

Abstract
A recent crystal structure of the precleaved HDV ribozyme along with biochemical data support a mechanism for phosphodiester bond self-cleavage in which C75 acts as a general acid and bound Mg2+ ion acts as a Lewis acid. Herein this precleaved crystal structure is used as the basis for quantum mechanical/molecular mechanical calculations. These calculations indicate that the self-cleavage reaction is concerted with a phosphorane-like transition state when a divalent ion, Mg2+ or Ca2+, is bound at the catalytic site but is sequential with a phosphorane intermediate when a monovalent ion, such as Na+, is at this site. Electrostatic potential calculations suggest that the divalent metal ion at the catalytic site lowers the pKa of C75, leading to the concerted mechanism in which the proton is partially transferred to the leaving group in the phosphorane-like transition state. These observations are consistent with experimental data, including pKa measurements, reaction kinetics, and proton inventories with divalent and monovalent ions.
Keywords: QM/MM, ribozyme, proton transfer, phosphorane intermediate
RNA enzymes, or ribozymes, participate in a number of biologically critical reactions, including tRNA maturation, splicing, and translation. The hepatitis delta virus (HDV) ribozyme is widespread in biology and functions in viral processing in human cells through its phosphodiester self-cleavage.1 The HDV ribozyme undergoes a reaction in which a phosphodiester bond is formed between O2′(U-1) and P(G1) and broken between P(G1) and O5′(G1). A recent structure of the HDV ribozyme trapped in the precleaved state was solved in the presence of Mg2+ with a C at position 75, and at low pH of 5.0 to maintain C75 in the protonated state.2 In this structure, an active site Mg2+ ion was clearly resolved. The structure, along with a variety of biochemical data, suggest a plausible mechanism for this reaction: C75 acts as a general acid and transfers a proton to the 5′-bridging oxygen leaving group of G1, and a catalytic Mg2+ ion serves as a Lewis acid to stabilize the 2′-oxyanion of U-1 (Figure 1).2,3 We recently conducted several molecular dynamics (MD) studies on this reaction,4,5 leading to two main conclusions: (1) the catalytic Mg2+ ion remains localized (chelated) near the reverse G25•U20 wobble, in contrast to a metal ion that is more diffuse near the standard G1•U37 wobble, and (2) protonation of C75 keeps the active site primed for the catalytic reaction through key hydrogen-bonding interactions and binding of Mg2+. To our knowledge, this precleaved crystal structure has not yet been used to investigate the mechanism of the self-cleavage reaction with theoretical methods.
Figure 1.

Schematic illustration of the QM region chosen for the QM/MM calculations used to generate the results in this study. The atoms in red are included in the QM region, and wavy lines indicate the QM/MM boundary. The region contains 87 QM atoms. The arrows describe the self-cleavage reaction.
In this Letter, we perform quantum mechanical/molecular mechanical (QM/MM) calculations based on this precleaved crystal structure to examine the self-cleavage mechanism of the HDV ribozyme. Our objective is to determine if this reaction is sequential, potentially involving a phosphorane intermediate, or concerted and to examine the role of the catalytic Mg2+ in the mechanism. Our calculations indicate that the self-cleavage reaction in the HDV ribozyme in the presence of bound Mg2+ is concerted with a phosphorane-like transition state and that the catalytic Mg2+ strongly influences the mechanism. We find that the mechanism becomes sequential when the catalytic Mg2+ is replaced by a monovalent ion but remains concerted when the catalytic Mg2+ is replaced by another divalent ion, Ca2+. Our analysis suggests that the divalent metal ion lowers the pKa of C75, leading to a concerted mechanism in which the proton is already partially transferred to the leaving group in the phosphorane-like transition state. These results are consistent with previous experimental mechanistic studies on the HDV ribozyme.6–9
Three prior quantum mechanical calculations were performed on the HDV ribozyme but differ fundamentally from this study in that they used a starting model based on the crystal structure of the HDV ribozyme with a C75U mutation,10 albeit mutated back to C in silico. This C75U mutant structure does not appear to be reflective of the catalytic conformation of the ribozyme for multiple reasons involving mis-positioning of functionalities and metal ions, as discussed elsewhere.11 Moreover, these studies have provided conflicting results regarding a stable phosphorane intermediate. In one study, density functional theory (DFT) was used to perform geometry optimizations on two cutoff models, each containing 42 non-hydrogen atoms, with the environment modeled as a dielectric continuum aqueous solvent.12 A stable phosphorane intermediate was found in a model with C75 acting as the general acid but not in a model with C75 acting as the general base, and analysis based on the energy barriers and estimated concentrations favored the C75 general acid mechanism. In a second study, DFT geometry optimizations of similar, relatively small model systems favored the C75 general acid mechanism but with no stable intermediate.13 In a third study, QM/MM calculations of the solvated ribozyme favored the C75 general base mechanism with no stable intermediate.14 Thus, there is no consensus among these three theoretical studies on the formation of stable intermediates or whether C75 serves as the general acid or base in the mechanism when the structure derived from the C75U mutant is probed. In addition, a recent molecular dynamics study of the HDV ribozyme based on the C75U structure supports C75 as the general acid.15
In contrast to these previous studies, the QM/MM calculations in the present study are based on the precleaved crystal structure (PDB ID 3NKB) solved with wild-type sequences (i.e., with the C75 present and protonated) and with Mg2+ bound.2 The upstream nucleotide and scissile phosphate were modeled as described previously.2 Specifically, the U-1 nucleotide was not fully resolved in that structure and was positioned based on analogy to the cleavage site of the hammerhead ribozyme, in conjunction with the available experimental electron density data for the HDV ribozyme. The resulting model of the precleaved HDV ribozyme contains 73 nucleotides. Hydrogen atoms were added to the entire structure using Accelrys Discover Studio Visualizer 2.0. C41 was protonated at N3 to maintain its hydrogen-bonded base triple,16 and C75 was protonated at N3, as suggested by pKa measurements on the precleaved form.8 The 11 Mg2+ ions resolved in the crystal structure were included, and the ribozyme was solvated with rigid TIP3P waters17 in a periodically replicated orthorhombic box. The system was neutralized with Na+ ions, and physiological monovalent ionic strength was added to the solvent to give ~0.15 M NaCl.
The starting configurations for the QM/MM optimizations were obtained from molecular dynamics (MD) simulations of the solvent and Na+ and Cl− ions according to the equilibration procedure described in the Supporting Information. This MD equilibration was performed with the Desmond18,19 program using the AMBER9920,21 force field. During equilibration, the ribozyme and the Mg2+ ions were fixed at the coordinates from the crystal structure. Following the MD equilibration, the solvent-equilibrated system was truncated by deleting all solvent molecules outside a cutoff distance of 10 Å from the ribozyme. In addition, the O2′(U-1) was deprotonated to drive its nucleophilic attack on the scissile phosphate in the subsequent QM/MM calculations. Note that this study does not focus on the mechanism of deprotonation of this oxygen or the identity of the general base.
The QM/MM calculations were performed using the Qsite program.22 The QM region was treated with density functional theory (DFT) at the B3LYP/6-31G** level of theory, including the LACVP* pseudopotential for the metal ions K+, Cs+ and Ca2+, with an ultrafine grid. The hydrogen-capping method was used for the residues at the boundary of the QM and MM regions.22 The MM region was described by the OPLS2005 force field, and a 100 Å residue-based nonbonded cutoff was used. Because we are interested mainly in the changes that occur in the active site, all atoms outside a 20 Å radius sphere centered at the scissile phosphate were held fixed throughout the QM/MM calculations, although their energetic influence on all atoms (QM and MM) was maintained throughout the study. Note that conformational changes beyond this sphere will therefore not be observed in these calculations. Prior to the full QM/MM geometry optimization, an initial MM geometry optimization of the atoms within this 20 Å radius sphere was performed with the QM region fixed.
Four different QM regions were considered for this system. Each of these regions contains the following key residues: C75 nucleobase, U-1 sugar, G1 phosphate (the scissile phosphate), U23 phosphate, and the active site Mg2+ ion with its three crystallographic waters. These residues, as well as the additional residues included in each region, are depicted in Figure S1. QM/MM geometry optimizations were performed with each of these QM regions to enable comparison of key distances and angles in the active site. Table S1 provides a comparison of the important active site distances of the ribozyme after QM/MM geometry optimization using the different QM regions. Based on this comparison, the QM region chosen for the subsequent calculations contained 87 atoms and consisted of the U-1 nucleotide, G1 sugar and scissile phosphate, protonated C75 nucleobase, U23 phosphate, C22 sugar, and catalytic Mg2+ ion with its three water ligands (Figure 1).
While these types of QM/MM calculations provide useful qualitative mechanistic information, they are also associated with known limitations. In complex systems, a large number of related minima and transition states (i.e., first-order saddle points) are present on the potential energy surface. The specific reactant, transition state, and product identified with QM/MM calculations depends on the starting configuration, which corresponds to the crystal structure coordinates of the precleaved ribozyme immersed in equilibrated solvent in the present work. The resulting reaction pathway is therefore not unique. Nonetheless, the underlying assumption of these calculations is that the qualitative mechanism is representative of a set of such reaction pathways. The energetics along the reaction pathway are not quantitatively meaningful because entropic contributions are neglected, and conformational sampling is required to average over the entire set of relevant reaction pathways. Future work will utilize ab initio molecular dynamics methods to generate free energy profiles along the dominant reaction coordinates23 or an approach such as transition path sampling to obtain additional information about the mechanisms and the relative rate constants.
In the present study, QM/MM calculations, in conjunction with experimental data from the literature, provide insight into the qualitative mechanism of the self-cleavage reaction of the HDV ribozyme. The specific objectives of this study were to determine if this self-cleavage mechanism is sequential, potentially with a phosphorane intermediate, or concerted and to understand the impact of the catalytic Mg2+ ion on the mechanism. The reactant (precleaved) and product (cleaved) structures were determined by placing the transferring hydrogen from the general acid C75 near its donor or acceptor, respectively, prior to the QM/MM geometry optimization using the methods described above. The reactant and product structures, which are minima on the QM/MM potential energy surface, are shown in Figures 2a and 2c, respectively. In the reactant structure, the proton (H3) is bonded to N3, and the P-O5′ bond is formed, while in the product structure, the proton is bonded to O5′, the P-O2′ bond is formed, and the P-O5′ bond is broken. The root-mean-square deviation (RMSD) between the starting structure and the optimized reactant structure was 0.41 Å. These structural differences arise predominantly from a decrease in the N3(C75)-O5′(G1) distance (i.e., the proton donor-acceptor distance) from 3.6 Å to 3.01 Å.
Figure 2.

Cleavage site for the (a) reactant, (b) transition state, and (c) product states obtained from the QM/MM optimizations. The active site Mg2+ (purple) is coordinated to the pro-RP oxygen of the scissile phosphate and the nucleophilic O2′ of U-1. In the transition state, the transferring hydrogen is depicted as a gray ball. Catalytically relevant bond distances are given.
Starting from the QM/MM-optimized reactant and product structures, we identified a transition state (TS) using the quasi-synchronous transit (QST)24 method and confirmed that the resulting stationary point has a single imaginary frequency. The transition state obtained has a phosphorane-like structure, with the scissile phosphate penta-coordinated, the P-O2′ bond partially formed, and the P-O5′ bond partially broken (Figure 2b). To confirm that this TS corresponds to the reactant and product of interest, we calculated the mass-weighted minimum energy path (MEP) from the TS back to the reactant and product structures along the intrinsic reaction coordinate (IRC). The energy along this MEP is depicted in Figure 3a. The calculated energy barriers along the MEP are 25 kcal/mol and 15 kcal/mol in the forward and reverse directions, respectively. We emphasize that these energies cannot be used to estimate rate constants because they do not include entropic effects and only represent a single reaction pathway. The RMSDs between the optimized TS and reactant and between the optimized product and reactant are 0.15 Å and 0.19 Å, respectively, suggesting only minor conformational changes along this portion of the overall reaction pathway.
Figure 3.

Minimum energy path (MEP) for the self-cleavage reaction obtained using QM/MM methods with Mg2+ at the catalytic site. The energy (a) and relevant distances (b) are shown along the intrinsic reaction coordinate.
According to the MEP shown in Figure 3, the mechanism of self-cleavage in the HDV ribozyme is a concerted reaction with a phosphorane-like TS (i.e. there is no intermediate along this reaction pathway). The relevant reaction coordinates in this process are the P(G1)-O5′(G1) and P(G1)-O2′(U-1) distances, as well as the difference between the N3(C75)-H3(C75) and O5′(G1)-H3(C75) distances. Figure 3b depicts the values of these distances along the IRC. Although the overall self-cleavage reaction is concerted, with a single TS connecting the reactant and product, it is asynchronous and can be visualized in three stages along the IRC. Initially the O2′ attacks the scissile phosphate to bring the local geometry into a favorable conformation for the subsequent proton transfer and the bond formation/cleavage processes. This initial attack is observed in the P-O2′ distance decreasing without much change in P-O5′ or proton transfer distances (Figure 3b). Next, the proton (H3) is transferred from N3(C75) to O5′(G1), which is observed in the (N3-H3)–(O5′-H3) distance changing significantly and the P(G1)-O2′(U-1) and P(G1)-O5′(G1) distances remaining virtually unchanged. In the last stage, the P(G1)-O2′(U-1) and P(G1)-O5′(G1) bonds are completely formed and broken, respectively.
Although no stable intermediate was observed along the reaction pathway depicted in Figure 3, suggesting a concerted mechanism, we cannot rule out the possibility that a sequential pathway also exists for this system. Formation of intermediate penta-coordinate phosphorane species has been proposed in some other ribozyme catalyzed mechanisms.12 We searched for these intermediate minima by performing a series of constrained optimizations along the P-O5′ and P-O2′ coordinates. These constrained optimizations were performed for three different configurations at the proton transfer interface corresponding to the reactant state, transition state, and product state. As shown in Figure S2, the three resulting constrained potential energy surfaces do not show evidence of additional minima corresponding to intermediate structures. Thus, although an exhaustive search of the many-dimensional potential energy surface is not possible, the evidence provided by our calculations is consistent with a concerted mechanism in the presence of a bound Mg2+ ion.
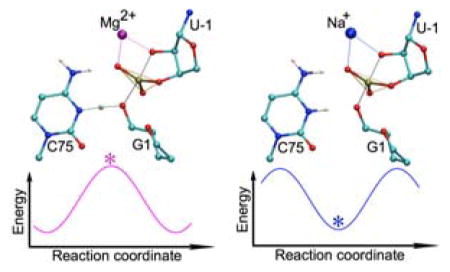
As mentioned above, the structure of the precleaved HDV ribozyme contains a catalytic Mg2+ ion interacting with one of the non-bridging (pro-RP) oxygens of the scissile phosphate. This Mg2+ ion is depicted as a purple sphere in Figure 2. We hypothesized that the absence of the phosphorane intermediate may be related to the presence of the Mg2+ ion at that position. Biochemical experiments indicate that the HDV ribozyme is active with a wide range of divalent ions, especially Ca2+,25,26 as well as monovalent ions alone at high concentrations.6,27 To explore our hypothesis, we replaced the catalytic Mg2+ ion with either a single monovalent ion or another divalent ion (Li+, Na+, K+, Cs+ or Ca2+) and investigated the possibility of a phosphorane intermediate for each metal ion. We also examined the case with no ion at the catalytic position. Our previous molecular dynamics simulations of the precleaved HDV ribozyme in the absence of the catalytic Mg2+ resolved in the crystal structure2 indicated that a single Na+ ion interacts diffusely with this catalytic site for the majority of the time but does not always remain in this region.5 These previous simulations support the replacement of the catalytic Mg2+ by a single monovalent ion and also justify exploring the case with no ion at the catalytic position.
For each of these modified systems, the starting point for the QM/MM optimization was the TS obtained for the original system, converted to the modified system by replacing the catalytic Mg2+ with another ion or by removing it. In each case, we searched for both a transition state and a minimum from this starting structure using a variety of constrained optimization procedures. The results are summarized in Table 1, with the active site structural details given in Table S2 and the relative energies of the stationary points given in Table S3. For all cases with a monovalent ion or no ion at the catalytic position, a phosphorane intermediate was obtained as a minimum on the potential energy surface. This intermediate was ~4–9 kcal/mol higher in energy than the reactant. In contrast, for both cases with a divalent ion at the catalytic position, no such intermediate was obtained (i.e., no minima other than the reactant and product states were found), but a phosphorane-like transition state was identified. The MEP with Ca2+ at the catalytic site is provided in Figure S5. Although we found a phosphorane intermediate for all cases with a monovalent ion at the catalytic position, we were unable to find a phosphorane-like transition state for these cases. In other words, for each case studied, we found either a phosphorane-like transition state or a phosphorane intermediate, but not both.
Table 1.
Results of exploring different cases of active site metal ions. Phosphorane intermediates were obtained in cases with monovalent ions at the catalytic position but not in cases with divalent ions at the same site.
| Metal ion at catalytic position | Phosphorane intermediate obtained | Phosphorane-like TS obtained |
|---|---|---|
| Mg2+ | No | Yes |
| Ca2+ | No | Yes |
| Li+ | Yes | No |
| Na+ | Yes | No |
| K+ | Yes | No |
| Cs+ | Yes | No |
| no metal ion | Yes | No |
A second active site Mg2+ ion has been suggested to interact with the standard G1•U37 wobble.28 Recent molecular dynamics studies suggest that this metal ion interacts diffusely with the standard wobble and plays a structural rather than catalytic role.4,5 In the calculations described above, only Na+ ions were interacting with this standard G•U wobble. We also performed calculations with a Mg2+ at the standard wobble, and the results were qualitatively the same as those obtained in its absence. Thus, this diffusely interacting Mg2+ ion does not appear to influence the qualitative mechanism of self-cleavage, providing further evidence that it does not play a significant catalytic role.
The structures of the phosphorane-like TS with a divalent ion at the catalytic position and the phosphorane intermediate with a monovalent ion at the catalytic position are very similar. The most prominent difference between these two structures is the position of H3(C75) (Figure S3, Table S2). In the phosphorane-like TS, this hydrogen is midway between N3(C75) and O5′(G1), and in the phosphorane intermediate, this hydrogen is still bonded to N3(C75). Quantitatively, the N3(C75)-H3(C75) and O5′(G1)-H3(C75) distances are 1.32 Å and 1.31 Å, respectively, for the phosphorane-like TS obtained with the catalytic Mg2+ and 1.06 Å and 1.86 Å, respectively, for the phosphorane intermediate with Na+ (Table S2). Note that the N3(C75)-O5′(G1) distance is ~0.3 Å smaller in the phosphorane-like TS than in the phosphorane intermediate. One plausible explanation for these structural differences is that the higher charge of the Mg2+ ion allows the divalent metal ion to withdraw more electron density from C75, thereby increasing the acidity of the proton H3(C75). Indeed, solution and crystallographic experiments reveal lowering of pKa values of C75 from 7.25 to 5.9 upon going from unbound to bound Mg2+, respectively, but maintaining of a high pKa value, near 7.5, in the presence of up to 1 M Na+.8,9,11 (Note that all of these pKa values are perturbed toward neutrality from the unshifted value of 4.2 for free cytosine, and hence favor general acid catalysis.)
The lower pKa value of C75 with bound divalent metal ions as compared to monovalent ions is supported by comparison of the electrostatic potentials generated for the reactant state with a Mg2+ ion or a Na+ ion at the catalytic position (Figure 4). This figure suggests that the electrostatic potential at C75 is more positive with the catalytic Mg2+ in the active site, resulting in a lower pKa value of C75. To determine whether this phenomenon was due to differences between the divalent and monovalent metal ions or between the optimized reactant structures, we also calculated the electrostatic potentials for each optimized structure with the catalytic Mg2+ replaced by Na+, or the bound Na+ replaced by Mg2+. These results, which are provided in Figure S4, indicate that the differences between the electrostatic potentials shown in Figure 4 are due to the presence of the divalent versus monovalent ion, rather than to structural differences.
Figure 4.

Molecular electrostatic potential mapped onto the electron density isosurface. The QM region is shown here for the reactant state with (a) a Mg2+ ion and (b) a Na+ ion at the catalytic position. In both panels, the scale is from −0.5 to 0.5 kT/e at 298 K. The dotted gray circle indicates the position of the metal ion in each case. These figures suggest that the Mg2+ ion at the catalytic site withdraws greater electron density from C75, making the residue more electron deficient and the transferring proton more acidic, as compared to the case of a Na+ ion at the catalytic site.
In conclusion, our QM/MM calculations indicate that the self-cleavage reaction of the HDV ribozyme is concerted with a phosphorane-like transition state in the presence of Mg2+. When the catalytic Mg2+ ion is replaced by a monovalent ion, the mechanism becomes sequential with a phosphorane intermediate. On the other hand, when this catalytic Mg2+ ion is replaced by another divalent ion, the mechanism remains concerted. These results are consistent with the experimental observations that the HDV ribozyme functions with similarly fast reaction kinetics with Mg2+ and Ca2+,25,26 suggesting that these reactions proceed by the same mechanism, but functions about 20-fold slower with Na+,29 suggesting that this reaction may proceed by an altered mechanism. In addition, proton inventory experiments support a stepwise reaction of the HDV ribozyme in the presence of Na+ but a concerted reaction in physiological concentrations of Mg2+.7,9 According to our calculations, the main structural difference between the phosphorane-like TS and the phosphorane intermediate is that the proton is partially transferred to the leaving group in the former but not in the latter. Our analysis suggests that the catalytic Mg2+ ion withdraws more electron density from C75 than does a monovalent metal ion bound at this site. As a result, the pKa value of C75 is lower in the presence of the divalent metal ion, leading to a concerted mechanism with a phosphorane-like transition state in which the C75 proton is partially transferred. This result is consistent with the experimental observation that the pKa value of C75 is lowered by more than 1 unit by bound Mg2+ but is not affected by high concentrations of Na+.8,9,11 Our studies herein suggest unique ways in which the HDV ribozyme uses communication between a metal ion and a nucleobase to help drive catalysis.
Supplementary Material
Acknowledgments
We gratefully acknowledge assistance from Prof. Barbara Golden, Prof. Michael Harris, Phil Hanoian, Dr. Alexander Soudackov, Chaehyuk Ko, and Dr. Victor Guallar. This project was supported by NIH grant GM56207 (S.H.-S.) and NIH grant GM095923 (P.C.B).
Footnotes
Supporting Information Available: Description of molecular dynamics equilibration procedure; analysis of QM regions considered for QM/MM calculations; potential energy surfaces obtained from constrained optimizations; comparison of structures and energetics of phosphorane-like transition states and phoshorane intermediates with divalent and monovalent ions, respectively, at catalytic site; analogous figure to Figure 3 with Ca2+ instead of Mg2+ at the catalytic site; analogous figure to Figure 4 with Mg2+ and Na+ ions exchanged. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Webb CHT, Luptak A. HDV-Like Self-Cleaving Ribozymes. RNA Biol. 2011;8 doi: 10.4161/rna.8.5.16226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JH, Yajima R, Chadalavada DM, Chase E, Bevilacqua PC, Golden BL. A 1.9 Å Crystal Structure of the HDV Ribozyme Precleavage Suggests Both Lewis Acid and General Acid Mechanisms Contribute to Phosphodiester Cleavage. Biochemistry. 2010;49:6508–6518. doi: 10.1021/bi100670p. [DOI] [PubMed] [Google Scholar]
- 3.Das S, Piccirilli J. General Acid Catalysis by the Hepatitis Delta Virus Ribozyme. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 4.Veeraraghavan N, Ganguly A, Chen JH, Bevilacqua PC, Hammes-Schiffer S, Golden BL. Metal Binding Motif in the Active Site of the HDV Ribozyme Binds Divalent and Monovalent Ions. Biochemistry. 2011;50:2672–2682. doi: 10.1021/bi2000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veeraraghavan N, Ganguly A, Golden BL, Bevilacqua PC, Hammes-Schiffer S. Mechanistic Strategies in the HDV Ribozyme: Chelated and Diffuse Metal Ion Interactions and Active Site Protonation. J Phys Chem B. 2011;115:8346–8357. doi: 10.1021/jp203202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakano S, Chadalavada DM, Bevilacqua PC. General Acid-Base Catalysis in the Mechanism of a Hepatitis Delta Virus Ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 7.Nakano S, Bevilacqua PC. Proton Inventory of the Genomic HDV Ribozyme in Mg2+-Containing Solutions. J Am Chem Soc. 2001;123:11333–11334. doi: 10.1021/ja0166850. [DOI] [PubMed] [Google Scholar]
- 8.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Direct Measurement of a pK(a) near Neutrality for the Catalytic Cytosine in the Genomic HDV Ribozyme Using Raman Crystallography. J Am Chem Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 9.Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: Solvent Isotope Effects and Proton Inventories in the Absence of Divalent Metal Ions Support C75 as the General Acid. J Am Chem Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- 10.Ke A, Zhou K, Ding F, Cate JHD, Doudna JA. A Conformational Switch Controls Hepatitis Delta Virus Ribozyme Catalysis. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 11.Nakano S, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: A Mutant of the C41 Motif Provides Insight into the Positioning and Thermodynamic Linkage of Metal Ions and Protons. Biochemistry. 2007;46:3001–3012. doi: 10.1021/bi061732s. [DOI] [PubMed] [Google Scholar]
- 12.Wei K, Liu L, Cheng YH, Fu Y, Guo QX. Theoretical Examination of Two Opposite Mechanisms Proposed for Hepatitis Delta Virus Ribozyme. J Phys Chem B. 2007;111:1514–1516. doi: 10.1021/jp070120u. [DOI] [PubMed] [Google Scholar]
- 13.Liu H, Robinet JJ, Ananvoranich S, Gauld JW. Density Functional Theory Investigation on the Mechanism of the Hepatitis Delta Virus Ribozyme. J Phys Chem B. 2007;111:439–445. doi: 10.1021/jp064292n. [DOI] [PubMed] [Google Scholar]
- 14.Banas P, Rulisek L, Hanosova V, Svozil D, Walter NG, Sponer J, Otyepka M. General Base Catalysis for Cleavage by the Active-Site Cytosine of the Hepatitis Delta Virus Ribozyme: QM/MM Calculations Establish Chemical Feasibility. J Phys Chem B. 2008;112:11177–11187. doi: 10.1021/jp802592z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TS, Giambasu GM, Harris ME, York DM. Characterization of the Structure and Dynamics of the HDV Ribozyme in Different Stages Along the Reaction Path. J Phys Chem Lett. 2011:2538–2543. doi: 10.1021/jz201106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veeraraghavan N, Bevilacqua PC, Hammes-Schiffer S. Long-Distance Communication in the HDV Ribozyme: Insights from Molecular Dynamics and Experiments. J Mol Biol. 2010;402:278–291. doi: 10.1016/j.jmb.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 18.Desmond Molecular Dynamics System. Vol. 2.2. D. E. Shaw Research; New York, NY: 2009. [Google Scholar]
- 19.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary IK, Moraes MA, Sacerdoti FD, et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. November 11–17; 2006. [Google Scholar]
- 20.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 21.Wang J, Cieplak P, Kollman PA. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J Comp Chem. 2000;21:1049–1074. [Google Scholar]
- 22.Qsite. 5.0 . Schrodinger, LLC; New York, NY: 2008. [Google Scholar]
- 23.Rosta E, Nowotny M, Yang W, Hummer G. Catalytic Mechanism of RNA Backbone Cleavage by Ribonuclease H from Quantum Mechanics/Molecular Mechanics Simulations. J Am Chem Soc. 2011;133:8934–8941. doi: 10.1021/ja200173a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlegel HB. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr J Chem. 1993;33:449–454. [Google Scholar]
- 25.Suh YA, Kumar PKR, Taira K, Nishikawa S. Self-Cleavage Activity of the Genomic HDV Ribozyme in the Presence of Various Divalent Metal Ions. Nucleic Acids Res. 1993;21:3277–3280. doi: 10.1093/nar/21.14.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano S, Cerrone A, Bevilacqua PC. Mechanistic Characterization of the HDV Genornic Ribozyme: Classifying the Catalytic and Structural Metal Ion Sites within a Multichannel Reaction Mechanism. Biochemistry. 2003;42:2982–2994. doi: 10.1021/bi026815x. [DOI] [PubMed] [Google Scholar]
- 27.Fedoruk-Wyszomirska A, Giel-Pietraszuk M, Wyszko E, Szymanski M, Ciesioka J, Barciszewska M, Barciszewski J. The Mechanism of Acidic Hydrolysis of Esters Explains the HDV Ribozyme Activity. Mol Biol Rep. 2009;36:1647–1650. doi: 10.1007/s11033-008-9364-7. [DOI] [PubMed] [Google Scholar]
- 28.Chen JH, Gong B, Bevilacqua PC, Carey P, Golden BL. A Catalytic Metal Ion Interacts with the Cleavage Site G Center Dot U Wobble in the HDV Ribozyme. Biochemistry. 2009;48:1498–1507. doi: 10.1021/bi8020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano S, Proctor DJ, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: Assessing the Catalytic and Structural Contributions of Divalent Metal Ions within a Multichannel Reaction Mechanism. Biochemistry. 2001;40:12022–12038. doi: 10.1021/bi011253n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.