murine 2-5A synthetase cDNA transfected in NIH-3T3 cells impairs EMCV but not VSV replication. Virology 179: 228–233 [DOI] [PubMed] [Google Scholar]
- 11. Eldin P., et al. 2009. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 90: 536–545 [DOI] [PubMed] [Google Scholar]
- 12. El Mchichi B., et al. 2010. SUMOylation promotes PML degradation during encephalomyocarditis virus infection. J. Virol. 84: 11634–11645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Everett R. D., Chelbi-Alix M. K. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89: 819–830 [DOI] [PubMed] [Google Scholar]
- 14. Fitzgerald K. A. 2011. The interferon inducible gene: viperin. J. Interferon Cytokine Res. 31: 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fogal V., et al. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19: 6185–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geoffroy M. C., Chelbi-Alix M. K. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31: 145–158 [DOI] [PubMed] [Google Scholar]
- 17. Haller O., Kochs G. 2011. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 31: 79–87 [DOI] [PubMed] [Google Scholar]
- 18. Hato S. V., et al. 2010. Differential IFN-alpha/beta production suppressing capacities of the leader proteins of mengovirus and foot-and-mouth disease virus. Cell. Microbiol. 12: 310–317 [DOI] [PubMed] [Google Scholar]
- 19. Ishov A. M., et al. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147: 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen K., Shiels C., Freemont P. S. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene 20: 7223–7233 [DOI] [PubMed] [Google Scholar]
- 21. Kamitani T., et al. 1998. Identification of three major sentrinization sites in PML. J. Biol. Chem. 273: 26675–26682 [DOI] [PubMed] [Google Scholar]
- 22. Kawai T., Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7: 131–137 [DOI] [PubMed] [Google Scholar]
- 23. Krieghoff-Henning E., Hofmann T. G. 2008. Role of nuclear bodies in apoptosis signalling. Biochim. Biophys. Acta 1783: 2185–2194 [DOI] [PubMed] [Google Scholar]
- 24. Kristiansen H., Gad H. H., Eskildsen-Larsen S., Despres P., Hartmann R. 2011. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 31: 41–47 [DOI] [PubMed] [Google Scholar]
- 25. Lallemand-Breitenbach V., et al. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 193: 1361–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meurs E. F., et al. 1992. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J. Virol. 66: 5805–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Negorev D., Maul G. G. 2001. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 20: 7234–7242 [DOI] [PubMed] [Google Scholar]
- 28. Ozato K., Shin D. M., Chang T. H., Morse III H. C. 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8: 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palmenberg A. C., et al. 1984. The nucleotide and deduced amino acid sequences of the encephalomyocarditis viral polyprotein coding region. Nucleic Acids Res. 12: 2969–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pampin M., Simonin Y., Blondel B., Percherancier Y., Chelbi-Alix M. K. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 80: 8582–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pichlmair A., Reis e Sousa C. 2007. Innate recognition of viruses. Immunity 27: 370–383 [DOI] [PubMed] [Google Scholar]
- 32. Pindel A., Sadler A. 2011. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 31: 59–70 [DOI] [PubMed] [Google Scholar]
- 33. Player M. R., Torrence P. F. 1998. The 2-5A system: modulation of viral and cellular processes through acceleration of RNA degradation. Pharmacol. Ther. 78: 55–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Regad T., Chelbi-Alix M. K. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20: 7274–7286 [DOI] [PubMed] [Google Scholar]
- 35. Regad T., et al. 2001. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 20: 3495–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reichelt M., et al. 2011. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog. 7: e1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sadler A. J., Williams B. R. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen M., et al. 2008. Picornavirus genome replication. Identification of the surface of the poliovirus (PV) 3C dimer that interacts with PV 3Dpol during VPg uridylylation and construction of a structural model for the PV 3C2-3Dpol complex. J. Biol. Chem. 283: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strauss D. M., Wuttke D. S. 2007. Characterization of protein-protein interactions critical for poliovirus replication: analysis of 3AB and VPg binding to the RNA-dependent RNA polymerase. J. Virol. 81: 6369–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshida H., et al. 2007. PML-retinoic acid receptor alpha inhibits PML IV enhancement of PU.1-induced C/EBPepsilon expression in myeloid differentiation. Mol. Cell. Biol. 27: 5819–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhong S., et al. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95: 2748–2752 [PubMed] [Google Scholar]
- 42. Zhou A., Paranjape J. M., Der S. D., Williams B. R., Silverman R. H. 1999. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology 258: 435–440 [DOI] [PubMed] [Google Scholar]