

Abstract
Herpes simplex virus 1 (HSV-1) glycoprotein K (gK) is expressed on virions and functions in entry, inasmuch as HSV-1(KOS) virions devoid of gK enter cells substantially slower than is the case for the parental KOS virus (T. P. Foster, G. V. Rybachuk, and K. G. Kousoulas, J. Virol. 75:12431–12438, 2001). Deletion of the amino-terminal 68-amino-acid (aa) portion of gK caused a reduction in efficiency and kinetics of virus entry similar to that of the gK-null virus in comparison to the HSV-1(F) parental virus. The UL20 membrane protein and gK were readily detected on double-gradient-purified virion preparations. Immuno-electron microscopy confirmed the presence of gK and UL20 on purified virions. Coimmunoprecipitation experiments using purified virions revealed that gK interacted with UL20, as has been shown in virus-infected cells (T. P. Foster, V. N. Chouljenko, and K. G. Kousoulas, J. Virol. 82:6310–6323, 2008). Scanning of the HSV-1(F) viral genome revealed the presence of a single putative tobacco etch virus (TEV) protease site within gD, while additional TEV predicted sites were found within the UL5 (helicase-primase helicase subunit), UL23 (thymidine kinase), UL25 (DNA packaging tegument protein), and UL52 (helicase-primase primase subunit) proteins. The recombinant virus gDΔTEV was engineered to eliminate the single predicted gD TEV protease site without appreciably affecting its replication characteristics. The mutant virus gK-V5-TEV was subsequently constructed by insertion of a gene sequence encoding a V5 epitope tag in frame with the TEV protease site immediately after gK amino acid 68. The gK-V5-TEV, R-gK-V5-TEV (revertant virus), and gDΔTEV viruses exhibited similar plaque morphologies and replication characteristics. Treatment of the gK-V5-TEV virions with TEV protease caused approximately 32 to 34% reduction of virus entry, while treatment of gDΔTEV virions caused slightly increased virus entry. These results provide direct evidence that the gK and UL20 proteins, which are genetically and functionally linked to gB-mediated virus-induced cell fusion, are structural components of virions and function in virus entry. Site-specific cleavage of viral glycoproteins on mature and fully infectious virions utilizing unique protease sites may serve as a generalizable method of uncoupling the roles of viral glycoproteins in virus entry and virion assembly.
INTRODUCTION
Membrane fusion phenomena are of paramount importance in the infectious life cycle of herpes simplex virus 1 (HSV-1). HSV-1 enters cells predominantly via fusion of its viral envelope with cellular plasma membranes in a pH-independent manner. An alternative pathway involves receptor-mediated endocytosis and fusion of the viral envelope with endocytic membranes facilitated by the low-pH environment of endosomes (30). The virus can spread from infected to uninfected cells by causing virus-induced cell fusion, allowing virions to enter uninfected cells without being exposed to extracellular spaces. These membrane fusion phenomena are known to be mediated by viral glycoproteins embedded in the viral envelope and expressed on infected cellular plasma membranes (reviewed in reference 33).
Virus entry into susceptible cells involves the coordinated functions of glycoproteins gD, gB, gH, and gL (3, 8, 21, 25), while a fifth glycoprotein, gC, is known to enhance initial binding of the virus to cellular membranes (20). The virion glycoproteins gB and gC bind to glycosaminoglycan (GAG) moieties of cell surface proteoglycans (20, 36). This initial attachment of virions to cellular membranes is thought to facilitate subsequent interaction of gD with one or more of its specific receptors, including the herpesvirus entry mediator (HVEM, or HveA), nectin-1 (HveC), or 3-O-sulfated heparan sulfate (17, 27, 35). Apparently, gB can also bind to additional receptors, including paired immunoglobulin-like type 2 receptor alpha (PILR-α), nonmuscle myosin heavy chain IIA (NMHC-IIA), and myelin-associated glycoprotein (MAG), that function in virion attachment and virus entry (1, 34, 38). Recent data support the hypothesis that gB is the sole fusion glycoprotein, since it is the only glycoprotein that possesses features of other known viral fusion glycoproteins, such as the well-characterized vesicular stomatitis virus (VSV) G glycoprotein (19, 32). Virus-induced cell fusion is thought to be mediated by a mechanism very similar to that occurring during fusion of the viral envelope with cellular membranes, inasmuch as the viral glycoproteins gD, gB, gH, and gL and the presence of viral receptors are also required for virus-induced cell fusion (2, 31, 40, 41). However, virus-induced cell fusion requires the presence of additional viral glycoproteins and membrane proteins, including gE, gI, gM, gK, and the UL20 and UL45 proteins (5, 7, 12, 18, 26, 43).
We reported that gK is a structural component of virions as a Golgi complex-dependent glycosylated species and functions in virus entry, inasmuch as virions lacking gK enter susceptible cells in cell culture substantially slower (15). Furthermore, we showed that HSV-1 gK and UL20 functionally and physically interact and that these interactions are absolutely necessary for their coordinate intracellular transport, cell surface expression, and functions in the HSV-1 life cycle (10, 14). Recently, we showed that a peptide composed of the amino-terminal 82 amino acids (aa) (including the predicted 30-aa signal sequence) of gK (gKa) complemented in trans for gB-mediated cell fusion (5). In addition, we demonstrated that UL20 and the gKa peptide physically interacted with gB and gH in infected cells, suggesting that gB-mediated virus-induced cell fusion is regulated via direct interactions with gK and UL20 (6).
In this work, we sought to confirm the presence of gK on purified virion particles and to assess whether the UL20 protein is also a structural component of virion particles in association with gK and function in virus entry. A substantial difficulty in assessing the role of gK in virus entry is the fact that a lack of gK leads to drastic defects in cytoplasmic virion envelopment and infectious virus production (11, 22, 23). We present here a generalizable strategy to modify/inactivate gK or potentially any other viral glycoprotein on mature virion particles, effectively uncoupling the roles of gK in cytoplasmic virion envelopment and virus entry.
MATERIALS AND METHODS
Cell culture.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Rockville, MD). VK302 cells permanently expressing gK were originally obtained from David Johnson (Oregon Health Sciences University, Portland, OR). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics.
Bioinformatics analysis of predicted protease sites within HSV-1 proteins.
The ExPASy PeptideCutter software tool (16), available at the ExPASy Bioinformatics Resource portal (http://web.expasy.org/peptide_cutter/), was utilized to predict potential tobacco etch virus (TEV) cleavage sites within each predicted amino acid sequence of the HSV-1(F) genome (GenBank accession number GU734771). The PeptideCutter algorithm predicted the presence of a single putative TEV site within gD at amino acid 235, while additional TEV predicted sites were found within the UL5 (helicase-primase helicase subunit; single TEV predicted site at amino acid 466), UL23 (thymidine kinase; two TEV predicted sites at amino acid 250 and 331), UL25 (DNA packaging tegument protein; one TEV site at amino acid 533), and UL52 (helicase-primase primase subunit; one TEV site at amino acid 184). Treatment of purified virions will access only structural envelope proteins, such as gD, and not any of the other nonstructural viral proteins containing TEV sites.
Construction of HSV-1 recombinant mutants.
The recombinant mutant YE102-VC1 was engineered to express gK and UL20 containing the V5 and 3×FLAG antigenic epitopes, respectively. Specifically, a DNA sequence encoding the V5 epitope and the enterokinase recognition site, Asp-Asp-Asp-Asp-Lys, was inserted in frame immediately after amino acid 68 of gK (including the predicted 30-aa signal sequence). The 3×FLAG epitope was inserted in frame at the amino terminal of UL20, as described earlier (24). All recombinant viruses were constructed using the markerless two-step red-mediated recombination mutagenesis system (42) on the bacterial artificial chromosome (BAC) plasmid pYEbac102 carrying the HSV-1(F) genome or the BAC plasmid pYK608 carrying the HSV-1(F) genome modified to express three fluorescent proteins as fusion proteins with gB, VP22, and VP26 (gifts from Yasushi Kawaguchi, University of Tokyo, Tokyo, Japan). The mutant virus gDΔTEV was constructed by altering the DNA sequence within the gD gene predicted to specify a TEV protease recognition site on the bacterial artificial chromosome YK608 genome. Specifically, gD amino acid 235 was changed from glutamine to asparagine (Fig. 1). The gDΔTEV virus was subsequently used to generate the gK-V5-TEV recombinant virus, which expressed gK containing the V5 epitope and the TEV protease recognition site ENLYFQG inserted in frame immediately after amino acid 68 of gK.
Fig. 1.

Schematic representation of mutant viral genomes. (A) HSV-1(F)-YE102-VC1 recombinant virus generated from pYEbac102, carrying an in-frame insertion of the V5 epitope and enterokinase recognition site inserted immediately after amino acid 68 of gK, as well as an in-frame insertion of the 3×FLAG epitope at the amino terminus of UL20. (B) Schematic representation of recombinant viruses constructed for TEV-specific proteolytic activity. The V5 epitope tag and the TEV protease site (ENLYFQG) were inserted immediately after amino acid 68 of gK to generate mutant virus gK-V5-TEV.
Virus entry assay.
The construction and characterization of the gK mutant virus gKΔ31-68 lacking 68 aa from its amino terminus (including the 30-aa signal sequence) was recently described (5). The relative entry efficiencies of the wild-type HSV-1(F) YE102, gK-null (ΔgK), and gKΔ31-68 viruses were tested on Vero cells by determining the number of cells expressing viral antigens after entry was allowed to occur for 1 h at 34°C. Specifically, viruses at a multiplicity of infection (MOI) of 1 were allowed to attach to Vero cell monolayers at 4°C for 1 h. The virus inoculum was subsequently removed, warm medium (34°C) was added, and the cultures were shifted to 34°C to allow virus penetration. After 1 h at 34°C, extracellular virions were inactivated by treatment with low-pH buffer (0.1 M glycine, pH 3.0), and cells were washed three times with phosphate-buffered saline (PBS). Twelve hours postinfection (hpi), the cells were fixed and stained with anti-ICP4 antibody (Virusys, Inc., Taneytown, MD). The relative efficiency of virus entry was calculated as the percentage of cells expressing ICP4 after low-pH treatment relative to results for PBS-treated cells by flow cytometry. Mean values and standard deviations of three independent experiments were calculated. Similarly, kinetics of virus entry were determined at 34°C by measuring the number of ICP4-positive Vero cells by flow cytometry at 12 hpi. The percent positive cells for each time point (10, 20, 30, and 60 min) was determined for a total number of 5,000 Vero cells infected at an MOI of 1.
Growth kinetics and plaque morphology of wild-type and mutant viruses.
Growth kinetics studies were performed essentially as we have described earlier (11, 13, 15). Briefly, viruses were adsorbed onto a 90%-confluent monolayer of Vero cells in a 12-well plate at 4°C for 1 h at a low multiplicity of infection (MOI = 0.2). Subsequently, cells were incubated at 37°C and 5% CO2 for 1 h to allow penetration of the virus. Any remaining extracellular virions were inactivated by briefly treating the cells with PBS at low pH (pH 3.0). Virus stocks were prepared from infected cells at 6, 12, 24, 36, and 48 hpi, and the virus titers were obtained by endpoint titration on Vero cell in triplicate. Viral plaques were stained using anti-HSV-1 polyclonal antibody, as described previously (5). Viral plaques were also visualized by fluorescence microscopy.
Virus entry kinetics of TEV protease-treated virions.
Partially purified virions (generated by concentrating viruses by ultracentrifugation of infected cell supernatants) were used for enzyme digestion studies. Equal PFU of recombinant mutant viruses gDΔTEV and gK-V5-TEV were treated as follows: (i) PBS alone (all PBS used for these experiments was at pH 6.8), (ii) PBS plus 1 unit of AcTEV protease (Invitrogen, Inc., Carlsbad, CA), and (iii) PBS plus 0.01% SDS-inactivated AcTEV protease. SDS-inactivated enzyme was passed through Zeba spin desalting columns (Thermo Fisher Scientific, Waltham, MA) to eliminate SDS. All treatments were kept at 30°C for 1 h, mixed with serum-free medium, and adsorbed to a 90%-confluent monolayer of Vero cells on a 12-well plate. The plates were rocked gently for 1 h at room temperature. The virus inoculum was removed, and the wells were overlaid with 1 ml of DMEM containing 1% methylcellulose and 2% fetal bovine serum (FBS). Cells were fixed at 48 hpi with ice-cold methanol for 10 min, and immunocytochemistry was performed as described above. The effect of TEV protease treatment on the kinetics of virus entry was performed essentially as described earlier, with the exception that viruses were treated with TEV protease or PBS and that the number of infectious virions was determined via limited dilution on Vero cells, as described previously (15).
TEV digestion of infected cell extracts and immunoblot assays.
Nearly confluent monolayers of Vero cells in a T-75 flask were infected with gK-V5-TEV virus. The cells were collected 48 hpi and lysed with mammalian protein extraction reagent (M-PER; Pierce, Rockford, IL) lysis buffer containing protease inhibitors and briefly sonicated. The gK-V5-TEV protein was immunoprecipitated with protein G magnetic beads bound to V5 antibody (Invitrogen, Carlsbad, CA). The eluted protein was treated with the AcTEV protease as suggested by the manufacturer (Invitrogen). Sample buffer containing 5% β-mercaptoethanol was added to the digested protein and heated at 55°C for 15 min. Proteins were resolved on 4 to 20% SDS-PAGE gel and immobilized on nitrocellulose membranes. Immunoblot assays were carried out using mouse anti-V5 antibody and goat anti-mouse horseradish peroxidase (HRP) light-chain secondary antibody (Abcam, Inc., Cambridge, MA).
Virus purification.
Wild-type HSV-1(F) pYEbac102 and double-labeled YE102-VC1 (gK-EK-V5; UL20-3×FLAG) virions were purified by two serial passages using iodixanol gradients. Briefly, supernatants and cells from 24 T-150 flasks of Vero cells infected with either pYEbac102 or YE102-VC1 were collected at 24 and 36 hpi, respectively, and treated with TNE (10 mM Tris; 500 mM NaCl; 1 mM EDTA; pH 7.5) buffer for 15 min at 4°C. Subsequently, the cell pellet was separated by centrifugation at 6,000 × g for 45 min at 4°C. Polyethylene glycol (PEG) (molecular weight, 8,000; 7% [wt/vol]) was added to the supernatant and kept on a stirrer overnight at 4°C. The next day, this solution was spun at 6,000 × g for 30 min at 4°C. The resulting pellet was resuspended in 10 ml of TNE buffer, loaded onto a 50 to 20% discontinuous iodixanol gradient, and spun at 141,000 × g for 3 h. The viral band from the 30 to 40% interface was collected, loaded onto a second 50 to 20% iodixanol step gradient, and centrifuged again at 141,000 × g for 3 h at 4°C. The resulting optically dense band was centrifuged through a 20% iodixanol cushion at 141,000 × g for 1 h at 4°C. The resulting pellet was resuspended in 250 μl of NP-40 lysis buffer (Invitrogen) and used for immunoprecipitation and immunoblot assays.
Immunoprecipitation and immunoblot assays.
The gK-V5-EK (gK tagged with V5 and enterokinase amino acid sequence) protein was immunoprecipitated with protein G magnetic Dynabeads bound to V5 antibody according to the manufacturer's instructions (Invitrogen). The protein was eluted from the magnetic beads in 40 μl of elution buffer and used for immunoblot assays. Immunoblot assays were carried out using anti-glyceraldehyde-3-phosphate-dehydrogenase (anti-GAPDH) (Chemicon, International Inc.), anti-gB, anti-gD, anti-gC (Virusys), anti-FLAG (Sigma-Aldrich, Inc., St. Louis, MO), anti-V5 antibody (Invitrogen), goat anti-mouse HRP against light-chain (Fab), heavy chain (Fc) whole mouse IgG molecule (Abcam), and anti-ICP8 monoclonal antibody (Abcam).
Immunogold labeling for TEM.
Semipurified virions immobilized on 400-mesh Butvar/carbon-coated nickel grids (Electron Microscopy Sciences, Inc., Hatfield, PA) were used to detect the presence of gK, gB, and UL20 on the virions using immunogold labeling, as has been described previously for bovine herpesvirus 1 (37). Briefly, 5 μl of semipurified virions suspension was adsorbed onto the coated surface of the grids for 30 min. The grids were then blocked using 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 5 min. Excess blocking buffer was removed using the tip of a filter paper, followed by incubation of the grids with 5 μl of mouse anti-V5 antibody (Invitrogen) at a dilution of 1:10,000 in 1% BSA in TBS for 30 min. This sample preparation step was followed by washing the grids with 1% BSA in TBS for 5 min. A 5-μl droplet of goat anti-mouse IgG (whole molecule)-Gold 10-nm colloidal gold (Sigma-Aldrich) at 1:80 dilution in 1% BSA in TBS was added on top of each grid and incubated for 30 min. The grids were then washed with 1% BSA in TBS by adding 5 μl on each grid, and excess liquid was removed with the tip of a filter paper. A 2% solution of sodium phosphotungstate, pH 6.8, was added as a final step for contrast purposes. Grids were desiccated and visualized by transmission electron microscopy (TEM).
RESULTS
The UL20 membrane protein and gK are structural components of HSV-1 virions.
To assess whether both gK and UL20 are present and interact as structural components of HSV-1 virions, the recombinant virus HSV-1(F)-YE102-VC1 was constructed by modifying the gK and UL20 amino acid sequences to contain V5 and 3×FLAG antigenic epitopes, respectively (Fig. 1A). The V5 epitope tag was inserted immediately after gK amino acid 68 (this amino acid numbering includes the predicted signal sequence), while the 3×FLAG epitope was inserted at the N terminus of the UL20 protein, as described previously for similar gene constructions (9, 10, 14) and detailed in Materials and Methods. The YE102-VC1 virus replicated efficiently in Vero cells; however, average plaque sizes were consistently smaller, by approximately 30%, than those of the parental HSV-1(F)-YE102 virus (Fig. 2A). Similarly, YE102-VC1 replicated slower than the YE102 parental virus, approaching viral titers that were approximately half a log lower than YE102 at 24 hpi (Fig. 2B). However, at longer times postinfection (36 to 48 hpi), titers of both viruses were similar (not shown).
Fig. 2.

Replication kinetics and plaque morphology of the YE102-VC1 virus in comparison to those of HSV-1(F)-YE102. (A) Representative viral plaques of HSV-1(F)-YE102 and HSV-1(F)-YE102-VC1 on Vero cells at 48 hpi. Viral plaques were visualized by phase-contrast microscopy after immunostaining with anti-HSV-1 polyclonal antibody. (B) One-step replication kinetics of HSV-1(F)-YE102 and HSV-1(F)-YE102-VC1 at an MOI of 0.2 on Vero cells. The x axis shows hours postinfection, and the y axis shows PFU/ml. All experiments were performed in triplicate. Error bars represent standard errors of the means.
YE102-VC1 virions were purified by sequential isopycnic density sedimentation in iodixanol discontinuous gradients as described in Materials and Methods. The cellular protein GAPDH was absent from gradient fractions containing infectious virions, indicating the relative absence of contaminating cellular proteins, while GAPDH was readily detected in control cellular extracts (Fig. 3, panel labeled α-GAPDH). Similarly, the nonstructural, immediate-early protein ICP8 was not detected in purified virion samples, while it was detected readily in samples obtained from infected cells (Fig. 3, panel labeled α-ICP8). Purified virions were tested for the presence of the viral glycoproteins gB, gD, gC, and gK and the membrane protein UL20 using monoclonal antibodies against gB, gD, gC, anti-V5 (gK), and anti-3×FLAG (UL20) in Western immunoblots. Monoclonal antibody to gB detected putative mature gB protein species migrating with an apparent molecular mass of approximately 120 kDa, while a relatively prominent gB-related species of approximately 45 kDa was also detected (Fig. 3, panel labeled α-gB). Monoclonal antibody to gD detected a single protein species migrating with an apparent molecular mass of approximately 55 kDa (Fig. 3, panel labeled α-gD), while antibody to gC detected a protein with an apparent molecular mass of 125 kDa. Anti-3×FLAG antibody readily detected the presence of the UL20 protein migrating with a characteristic ladder pattern, with the most prominent protein species migrating with an apparent molecular mass of approximately 27 kDa, in agreement with our previous findings (6), as well as UL20-associated protein species migrating with apparent molecular masses of approximately 35 and 45 kDa (Fig. 3, panel α-FLAG). The anti-V5 antibody detected the presence of the V5-tagged gK-associated protein species migrating with an apparent molecular mass of approximately 65 to 75 kDa, which was more than twice the apparent molecular mass of glycosylated species of gK previously detected in infected cells (5, 6). The anti-V5 (gK) antibody immunoprecipitated gK migrating with an apparent molecular mass of 125 kDa and coimmunoprecipitated UL20 migrating at approximately 27 kDa, which was detected with the anti-3×FLAG antibody (Fig. 3, panel labeled IP:V5Ab).
Fig. 3.
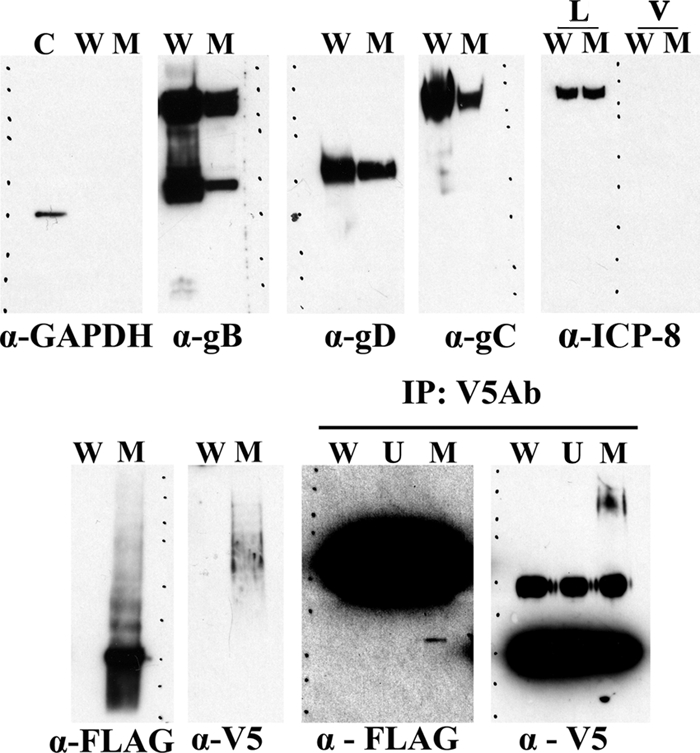
Detection of gK and UL20 on gradient-purified virions. Western immunoblot analysis of double-gradient-purified virions was performed using anti-V5 (gK) and anti-FLAG (UL20) antibodies. Individual lanes are labeled as follows: C, positive control for GAPDH; W, lysates from purified wild-type virus [HSV-1(F)-YE102]; M, lysates from purified mutant virus YE102-VC1; L, lysates from infected Vero cells; V, samples derived from purified virions. Individual blots were probed with anti-GAPDH, anti-ICP8, anti-gB, anti-gD, anti-gC, anti-FLAG (UL20), or anti-V5 (gK) antibody as indicated. The panel labeled at the top as IP:V5Ab shows immunoblots of immunoprecipitations with anti-V5 (gK) antibody probed with either anti-FLAG (UL20) or anti-V5 (gK) antibodies. In this panel, lanes are immunoprecipitates from mock-infected Vero cells (U), HSV-1(F)-YE102 purified virions (W), or YE102-VC1 purified virions (M). Molecular mass standards are shown with dots on each panel (250, 150, 100, 75, 50, 37, 25, 20, 15, and 10 kDa; Precision Plus protein standards; Bio-Rad). HRP-conjugated goat anti-mouse (HRP-GAb; IgG) was used for all data, except data shown on the panel labeled IP:V5Ab, wherein the F(ab)2- and Fc-purified portions of the HRP-GAb IgG were used for the α-V5 and α-FLAG panels, respectively.
The presence of gK on virion particles was further assessed using colloidal gold immuno-electron microscopy (Fig. 4). More than 100 virion particles for each virus were visually scanned for the presence of gold particles. V5-tagged gK was readily detected on most virion envelopes (>90 out of 100 visualized virions contained gold particles) of recombinant viruses VC1 and gK-V5-TEV, both of which contained the V5 epitope tag inserted after gK amino acid 68 (Fig. 4B and C), while the antibody did not react with the VC1 parental virus YE102 (none of 100 visualized virions contained gold particles) (Fig. 4A). Similarly, anti-gB monoclonal antibody readily detected gB on most visualized virions (not shown).
Fig. 4.

Transmission immunogold electron microscopy to detect gK and gB on virion particles. (A) HSV-1(F)-YE102 virions stained with anti-gK (anti-V5) antibody; (B) YE102-VC1 stained with anti-gK (anti-V5) antibody; (C) gK-V5-TEV stained with anti-gK (anti-V5) antibody.
The amino terminus of gK functions in virus entry.
Previously, we reported that deletion of 68 aa from the amino terminus of gK (including the 30-aa signal sequence; actual deletion of 38 aa after cleavage of the predicted signal sequence) allowed efficient virus envelopment and infectious virus production, while gB-mediated virus-induced cell fusion was abolished (5). The entry efficiency of the recombinant virus gKΔ31-68, which lacks the first 68 aa of gK, was compared to those of HSV-1(F; YE102) and ΔgK viruses. The construction and characterization of these viruses have been described previously (5). Flow cytometry was utilized to determine the relative efficiencies and kinetics of entry of these viruses into Vero cells (see Materials and Methods). Both ΔgK and gKΔ31-68 entered Vero cells less efficiently than their parental virus, YE102 (Fig. 5A and B).
Fig. 5.

Assessment of the efficiencies of gK-null and gKΔ31–68 virus entry into Vero cells. (A) The relative efficiency of entry is shown as the percentage of cells expressing ICP4 at 12 hpi. Mean values and standard deviations for three independent experiments were calculated as shown. (B) Representative kinetics of virus HSV-1(F), gK-null, and gKΔ31-68 entry into Vero cells. Vero cell monolayers were infected with an MOI of 1. At different times postentry at 34°C (10, 20, 30, and 60 min), viruses that had not entered yet were inactivated with low pH, and the kinetics of virus entry were determined by monitoring ICP4 expression at 12 hpi using flow cytometry. For each sample, the percent ICP4-positive cells in a population of 5,000 Vero cells was measured.
To further confirm that deletion of the amino-terminal 68 aa of gK caused defects in virus entry, we utilized incorporation of a unique TEV protease site within gK to selectively delete the amino terminus of gK from mature virions. Scanning of the HSV-1(F) viral genome revealed the presence of a single putative TEV site within gD, while additional TEV predicted sites were found within the UL5 (helicase-primase helicase subunit), UL23 (thymidine kinase), UL25 (DNA packaging tegument protein), and UL52 (helicase-primase primase subunit) (see Materials and Methods). The recombinant virus gDΔTEV was constructed via site-directed mutagenesis of the YK608 genome, changing the single putative gDTEV site from PQAYQQG to PQAYQNG (Q235 to N235; bold and underlined). Subsequently, the recombinant virus gK-V5-TEV was constructed by inserting a 19-aa gene segment coding for the V5 epitope tag fused in frame with the predicted TEV protease site (ENLYFQG), inserted immediately after gK amino acid 68. Both gDΔTEV and gK-V5-TEV viruses produced average-size plaques that were approximately 30% smaller than those of the YK608 parental virus at 48 hpi, while all viruses expressed the characteristic fluorescence pattern of green, red, and cyan due to fluorescent proteins fused in frame to the VP26, VP22, and gB viral proteins, respectively (Fig. 6A) (39). The gDΔTEV and gK-V5-TEV viruses replicated efficiently, although slower in Vero cells than was the case for their parental wild-type virus, YK608, while the two mutant viruses approached similar viral titers at 48 hpi (Fig. 6B). A gK-V5-TEV revertant (R-gK-V5-TEV) virus was generated by replacing the gK-V5-TEV gene with the wild-type HSV-1(F) gK gene using two-step red-mediated recombination mutagenesis. The R-gK-V5-TEV virus exhibited plaque morphology and growth characteristics similar to those of the gDΔTEV virus, suggesting that gK-V5-TEV did not contain any secondary site mutations (not shown).
Fig. 6.

Construction and characterization of recombinant viruses containing TEV modifications. (A) Representative plaque morphology of the gDΔTEV and gK-V5-TEV viruses in comparison to those of their parental HSV-1(F)-YK608 virus at 48 hpi. Viral plaques were visualized after immunohistochemical staining using phase-contrast microscopy and by fluorescence microscopy. (B) Replication kinetics of HSV-1(F) YK608, gDΔTEV, and gK-V5-TEV viruses on Vero cells infected with an MOI of 0.2. The x axis shows hours postinfection, and the y axis shows PFU/ml. All experiments were performed in triplicate. Error bars represent standard errors of the means.
To ascertain whether the TEV protease was active against the gK-V5-TEV tagged protein, immunoprecipitates of gK prepared from gK-V5-TEV virus-infected cells were treated with the TEV protease as described in Materials and Methods. Treatment of cellular extracts with the TEV protease caused a substantial reduction in the amount of mature gK migrating with the expected molecular mass of approximately 40 kDa in comparison to results for phosphate-buffered saline (PBS)-treated samples. In contrast, inactivated TEV protease failed to reduce the amount of mature gK (Fig. 7A and B).
Fig. 7.

Treatment of infected cell extracts. (A) Cell extracts from gK-V5-TEV-infected cells were immunoprecipitated with anti-V5 antibody and mock or TEV treated for 1 h at 30°C. Immunoprecipitates were tested for the presence of gK using anti-V5 antibody. Lane 1 (PBS), cell extracts mock digested in PBS buffer; lane 2 (PBS + E), cell extracts digested with AcTEV in PBS; lane 3 [PBS + INACT.E (CP)], PBS plus inactivated enzyme passed through a desalting column; lane 4 [PBS + E (CP)], PBS plus enzyme passed through a desalting column. (B) Densitometric analysis of immunoblots shown in panel A.
To assess whether TEV treatment of recombinant viruses affected virus entry, partially purified and concentrated virion stocks were treated with the TEV protease and viral infectivity was assessed by determining the number of PFU produced after TEV treatment on Vero cells, as described in Materials and Methods. TEV treatment of gK-V5-TEV virions resulted in a 34% or 32% reduction in virus infectivity compared to results for treatment with inactivated TEV protease or PBS (respectively) (Fig. 8A and B). This reduction in virus entry can be appreciated even more when compared to results for TEV treatment of gDΔTEV, which appeared to be modestly protected after TEV treatment, exhibiting a 14% or 10% increase in viral infectivity compared to inactivated TEV protease or PBS treatment, respectively. Similar results were obtained when kinetics of viral entry were obtained by inactivating virion remaining on the cell surface at different times using the low-pH inactivation protocol (see Materials and Methods). TEV treatment resulted in a consistent reduction of gK-V5-TEV virion entry at different time points, while it did not have any appreciable effect on gDΔTEV virion entry (Fig. 8C and D).
Fig. 8.

The effect of TEV protease treatment on virion infectivity. gDΔTEV (A) or gK-V5-TEV (B) virus obtained from supernatants of infected cells was treated with PBS, AcTEV protease (PBS + E), or SDS-inactivated AcTEV protease [PBS + INACT.E (CP)], and the numbers of resultant PFU were determined. Similarly, the relative kinetics of viral entry were determined for gDΔTEV (C) or gK-V5-TEV (D) in the presence (PBS + E) or absence (PBS) of the AcTEV protease. These experiments were done in triplicate, and error bars represent standard errors of the means. Asterisks in panel B designate statistically significant differences as calculated by one-way analysis of variance (ANOVA) (P < 0.05).
DISCUSSION
HSV-1 enters by fusion of the viral envelope with cellular membranes and can spread from infected to uninfected cells via virus-induced cell fusion. Both membrane fusion phenomena are thought to be mediated by similar mechanisms involving glycoprotein gB as the sole fusogen, which is regulated by interactions with other viral glycoproteins, membrane proteins, and cellular receptors. Viral glycoproteins and membrane proteins, including gK, gM, gE, gI, and the UL20 membrane protein, have been cast as nonessential for virus entry, while it is largely accepted that they are absolutely essential for virus-induced cell fusion. We report here that both gK and UL20 are structural components of the virion particle and interact with each other, as has been found in infected cellular membranes. Furthermore, we utilized protease-specific cleavage of gK on the mature virion particle to demonstrate that the amino terminus of gK functions in virus entry, in agreement with its role in virus-induced cell fusion (5).
We have shown previously that both gK and UL20 can be modified via insertion of small antigenic epitope tags without drastically affecting their structure and functions in the life cycle of the virus (9, 15). To confirm the presence of gK and detect UL20 on purified virions, the recombinant virus YE102-VC1 was constructed to express gK tagged with the V5 amino acid sequence immediately followed by a 5-aa sequence predicted to be a cleavage site of the enterokinase protease. This 19-aa sequence was inserted in frame after gK amino acid 68 (threonine), while the previously reported V5-alone insertion was immediately after amino acid 69 (histidine) (9). The YE102-VC1 virus was also modified to include a 3×FLAG epitope tag at the amino terminus of UL20, as described previously (10, 14). The VC-1 virus replicated less efficiently in Vero cells and formed smaller plaques than its parental virus, YE102, suggesting that the presence of both gK and UL20 modifications only mildly inhibited virus growth and spread. VC-1 replication and plaque morphology were partially complemented on either gK-expressing VK302 cells or UL20-expressing FRT cells, suggesting that both gK and UL20 modifications were responsible for the mild reduction in infectious virus production and virus spread exhibited by the YE102-VC1 virus (not shown).
UL20 and gK were detected on highly purified virions, as evidenced by the absence of the cellular protein GAPDH and the nonstructural ICP8 viral protein. V5 (gK) antibody immunoprecipitates probed with either anti-V5 antibody or anti-FLAG (UL20) antibodies revealed the presence of UL20 and gK of approximately 27 kDa and 125 kDa, respectively. This 125-kDa apparent molecular mass of gK was much larger than the monomeric form of gK (37 kDa), although potentially dimeric gK protein species of approximately 65 kDa have been detected previously in infected cells (10). These results suggest that UL20 interacted predominantly with multimeric (tetrameric) forms of gK in the virion particle. These multimeric forms of gK were probably not readily detected in infected cell lysates because they could not enter gels in the presence of other cellular proteins. Both gK and UL20 are very sensitive to temperature-dependent aggregation, and samples containing gK and/or UL20 cannot be boiled because they form aggregates that do not enter separating gels. The larger amount of total protein present in gK and UL20 samples obtained from infected cells exacerbates this phenomenon (unpublished observations). The presence of gK on purified virions was confirmed using transmission immuno-electron microscopy. The UL20 protein was also readily detected on virions by TEM, although the anti-UL20 antibody was directed against the amino terminus of UL20, predicted to be located on the inner side of the viral envelope. This staining was probably due to partial breakage of virion envelopes that exposed the UL20 epitope to the antibody (not shown). Collectively, these data support our hypothesis that gK is present in the viral envelope as an interactive complex with UL20, in agreement with our previous findings in infected cells (10, 14).
We showed previously that HSV-1(KOS) gK-null virus entered Vero cells substantially slower than its parental KOS strain (15). Because gK is required for efficient cytoplasmic virion envelopment and infectious virus production, viral entry experiments with gK-null viruses reflect the ability of only a very small portion of potentially aberrantly enveloped virions to enter cells, substantially complicating interpretation of viral entry results. Virus entry experiments with the gKΔ31-68 virus, which carries a deletion of the first 68 aa (38-aa deletion after the signal sequence of 30 aa) of gK, revealed that it entered Vero cells substantially less efficiently than the parental virus, YE102, confirming that the amino terminus of gK is involved in virus entry, as has been found for virus-induced cell fusion (5, 6).
The purpose for engineering the enterokinase site within the amino terminus of gK was to assess whether treatment with enterokinase affected virus entry. Unfortunately, enterokinase treatment of virions did not affect virus entry. Optimum enterokinase protease activity required prolonged incubation at 37°C in the presence of detergent, which was incompatible with the use of this enzyme in virus entry experiments (not shown). As an alternative strategy, we chose to construct recombinant viruses containing TEV protease recognition sites. TEV protease is highly stable at temperatures lower than 37°C and a wide range of pH (6.0 to 8.0) conditions (29). Scanning of the HSV-1(F) genome for potential TEV protease sites revealed the presence of a single putative TEV site within gD among all viral glycoproteins, while additional sites were located within the UL5, UL23, UL25, and UL52 genes. The putative TEV protease site in gD of HSV-1(F) YK608 virus was inactivated by a single amino acid change, glutamine to asparagine, at amino acid position 235. The gD amino acid sequence extending from amino acid 234 to 244 was shown to be highly important for infectious virus production (28). Furthermore, insertion of the amino acid segment REDLP at gD amino acid 235 abrogated viral infectivity and resulted in the loss of binding of monoclonal antibody AP7, which is known to target the discontinuous neutralizing epitope on gD (4). However, the single-amino-acid replacement (glutamine to asparagine) in gD specified by the gDΔTEV virus was apparently well tolerated, since it caused only a modest reduction in the average plaque size and infectious virus production, suggesting that this amino acid position does not drastically affect gD functions.
TEV protease treatment of gK-V5-TEV virions resulted in a more than 30% reduction in virus infectivity compared to results for treatment with either inactivated TEV protease or PBS. This reduction in virus entry can be appreciated even more when compared to TEV protease treatment of the parental gDΔTEV virions, which appeared to slightly enhance virus entry. The inhibition level of gK-V5-TEV entry after TEV protease is in agreement with the observed reduction of gKΔ31-68 virus entry, suggesting that the amino terminus of gK is contributing to virus entry. We have shown previously that the amino-terminal 82-aa region of gK binds to the amino terminus of gB and that this interaction appeared to be important in infectious virus production and virus-induced cell fusion. Therefore, the observed reduction in virus entry after cleavage of the amino-terminal 68 aa of gK is consistent with the hypothesis that this gK peptide may bind gB within the virion particle and modify its ability to mediate viral-envelope-to-cellular-membrane fusion during virus entry.
The results presented herein provide additional evidence that gK and its interacting partner UL20 are structural components of the virion particle and function in virus entry. In this regard, these results support the hypothesis that viral-envelope-to-cell-membrane fusion is similar to virus-induced cell fusion. It is highly likely that functional interactions among viral glycoproteins necessary for virus-induced cell fusion are conserved within the viral envelope, suggesting that deletion of viral glycoproteins other than gB, gD, gH, and gL may affect viral entry, as has been shown here for gK. UL20, gK, and other membrane proteins embedded in the viral envelope may function to optimize gB's fusogenicity for yet-unknown reasons, one of which may be the optimal recognition of various receptors and entry into different types of cells.
The use of specific protease cleavage sites, such as the TEV protease site described here, provides a methodology to investigate the role of viral glycoproteins in virus entry independently of their role in virion assembly. In addition, site-specific cleavage of viral glycoproteins may provide a means of probing dynamic conformational changes of viral glycoproteins during virus entry and virus-induced cell fusion.
ACKNOWLEDGMENTS
This work was supported by grant AI43000 from the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (to K.G.K.), and Core Laboratories supported by the NIH National Center for Research Resources, grant P20 RR020159.
We acknowledge Greg McCormick for assistance with the TEM experiments.
Footnotes
Published ahead of print on 12 October 2011.
REFERENCES
- 1. Arii J., et al. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 2. Atanasiu D., Saw W. T., Cohen G. H., Eisenberg R. J. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cai W. H., Gu B., Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiang H. Y., Cohen G. H., Eisenberg R. J. 1994. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 68:2529–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chouljenko V. N., Iyer A. V., Chowdhury S., Chouljenko D. V., Kousoulas K. G. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J. Virol. 83:12301–12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chouljenko V. N., Iyer A. V., Chowdhury S., Kim J., Kousoulas K. G. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 84:8596–8606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davis-Poynter N., Bell S., Minson T., Browne H. 1994. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J. Virol. 68:7586–7590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai P. J., Schaffer P. A., Minson A. C. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J. Gen. Virol. 69:1147–1156 [DOI] [PubMed] [Google Scholar]
- 9. Foster T. P., Alvarez X., Kousoulas K. G. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 77:499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Foster T. P., Chouljenko V. N., Kousoulas K. G. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 82:6310–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foster T. P., Kousoulas K. G. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Foster T. P., Melancon J. M., Baines J. D., Kousoulas K. G. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 78:5347–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foster T. P., Melancon J. M., Kousoulas K. G. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18–29 [DOI] [PubMed] [Google Scholar]
- 14. Foster T. P., Melancon J. M., Olivier T. L., Kousoulas K. G. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 78:13262–13277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foster T. P., Rybachuk G. V., Kousoulas K. G. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 75:12431–12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gasteiger E., et al. 2005. Identification and analysis tools on the ExPASy server, p. 571–607 In Walker J. M. (ed.), The proteomics protocols handbook. Humana Press, Totowa, NJ [Google Scholar]
- 17. Geraghty R. J., Krummenacher C., Cohen G. H., Eisenberg R. J., Spear P. G. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 18. Haanes E. J., Nelson C. M., Soule C. L., Goodman J. L. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J. Virol. 68:5825–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heldwein E. E., et al. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 20. Herold B. C., WuDunn D., Soltys N., Spear P. G. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hutchinson L., et al. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 66:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hutchinson L., Johnson D. C. 1995. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 69:5401–5413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jayachandra S., Baghian A., Kousoulas K. G. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 71:5012–5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee H. C., Chouljenko V. N., Chouljenko D. V., Boudreaux M. J., Kousoulas K. G. 2009. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J. Virol. 83:6115–6124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ligas M. W., Johnson D. C. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Melancon J. M., Fulmer P. A., Kousoulas K. G. 2007. The herpes simplex virus UL20 protein functions in glycoprotein K (gK) intracellular transport and virus-induced cell fusion are independent of UL20 functions in cytoplasmic virion envelopment. Virol. J. 4:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montgomery R. I., Warner M. S., Lum B. J., Spear P. G. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 28. Muggeridge M. I., Wilcox W. C., Cohen G. H., Eisenberg R. J. 1990. Identification of a site on herpes simplex virus type 1 glycoprotein D that is essential for infectivity. J. Virol. 64:3617–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nallamsetty S., et al. 2004. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr. Purif. 38:108–115 [DOI] [PubMed] [Google Scholar]
- 30. Nicola A. V., McEvoy A. M., Straus S. E. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pertel P. E., Fridberg A., Parish M. L., Spear P. G. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 32. Roche S., Bressanelli S., Rey F. A., Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191 [DOI] [PubMed] [Google Scholar]
- 33. Roizman B., Knipe D. M. 2001. Herpes simplex viruses and their replication, p. 2399–2459 In Knipe D. M., Howley P. M. (ed.), Fields virology, 3rd ed., vol. 2 Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 34. Satoh T., et al. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shukla D., et al. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 36. Shukla D., Spear P. G. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simeonov K., Pollianova M., Jordanova P., Roussev R. 2007. Protein A-gold immunoelectron microscope analysis of the antigen activity of the structural components of bovine herpesvirus-4 (BHV-4) particles and surface antigen expression of cells infected with BHV-4. Bull. Vet. Inst. Pulawy 51:203–206 [Google Scholar]
- 38. Suenaga T., et al. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugimoto K., et al. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terry-Allison T., Montgomery R. I., Warner M. S., Geraghty R. J., Spear P. G. 2001. Contributions of gD receptors and glycosaminoglycan sulfation to cell fusion mediated by herpes simplex virus 1. Virus Res. 74:39–45 [DOI] [PubMed] [Google Scholar]
- 41. Terry-Allison T., et al. 1998. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J. Virol. 72:5802–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tischer B. K., von Einem J., Kaufer B., Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 43. Visalli R. J., Brandt C. R. 1991. The HSV-1 UL45 gene product is not required for growth in Vero cells. Virology 185:419–423 [DOI] [PubMed] [Google Scholar]